

Summary
Biogenesis of mitochondrial outer membrane proteins involves their integration into the lipid bilayer. Among these proteins are those that form a single-span topology, but our understanding of their biogenesis is scarce. In this study, we found that the MIM complex is required for the membrane insertion of some single-span proteins. However, other such proteins integrate into the membrane in a MIM-independent manner. Moreover, the biogenesis of the studied proteins was dependent to a variable degree on the TOM receptors Tom20 and Tom70. We found that Atg32 C-terminal domain mediates dependency on Tom20, whereas the cytosolic domains of Atg32 and Gem1 facilitate MIM involvement. Collectively, our findings (1) enlarge the repertoire of MIM substrates to include also tail-anchored proteins, (2) provide new mechanistic insights to the functions of the MIM complex and TOM import receptors, and (3) demonstrate that the biogenesis of MOM single-span proteins shows variable dependence on import factors.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology, Functional Aspects of Cell Biology
Graphical Abstract
Highlights
-
•
The single-span proteins Atg32 and Msp1 are new substrates of the MIM complex
-
•
Different domains of Atg32 mediate dependency on either Tom20 or MIM components
-
•
The MIM complex facilitates the membrane insertion of the tail-anchored protein Gem1
-
•
The membrane integration of Mcr1 and Fis1 is mainly MIM independent
Biological Sciences; Molecular Biology; Cell Biology; Functional Aspects of Cell Biology
Introduction
Mitochondria harbor between 800 (in budding yeast) to 1,500 (in mammals) different proteins, and their outer membrane is estimated to harbor several dozens of those proteins. Such mitochondrial outer membrane (MOM) proteins include enzymes, components of protein import complexes, metabolites transporters, and factors that mediate mitochondrial fusion, fission, and motility. The biogenesis of the MOM requires targeting of newly synthesized proteins to the organelle and their integration into the lipid bilayer. An important group among MOM proteins are those that span the membrane once with a single α-helical segment. Like all other MOM proteins, these single-span proteins are nuclear encoded and synthesized on cytosolic ribosomes (Dukanovic and Rapaport, 2011, Endo and Yamano, 2009, Wiedemann and Pfanner, 2017). Therefore, they must bear appropriate signals that ensure both their correct targeting to the organelle and their membrane integration by mitochondrial import components. None of these proteins contains a canonical cleavable N-terminal presequence, and instead, they carry internal targeting and sorting signals. Despite their well-recognized importance, the diverse molecular mechanisms by which such MOM proteins are specifically targeted to the organelle and inserted into their target membrane remain incompletely defined.
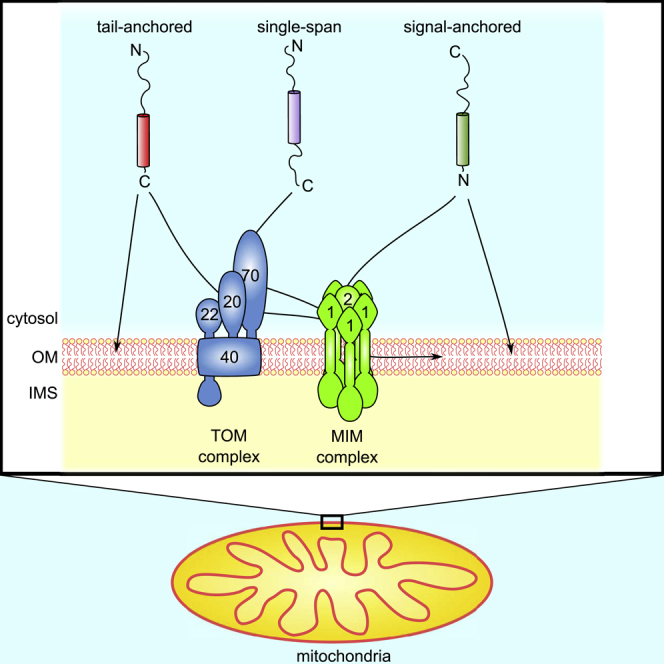
Depending on their orientation, single-span MOM proteins can be classified into three groups: the first two are signal- and tail-anchored proteins, which face the intermembrane space (IMS) with either the amino- or carboxyl-terminus, respectively. These proteins typically expose the bulk of the protein to the cytosol and only a very short segment faces the IMS. A third subclass of single-span proteins exposes soluble domains toward both the IMS and the cytosol, with the N terminus facing the cytosol.
Mitochondrial signal-anchored proteins in fungi include, according to current knowledge, the TOM receptor components, Tom20 and Tom70, and three additional proteins: OM45, Msp1, and the MOM isoform of Mcr1 (Mcr1mom). The targeting signal of these proteins consists of the transmembrane segment (TMS) and positively charged flanking regions (Dukanovic and Rapaport, 2011, Ellenrieder et al., 2015). Despite the common structural characteristics, it appears as if signal-anchored proteins do not share a common pathway of targeting and membrane integration. Mcr1mom and Msp1 follow a still uncharacterized membrane integration pathway that seems for Mcr1mom to be recapitulated with pure lipid vesicles (Meineke et al., 2008), whereas the TOM receptors use a pathway where preexisting TOM complexes play an undefined role (Ahting et al., 2005). Furthermore, the MOM proteins, Mim1 and Mim2, that form together the MIM complex promote both insertion of Tom20 and Tom70 into the MOM and their final assembly into the TOM core complex (Becker et al., 2008, Dimmer et al., 2012, Hulett et al., 2008, Ishikawa et al., 2004, Waizenegger et al., 2005). OM45, which exposes its soluble domain to the IMS, follows a unique membrane assembly pathway that involves crossing the outer membrane via the TOM complex and then insertion into the MOM from the IMS side with a possible contribution of the MIM complex (Song et al., 2014, Wenz et al., 2014).
In comparison with such detailed analysis of the insertion pathway of some signal-anchored proteins, much less is known about the mechanisms by which tail-anchored (TA) proteins are targeted to and integrated into the MOM. As for the signal-anchored proteins, it seems that these proteins do not follow a unified biogenesis pathway. On the one hand, the steady-state levels of yeast mitochondrial TA proteins such as the small TOM complex subunits, Tom5, Tom6, and Tom7, were reduced upon deletion of Mim1, Mas37/Sam37, and TOM receptors (Becker et al., 2008, Horie et al., 2002, Setoguchi et al., 2006, Stojanovski et al., 2007, Thornton et al., 2010). On the other hand, in mammalian cells import of other TA proteins like Omp25, Bak, and Bcl-XL was completely independent of the TOM subunits and of any protease accessible protein on the surface of the organelle (Horie et al., 2003, Setoguchi et al., 2006). Likewise, the biogenesis of Fis1 in yeast cells remained unchanged upon deletion of subunits of the import complexes TOM or TOB/SAM and by digestion of any protease-accessible protein on the mitochondrial surface. Furthermore, in vitro import assays demonstrated that Fis1 can insert into pure lipid vesicles in an unassisted manner (Kemper et al., 2008). We previously proposed that the specificity of such an insertion could be mediated by the low ergosterol levels of the MOM. In line with this proposal, reduction of ergosterol levels in ER membranes resulted in mislocalization of Fis1 to the ER (Krumpe et al., 2012). Although these previous results raise doubts about the necessity of import factors for the membrane integration of Fis1, they do not address the biogenesis pathway of other TA proteins like Gem1.
The biogenesis of single-span proteins exposing domains toward both sides of the membrane is even less understood. Proteins belonging to this group in yeast MOM are Mim1, Mim2, Atg32, and Tom22. The only protein from this group whose membrane integration process was studied is Tom22, which was reported to require TOM import receptors for its own import as well as the TOB complex and the MOM protein Mdm10 (Court et al., 1996, Thornton et al., 2010). However, since Tom22 is a core component of the TOM complex, its biogenesis mechanism probably reflects a specific case and does not provide a general model for other proteins from this group.
In the current study, we addressed some of the open questions regarding the biogenesis of single-span MOM proteins. We observed that the biogenesis of these proteins is variably dependent on import factors like the MIM complex or the TOM receptors. Furthermore, by constructing hybrid proteins composed of defined domains of these proteins, we could dissect the determinants that cause a variable dependency.
Results
The Membrane Integration of Signal-Anchored Proteins Variably Depends on MIM Components
To better characterize the pathways that culminate in the integration of signal-anchored proteins into the MOM, we chose two model proteins that in contrast to the previously established MIM substrates Tom20 and Tom70 are not subunits of the TOM complex. The first is the MOM isoform of Mcr1 (Mcr1mom, Figure S1A). We monitored the levels of Mcr1mom in the crude mitochondrial fraction from the deletion strains of MIM1, MIM2, or the TOM receptors TOM20 and TOM70/TOM71. As we observed before (Meineke et al., 2008), the absence of the TOM receptors did not cause a reduction in the levels of Mcr1mom. Actually, its relative levels were even enhanced in the absence of Tom20 (Figures S1B and S1C). Similarly, the relative amounts of Mcr1mom in cells lacking either Mim1 or Mim2 were higher than in control organelles (Figures S1B and S1C). We suggest that this apparent increase results from loading a fixed amount of mitochondrial proteins in each lane. Accordingly, reduction in the relative amounts of certain proteins in these altered organelles leads to an apparent increase in the relative levels of other proteins. To substantiate our results, we isolated mitochondria from either wild-type (WT) or cells lacking both Mim1 and Mim2 and monitored the levels of Mcr1mom in these organelles. The levels of Mcr1mom were unaltered in the cells lacking the MIM complex (Figures S1D and S1E). These findings demonstrate that the MOM isoform of Mcr1 can integrate into its target membrane in a process that is independent of the MIM complex or import receptors.
Next, we turned to monitor the mitochondrial steady-state levels of the other model protein, namely, the ATPase Msp1 (Figure 1A). Of note, the absence of the TOM receptors or of Mas37, a subunit of the TOB complex, which mediates membrane integration of β-barrel proteins, did not cause major alterations in the steady-state levels of Msp1, and interestingly, its relative levels were even increased in the absence of Tom20 (Figures 1B and 1C). In contrast, Msp1 levels in the mitochondria isolated from cells deleted for MIM1, MIM2, or both genes were highly reduced as compared with control samples. We further confirmed the dependency on the MIM complex by importing in vitro radiolabeled Msp1 molecules into organelles isolated from either wild-type or mim1/2Δ double deletion strain. The proper insertion of the newly synthesized Msp1 molecules into the MOM was verified by their resistance to alkaline extraction. This assay demonstrated that mitochondria isolated from the mutated cells had significantly lower capacity to integrate Msp1 into their membrane (Figures 1D and 1E).
Figure 1.

The MIM Complex Is Required for the Biogenesis of Msp1
(A) Schematic depiction of Msp1 topology.
(B) Mitochondria isolated from either the indicated deletion or their respective WT cells were analyzed by SDS-PAGE and immunodecoration with antibodies against the indicated proteins. Staining with Ponceau S is shown as a loading control.
(C) Msp1 levels were quantified and normalized to the intensities of the Ponceau S staining. The values in the corresponding WT cells were set to 100%. The bar diagram shows the average ± SD of at least three independent experiments.
(D) Radiolabeled Msp1 was imported in vitro for the indicated time periods into mitochondria isolated from either WT or mim1/2Δ cells. After import, mitochondria were subjected to alkaline extraction and the pellet was analyzed by SDS-PAGE and autoradiography.
(E) Quantification of the band corresponding to Msp1 in experiments as in (D). Import into mitochondria from WT cells after 20 min was set to 100%. The graph represents the mean values ± SD of three independent experiments. See also Figure S1.
Since it was shown that the TMS of Msp1 mediates the intracellular targeting of the protein (Wohlever et al., 2017), we wondered whether this part of Msp1 is responsible for the dependency on the MIM complex. To address this point, we constructed a fusion protein composed of the TMS (a.a. residues 1–32) of Msp1 fused N terminally to a GFP moiety (Msp1(TMS)-GFP) and introduced it into WT cells. First, we monitored the subcellular localization of this fusion protein by fluorescence microscopy and noted its complete co-localization with a mitochondrial marker protein (MTS-RFP, Figure 2A). Thus, it seems that the TMS of Msp1 is sufficient for mitochondrial targeting.
Figure 2.

The TMS of Msp1 Is Sufficient for Recognition by the MIM Complex
(A) Yeast WT cells harboring mitochondrial targeted RFP (MTS-RFP) and expressing either Msp1(TMS)-GFP or GFP alone as a control were visualized by fluorescence microscopy.
(B) Whole-cell lysate (WCL) and fractions corresponding to cytosol (cyt), microsomes (ER), and mitochondria (mito) from the indicated strains expressing Msp1(TMS)-GFP were analyzed by SDS-PAGE and immunodecoration. The fusion protein was detected by an anti-GFP antibody, whereas the endogenous Msp1 protein by anti-Msp1 antibody. Tom20 and Tom70 are known MIM substrate proteins. Hexokinase (Hxk1) is a cytosolic marker, whereas Erv2 is an ER protein. Ponceau S staining of the membrane is shown as a loading control.
Next, we transformed this construct into cells deleted for the single MIM components or for both genes and obtained from the transformed cells fractions corresponding to cytosol, ER, and mitochondria. Analysis of these fractions by SDS-PAGE followed by immunodetection revealed that the deletion of the MIM components caused a reduction in the levels of the fusion protein in the mitochondrial fraction of the mutated cells concomitantly with appearance of Msp1(TMS)-GFP in the ER fraction (Figure 2B). Of note, the absence of a functional MIM complex resulted in the mislocalization of the known MIM substrates Tom20 and Tom70 to the ER, whereas the β-barrel MOM protein Tom40 or the inner membrane protein Pic2 was detected only in the mitochondrial fraction (Figure 2B). These findings indicate that Msp1 is a new substrate of the MIM complex and its TMS is sufficient for this reliance. Moreover, the MIM complex seems to be required for the correct mitochondrial location of the protein.
Atg32 Is a New Substrate of the MIM Complex
Atg32 is a receptor for mitophagy factors that exposes domains to both sides of the MOM (Figure 3A). To study the requirements for its membrane integration, we employed a functional version where an HA-tag was placed within the cytosolic domain (Okamoto et al., 2009). We introduced this construct into WT and various mutated strains and monitored its steady-state levels in the crude mitochondrial fractions of these cells. We noticed that the levels of Atg32 were dramatically compromised upon the deletion of Mim1, Mim2, and Tom20 and moderately reduced by the absence of Tom70 (Figures 3B and 3C). Atg32 has a similar topology to the TOM component Tom22, which was reported to require the TOB/SAM complex for its proper biogenesis (Thornton et al., 2010). To test whether Atg32 shares such TOB dependency, we monitored its steady-state levels in a strain lacking Mas37, a non-essential component of the TOB complex. As reported, the levels of Tom22 were highly reduced in crude mitochondria isolated from this strain, but the amounts of Atg32 were unaffected, even when the cells were grown at 37°C (Figure 3D). Thus, we concluded that Atg32 does not require the TOB complex for proper membrane integration.
Figure 3.

Atg32 Biogenesis Requires the MIM Complex and the TOM Receptors
(A) Schematic representation of the topology of HA-Atg32.
(B) Crude mitochondrial fractions were obtained from mim1Δ, mim2Δ, tom20Δ, tom70/71Δ, and their respective WT strains containing a plasmid encoding HA-Atg32. Samples were analyzed by SDS-PAGE and immunodecoration with anti-HA and the other indicated antibodies.
(C) Quantification of the relative amounts of HA-Atg32 from at least three independent experiments as in (B). The levels were normalized to the intensities of the Ponceau S staining and presented as percentage of the levels in the corresponding WT samples. The graph represents average ± SD.
(D) Crude mitochondrial fractions were obtained from mas37Δ and its respective WT strains after growth at either 24°C or 37°C. Both cell types contained a plasmid encoding HA-Atg32. Samples were analyzed by SDS-PAGE and immunodecoration with anti-HA and the other indicated antibodies.
(E) WT, mim1Δ, and mim2Δ cells expressing HA-Atg32 were grown to logarithmic phase, treated at time = 0 with cycloheximide (CHX), and then collected after the indicated time intervals. Crude mitochondria were isolated and analyzed by SDS-PAGE and immunodecoration with antibodies against the HA-tag and the indicated proteins.
(F and G) Quantifications of the levels of HA-Atg32 (F) and Fis1 (G) from three independent experiments as in (E) using Tom40 as loading control. The values at time = 0 in each strain were set to 100%. Error bars correspond to ± SD.
See also Figures S2 and S4.
Since the steady-state levels of proteins reflect the combined outcome of biogenesis and degradation, we wondered whether the absence of the Mim proteins affects also the life span of Atg32. To this end, we add to WT or mim single deletion strains cycloheximide (CHX), which is known to stop new synthesis of proteins, and monitored the levels of Atg32 after different time intervals. The results of this assay revealed that the absence of either Mim1 or Mim2 shortened significantly the life span of Atg32, whereas it did not affect the stability of the control TA protein Fis1 (Figures 3E–3G). We propose that the absence of the MIM complex slows down the integration of newly synthesized Atg32 molecules into the MOM. This interference in the import process causes then accumulation of the non-imported molecules in the cytosol and subsequently their elimination. Taken together, these findings establish Atg32 as a novel substrate of the MIM pathway.
Since Atg32 showed a dependency on both the MIM complex and Tom70/71, we wondered whether these factors are involved in the same import pathway or in two parallel ones. To address this question, we overexpressed Mim1 in cells lacking both Tom70 and its paralog Tom71. We observed that these elevated amounts of Mim1 could not enhance the reduced levels of Atg32 in tom70/71Δ cells (Figure S2). These results might suggest that Tom70 is involved in the same pathway as the MIM complex, as already shown for the insertion of multi-span proteins (Becker et al., 2011, Papic et al., 2011), and reduced recognition by Tom70 cannot be compensated by more efficient MIM-mediated membrane insertion. However, this option should be taken with caution since it is not clear whether, in the absence of elevated levels of Mim2, the excess amounts of Mim1 molecules assemble into functional complexes.
The MIM Complex Contributes to the Membrane Integration of the Tail-Anchored Protein Gem1
Previous results suggested that Fis1 can assemble into the MOM in an unassisted manner (Kemper et al., 2008). We next aimed to test its dependency on the MIM complex and to compare its behavior with another TA protein, Gem1 (Figure 4A). To that goal, we monitored the steady-state levels of Fis1 and an N-terminally HA-tagged version of Gem1 in crude mitochondrial fractions of various mutated cells. Of note, the HA-tagged version of Gem1 is functional as we confirmed its ability to complement the growth defect resulting from the absence of chromosomally encoded Gem1 (Figure S3A). The relative levels of Fis1 were actually higher in the MIM mutant cells or unaffected in cells lacking the TOM receptors, whereas those of Gem1 were moderately reduced in cells lacking Mim1 or Mim2 and even more hampered in tom20Δ cells (Figures 4B and 4C). To verify that the difference in behavior of both proteins does not result from the presence of the N terminal HA-tag on one of them, we monitored the behavior of HA-tagged Fis1 and noticed that, like the native protein, it showed independency of the examined import factors (Figures S3B and S3C).
Figure 4.

The Biogenesis of Gem1 Depends on the MIM Complex and on Tom20
(A) Schematic representation of the topologies of HA-Gem1 and Fis1.
(B) Crude mitochondria were isolated from the indicated deletion and their respective WT strains containing a plasmid encoding HA-Gem1. Samples were analyzed by SDS-PAGE and immunodecoration with anti-HA and the indicated antibodies.
(C) Quantification of the relative amounts of HA-Gem1 and Fis1. The intensity of the bands from at least three independent experiments as the one presented in (B) were quantified and normalized to the Ponceau S signal. The levels in the corresponding WT cells were set to 100%. Error bars represent ±SD.
(D) Whole-cell lysate (WCL) and fractions corresponding to cytosol (cyt), microsomes (ER), and mitochondria (mito) from the indicated cells were analyzed by SDS-PAGE and immunodecoration. Tom20 and Tom70 are known MIM substrate proteins. Tom40 is a mitochondrial β-barrel protein, Hexokinase (Hxk1) is a cytosolic marker, whereas Erv2 is an ER protein. Ponceau S staining is shown as a loading control.
(E) WT, mim1Δ, and mim2Δ cells expressing HA-Gem1 were grown to logarithmic phase, treated at time = 0 with CHX, and then collected after the indicated time intervals. Crude mitochondria were isolated and then analyzed by SDS-PAGE and immunodecoration with anti-HA and the indicated antibodies.
(F) Quantifications of the levels of HA-Gem1 from three independent experiments as in (E) using Tom40 as loading control. The values at time = 0 in each strain were set to 100%. Error bars correspond to ±SD.
See also Figures S3 and S4.
Despite the higher relative levels of Fis1 in the crude mitochondrial fractions of cells deleted for MIM components (Figures 4B and 4C), when we performed subcellular fractionations of these cells, we actually observed a minor population of the Fis1 molecules in the ER (Figure 4D). Of note, this mistargeting involves only a tiny fraction of the Fis1 molecules and is in clear contrast to the massive ER mislocalization of the canonical MIM substrate Tom70 in these mutated cells (Figure 4D). Taken together, these results suggest that Gem1 does not absolutely depend on the MIM pathway for correct biogenesis but might use it when it is available. In contrast, the membrane integration of Fis1 into the MOM seems to be basically MIM independent, although the Mim proteins might slightly contribute to the fidelity of the correct targeting.
Next, we examined the life span of Gem1 in the mim single deletion strains by addition of CHX. In agreement with the aforementioned experiments, the initial levels of Gem1 in the mutated cells were lower than those in the control cells. However, the turnover of the protein was similar in all tested cells (Figures 4E and 4F). Thus, it seems that the absence of a functional MIM complex has no effect on the stability of the TA proteins Fis1 and Gem1 (Figures 3G and 4F).
pATOM36 Can Replace the MIM Complex in Mediating the Import of MOM Proteins
We previously found that the Trypanosoma brucei protein pATOM36 can functionally compensate the absence of the MIM complex (Vitali et al., 2018). Hence, we next aimed to test whether this protein can also replace the MIM function in facilitating the membrane integration of the two new substrates that we identified, Atg32 and Gem1. To that end, we compared the steady-state levels of Atg32 and Gem1 in the crude mitochondrial fraction of WT and single mim deletion strains that were transformed with an empty vector with those transformed with a vector encoding pATOM36. The results depicted in Figure S4 illustrate that pATOM36 can indeed reverse the reduction in the steady-state levels of these MIM substrates. These results further support the identification of pATOM36 as the functional equivalent of the MIM complex.
Different Domains of Atg32 Mediate Dependency on either Tom20 or MIM Components
Our results so far indicate that different single-span proteins depend to a variable extent on the MIM complex. To better understand the reason for this variability, we constructed a C-terminally truncated version of Atg32 (HA-Atg32(ΔC)) and two fusion proteins that are composed of the tail-anchor segment of Fis1 fused to the cytosolic domain of either Atg32 or Gem1 [HA-Atg32(cyt)-Fis1(TMS) or HA-Gem1(cyt)-Fis1(TMS), respectively] (Figure 5A). Of note, the latter construct has probably native-like topology as it can rescue the growth phenotype of cells lacking native Gem1 (Figure S3A). Next, we monitored the steady-state levels of these proteins in the crude mitochondrial fraction of control cells or cells deleted for selected import components. We observed that removal of the C-terminal domain of Atg32 eliminated the dependency of the protein on Tom20, whereas the reduced levels in the absence of MIM components or Tom70 remained (compare Figures 5B and 5C with Figures 3B and 3C). These findings suggest that the dependency on Tom20 is mediated by the IMS part of Atg32, whereas that on the MIM complex by the other parts of the protein.
Figure 5.

The Cytosolic Domain of Atg32 and Gem1 Dictates the Dependency on the MIM Complex
(A) Schematic representation of the topology of HA-Atg32(ΔC), HA-Atg32(cyt)-Fis1(TMS), and HA-Gem1(cyt)-Fis1(TMS).
(B, D, and F) Crude mitochondria were extracted from the indicated strains containing a plasmid expressing HA-Atg32(ΔC) (B), HA-Atg32(cyt)-Fis1(TMS) (D), or HA-Gem1(cyt)-Fis1(TMS) (F). Samples were analyzed by SDS-PAGE and immunodecoration with anti-HA and the indicated antibodies.
(C, E, and G) The intensities of the bands corresponding to the HA signal from at least three independent experiments as in (B), (D), or (F) were quantified and normalized with the signal of the Ponceau S staining. The relative intensity, as compared with the corresponding WT strain, of HA-Atg32(ΔC) (C), HA-Atg32(cyt)-Fis1(TMS) (E), and HA-Gem1(cyt)-Fis1(TMS) (G) are depicted. Error bars represent ±SD.
To narrow down these parts, we analyzed the fusion protein that is composed of the TMS of Fis1 and the cytosolic domain of Atg32. Remarkably, this construct presented similar dependency on Mim1/2 and Tom70 as the truncated version of Atg32 (Figures 5D and 5E). As the membrane integration of the TMS segment of Fis1 appears to be independent of the MIM complex, these findings suggest the surprising possibility that actually the cytosolic domain of Atg32 is responsible for the dependency on these proteins.
To better understand which parts of the single-span proteins facilitate reliance on the import factors, we examined also the construct where the Fis1 TMS was fused to the cytosolic domain of Gem1. Notably, this construct behaved similar to Gem1 and did not resemble the MIM independency of Fis1 (compare Figures 5F and 5G with Figures 4B and 4C). Thus, these results also support the assumption that the cytosolic domain of some single-span proteins dictate largely the insertion pathway that these proteins follow.
Since we suspected that the IMS domain of Atg32 mediates Tom20-dependency, we wished to study this possibility in more detail. To that aim, we constructed a fusion protein where the IMS domain of Atg32 is fused C terminally to Fis1 (HA-Fis1-Atg32(IMSD)), Figure 6A. Analysis of the steady-state levels of this fusion protein indicated that, like Fis1, it was Tom70- and MIM-independent, but similarly to Atg32 it showed dependency on Tom20 (Figures 6B and 6C). Hence, these findings indicate that the IMS domain of Atg32 mediates the requirement for Tom20.
Figure 6.

Tom20 Is Required for the Biogenesis of Hybrid Proteins Containing the IMS Domain of Atg32
(A) Schematic representation of the topology of HA-Fis1-Atg32(IMSD) and HA-Fis1(cyt)-Atg32(TMS + IMSD).
(B and D) Crude mitochondrial fractions isolated from the indicated strains containing a plasmid expressing either HA-Fis1-Atg32(IMSD) (B) or HA-Fis1(cyt)-Atg32(TMS + IMSD) (D) were analyzed by SDS-PAGE and immunodecoration with anti-HA and the indicated antibodies.
(C and E) The intensity of the bands corresponding to HA-Fis1-Atg32(IMSD) (C) or HA-Fis1(cyt)-Atg32(TMS + IMSD) (E) from at least three independent experiments as in (B) or (D), respectively, were quantified and normalized to the intensity of the Ponceau S. The levels are presented as percentage of the corresponding WT samples and error bars represent ±SD.
To further substantiate these conclusions, we fused the cytosolic domain of Fis1 to the TMS and IMS domains of Atg32 (HA-Fis1(cyt)-Atg32(TMS + IMSD), Figure 6A). Importantly, this construct showed strong dependency on Tom20 and only moderate requirement for Mim1. No significant dependency on Mim2 or Tom70 was observed (Figures 6D and 6E). Collectively, these results support the findings that the C-terminal domain of Atg32 mediates dependency on Tom20, whereas the cytosolic domains of Atg32, and to a lesser extent that of Gem1, have a major contribution to the requirement of the Mim proteins.
Discussion
The biogenesis of newly synthesized MOM proteins involves their synthesis on cytosolic ribosomes, possible association with cytosolic factors to maintain them in import competent conformation, targeting to the surface of the organelle, and finally integration into the membrane. In the current study, we investigated which proteins on the surface of mitochondria are involved in the targeting and membrane assembly of single-span proteins. Remarkably, we observed that, although these proteins share some structural features, each one of them depends to a different extent on the presence of import receptors and an insertase complex.
Some proteins like the MOM isoform of Mcr1 or Fis1 hardly, if at all, require the assistance of import factors. In contrast, the signal-anchored proteins Msp1 and the single-span protein Atg32 rely heavily on the presence of the MIM subunits and the latter protein depends also on Tom20 and Tom70. Yet, other proteins like Gem1 are only moderately dependent on the presence of the MIM complex and appear to require Tom20. Thus, it seems that yet undefined determinants of the substrate proteins govern their distinct requirements for support. Such determinants can be length and/or average hydrophobicity of the TMS, net charge of its flanking regions, the tendency of the TMS to form helical structure, the presence of a soluble domain in the IMS, or various parameters of the cytosolic domain. Inspection of the hydrophobicity and the flanking charges did not reveal a clear correlation between these parameters and the behavior of the proteins (Figure 7). Most likely, rather than a single parameter a combination of several of the aforementioned determinants dictate the behavior of a certain protein.
Figure 7.

The Biogenesis of the Tested Proteins Depends on Their Domain Composition
Left: Schematic representation of the analyzed proteins. The topology of the proteins is depicted (cyt, cytosolic domain; TMS, transmembrane segment; IMS, intermembrane space domain). In addition, the grand average of hydropathicity (GRAVY) value of the corresponding TMS, the length of the soluble domains, and the charges of the IMS segment are indicated.
Right: The average values of the steady-state levels measured for each protein in the indicated deletion strains is depicted with a color-coded system, which is explained at the bottom.
A link that we could identify is a Tom20 requirement that is mediated by the IMS domain of Atg32. All fusion proteins harboring this domain showed dependency on Tom20, and Atg32 variant lacking it lost this requirement. Tom20 can recognize directly this domain and/or the IMS domain is associated in the cytosol with a chaperone and Tom20 serves as the receptor for this chaperone-substrate complex. Regardless of the correct possibility, the biogenesis pathway of Atg32 and Gem1 provides a unique example where targeting to the organelle is facilitated by Tom20, whereas the membrane integration is mediated by the MIM complex. So far, known substrates of the MIM pathway, like multi-span helical proteins, were reported to interact with Tom70 prior to their engagement by the MIM complex (Becker et al., 2011, Papic et al., 2011); however, Atg32 is only moderately dependent on Tom70. Hence, this study indicates a novel cooperation between Tom20 and the MIM complex.
Another surprising finding of our study is the contribution of the cytosolic domains of the substrate proteins Atg32 and Gem1 to the dependency on the MIM complex. Since these domains remain in the cytosol and do not have to cross the MOM, their effect might be via their influence on the availability of the TMS for recognition and insertion by the MIM complex. Alternatively, the MIM complex may help in obtaining the correct conformation of the cytosolic domain by interacting with hydrophobic segments, which in the final conformation will be buried within this domain. A third possibility is that, as discussed above for the involvement of Tom20, the cytosolic domain of Atg32 or Gem1 is stabilized by cytosolic factor(s) and the MIM complex functions as a receptor for such factors. Actually, a study by Vögtle et al. support the latter option as it suggested that, at least for the multi-span protein Ugo1, the MIM complex functions more as a receptor than as an insertase (Vogtle et al., 2015).
The results of the current study substantiate that the membrane integration of the MOM proteins Fis1 and Mcr1 is basically MIM independent. Of note, although the MIM complex does not appear to affect the mitochondrial levels of Fis1, it seems that in its absence more Fis1 molecules are mis-targeted to the ER. One explanation for this observation is a potential involvement of the MIM complex in a quality control process that re-targets mitochondrial proteins, which were wrongly associated with the ER, to the mitochondria. It might be that the MIM complex is not required for the direct insertion of newly synthesized Fis1 molecules from the cytosol, but, due to unclear reason, it does facilitate the re-targeting of those molecules that were initially associated with the ER. Such an ER surface retrieval pathway was described recently for several mitochondrial proteins (Hansen et al., 2018).
Our findings also establish Atg32, Msp1, and Gem1 as novel substrates of the MIM pathway. However, also for these clear substrates, MIM deletion reduces the extent of membrane integration but still allows a considerable portion of these newly synthesized proteins to associate with the MOM. Thus, it is quite obvious that an alternative membrane insertion pathway must exist. Such an alternative route might involve a yet unknown protein factor or the lipid core of the membrane as it was reported that the optimal integration of helical proteins into the MOM requires low ergosterol content and the presence of cardiolipin and phosphatidic acid (Kemper et al., 2008, Sauerwald et al., 2015, Vogtle et al., 2015). A similar multi pathway scenario is also discussed for the membrane integration of TA proteins into the ER membrane where at least four, partially overlapping, potential pathways are postulated (Borgese et al., 2019).
Collectively, our results could be explained by a variable dependency on the MIM complex that is dictated by yet undefined combination of determinants. At one edge of the spectrum are proteins like Fis1 and Mcr1 that can be inserted into the MOM in an unassisted manner. On the other side are those proteins like Msp1 and Atg32 that heavily rely on the help of the MIM insertase, and in between these poles are substrates like Gem1 that are not absolutely dependent on the MIM machinery but probably can utilize it when it is present. Moreover, the import of single-span MOM proteins is further influenced by cytosolic factors and the lipid composition of the membrane. This collection of, partially unknown, parameters makes any attempt to predict which pathway a certain protein will follow a very demanding task.
Limitations of the Study
This study used Baker's yeast as a model organism. Higher eukaryotes do not have a MIM complex but rather, yet unidentified, equivalent factors. Hence, is unclear whether the various import requirements that were identified for the different utilized substrate proteins are valid also in higher eukaryotes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank E. Kracker for excellent technical assistance, K.S. Dimmer for helpful discussions, K. Okamoto for constructs and antibodies, and T. Endo for antibodies. This work was supported by the Deutsche Forschungsgemeinschaft (RA 1028/7-2 and 10-1 to D.R.) and the ITN TAMPting to D.G.V., B.C., and D.R. (funded by the People Progamme [Marie Curie Actions] of the European Union’s Seventh Framework Programme [FP7/2007–2013/] under the Research Executive Agency [grant number 607072]). L.D. is supported by a long-term fellowship from the Minerva Foundation.
Author Contributions
D.G.V., L.D., and B.A.C. designed and conducted experiments. A.K. conducted experiments. D.R. designed experiments and analyzed data. D.G.V. and D.R. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: January 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.100779.
Supplemental Information
References
- Ahting U., Waizenegger T., Neupert W., Rapaport D. Signal-anchored proteins follow a unique insertion pathway into the outer membrane of mitochondria. J. Biol. Chem. 2005;280:48–53. doi: 10.1074/jbc.M410905200. [DOI] [PubMed] [Google Scholar]
- Becker T., Pfannschmidt S., Guiard B., Stojanovski D., Milenkovic D., Kutik S., Pfanner N., Meisinger C., Wiedemann N. Biogenesis of the mitochondrial TOM complex: Mim1 promotes insertion and assembly of signal-anchored receptors. J. Biol. Chem. 2008;283:120–127. doi: 10.1074/jbc.M706997200. [DOI] [PubMed] [Google Scholar]
- Becker T., Wenz L.S., Kruger V., Lehmann W., Muller J.M., Goroncy L., Zufall N., Lithgow T., Guiard B., Chacinska A. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 2011;194:387–395. doi: 10.1083/jcb.201102044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgese N., Coy-Vergara J., Colombo S.F., Schwappach B. The ways of tails: the GET pathway and more. Protein J. 2019;38:289–305. doi: 10.1007/s10930-019-09845-4. [DOI] [PubMed] [Google Scholar]
- Court D.A., Nargang F.E., Steiner H., Hodges R.S., Neupert W., Lill R. Role of the intermembrane space domain of the preprotein receptor Tom22 in protein import into mitochondria. Mol. Cell. Biol. 1996;16:4035–4042. doi: 10.1128/mcb.16.8.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer K.S., Papic D., Schumann B., Sperl D., Krumpe K., Walther D.M., Rapaport D. A crucial role for Mim2 in the biogenesis of mitochondrial outer membrane proteins. J. Cell Sci. 2012;125:3464–3473. doi: 10.1242/jcs.103804. [DOI] [PubMed] [Google Scholar]
- Dukanovic J., Rapaport D. Multiple pathways in the integration of proteins into the mitochondrial outer membrane. Biochim. Biophys. Acta. 2011;1808:971–980. doi: 10.1016/j.bbamem.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Ellenrieder L., Martensson C.U., Becker T. Biogenesis of mitochondrial outer membrane proteins, problems and diseases. Biol. Chem. 2015;396:1199–1213. doi: 10.1515/hsz-2015-0170. [DOI] [PubMed] [Google Scholar]
- Endo T., Yamano K. Multiple pathways for mitochondrial protein traffic. Biol. Chem. 2009;390:723–730. doi: 10.1515/BC.2009.087. [DOI] [PubMed] [Google Scholar]
- Hansen K.G., Aviram N., Laborenz J., Bibi C., Meyer M., Spang A., Schuldiner M., Herrmann J.M. An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science. 2018;361:1118–1122. doi: 10.1126/science.aar8174. [DOI] [PubMed] [Google Scholar]
- Horie C., Suzuki H., Sakaguchi M., Mihara K. Characterization of signal that directs C-tail-anchored proteins to mammalian mitochondrial outer membrane. Mol. Biol. Cell. 2002;13:1615–1625. doi: 10.1091/mbc.01-12-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie C., Suzuki H., Sakaguchi M., Mihara K. Targeting and assembly of mitochondrial tail-anchored protein Tom5 to the TOM complex depend on a signal distinct from that of tail-anchored proteins dispersed in the membrane. J. Biol. Chem. 2003;278:41462–41471. doi: 10.1074/jbc.M307047200. [DOI] [PubMed] [Google Scholar]
- Hulett J.M., Lueder F., Chan N.C., Perry A.J., Wolynec P., Likic V.A., Gooley P.R., Lithgow T. The transmembrane segment of Tom20 is recognized by Mim1 for docking to the mitochondrial TOM complex. J. Mol. Biol. 2008;376:694–704. doi: 10.1016/j.jmb.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Ishikawa D., Yamamoto H., Tamura Y., Moritoh K., Endo T. Two novel proteins in the mitochondrial outer membrane mediate β-barrel protein assembly. J. Cell Biol. 2004;166:621–627. doi: 10.1083/jcb.200405138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C., Habib S.J., Engl G., Heckmeyer P., Dimmer K.S., Rapaport D. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J. Cell Sci. 2008;121:1990–1998. doi: 10.1242/jcs.024034. [DOI] [PubMed] [Google Scholar]
- Krumpe K., Frumkin I., Herzig Y., Rimon N., Özbalci C., Brügger B., Rapaport D., Schuldiner M. Ergosterol content specifies targeting of tail-anchored proteins to mitochondrial outer membranes. Mol. Biol. Cell. 2012;23:3927–3935. doi: 10.1091/mbc.E11-12-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meineke B., Engl G., Kemper C., Vasiljev-Neumeyer A., Paulitschke H., Rapaport D. The outer membrane form of the mitochondrial protein Mcr1 follows a TOM-independent membrane insertion pathway. FEBS Lett. 2008;582:855–860. doi: 10.1016/j.febslet.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Kondo-Okamoto N., Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Papic D., Krumpe K., Dukanovic J., Dimmer K.S., Rapaport D. Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1-dependent import pathway. J. Cell Biol. 2011;194:397–405. doi: 10.1083/jcb.201102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerwald J., Jores T., Eisenberg-Bord M., Chuartzman S.G., Schuldiner M., Rapaport D. Genome-wide screens in Saccharomyces cerevisiae highlight a role for cardiolipin in biogenesis of mitochondrial outer membrane multispan proteins. Mol. Cell. Biol. 2015;35:3200–3211. doi: 10.1128/MCB.00107-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi K., Otera H., Mihara K. Cytosolic factor- and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. EMBO J. 2006;25:5635–5647. doi: 10.1038/sj.emboj.7601438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Tamura Y., Yoshihisa T., Endo T. A novel import route for an N-anchor mitochondrial outer membrane protein aided by the TIM23 complex. EMBO Rep. 2014;15:670–677. doi: 10.1002/embr.201338142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D., Guiard B., Kozjak-Pavlovic V., Pfanner N., Meisinger C. Alternative function for the mitochondrial SAM complex in biogenesis of α-helical TOM proteins. J. Cell Biol. 2007;179:881–893. doi: 10.1083/jcb.200706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton N., Stroud D.A., Milenkovic D., Guiard B., Pfanner N., Becker T. Two modular forms of the mitochondrial sorting and assembly machinery are involved in biogenesis of α-helical outer membrane proteins. J. Mol. Biol. 2010;396:540–549. doi: 10.1016/j.jmb.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Vitali D.G., Kaser S., Kolb A., Dimmer K.S., Schneider A., Rapaport D. Independent evolution of functionally exchangeable mitochondrial outer membrane import complexes. Elife. 2018;7:e34488. doi: 10.7554/eLife.34488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtle F.N., Keller M., Taskin A.A., Horvath S.E., Guan X.L., Prinz C., Opalinska M., Zorzin C., van der Laan M., Wenk M.R. The fusogenic lipid phosphatidic acid promotes the biogenesis of mitochondrial outer membrane protein Ugo1. J. Cell Biol. 2015;210:951–960. doi: 10.1083/jcb.201506085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger T., Schmitt S., Zivkovic J., Neupert W., Rapaport D. Mim1, a protein required for the assembly of the TOM complex of mitochondria. EMBO Rep. 2005;6:57–62. doi: 10.1038/sj.embor.7400318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz L.S., Opalinski L., Schuler M.H., Ellenrieder L., Ieva R., Bottinger L., Qiu J., van der Laan M., Wiedemann N., Guiard B. The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. EMBO Rep. 2014;15:678–685. doi: 10.1002/embr.201338144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N., Pfanner N. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017;86:685–714. doi: 10.1146/annurev-biochem-060815-014352. [DOI] [PubMed] [Google Scholar]
- Wohlever M.L., Mateja A., McGilvray P.T., Day K.J., Keenan R.J. Msp1 is a membrane protein dislocase for tail-anchored proteins. Mol. Cell. 2017;67:194–202. doi: 10.1016/j.molcel.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.