

Abstract
Rosiglitazone is a synthetic ligand of peroxisome proliferator-activated receptor γ (PPARγ), and it can induce apoptosis and autophagy in a variety of cancer cells. In the present study, we aimed to investigate the influence of rosiglitazone on the proliferation and apoptosis of the 5637 and T24 human bladder cancer cell lines. The results demonstrated that the level of growth inhibition rate was gradually increased by treating the 5637 and T24 cells with higher doses of rosiglitazone and longer incubation time. Rosiglitazone exerted a potent inhibiting effect on migration of the 5637 and T24 cell lines. Moreover, rosiglitazone exerted a antineoplastic activity by inducing apoptosis and cell cycle arrest. Furthermore, treatment with rosiglitazone led to decrease the anti-apoptotic protein Bcl-2 level and increase the pro-apoptotic protein caspase 3 level in 5637 and T24 cells. Importantly, the protein expression of PPAR γ was significantly increased in the present of rosiglitazone in 5637 and T24 cells as compared to control group. In conclusion, the present study demonstrates that rosiglitazone has a potential antineoplastic activity in human bladder cancer cell lines, and the underlying mechanism was mediated, at least partially, through regulation of apoptosis-related protein and PPAR γ expression.
Keywords: Rosiglitazone, bladder cancer, peroxisome proliferator-activated receptor γ (PPAR γ)
Introduction
Bladder cancer is one of the most commonly diagnosed malignancy and is a major cause of morbidity and mortality in the worldwide [1]. In United States, more than 74,000 newly diagnosed cases and 16,000 deaths are confirmed in 2015 [2]. In China, with an expected 80,500 newly diagnosed cases and 32,900 deaths are predicted in 2015 [3]. In clinical treatment, chemotherapy is still the standard of care, however, patient outcomes is not effective or is poorly tolerated [4]. In fact, there have been no major advances for the treatment of bladder cancer in the last few decades [4,5].
Rosiglitazone is known to activate peroxisome proliferator activated receptor γ (PPAR γ) and is used in the treatment of type II diabetes [6]. PPAR γ is a ligand-activated transcription factor that regulates growth and differentiation within normal and cancer cell [7]. A growing number of studies have demonstrated that rosiglitazone has been used to suppress cancer cell growth and induce cell apoptosis, including renal cancer cell [8], adrenocortical cancer cell [7,9], non small cell lung cancer cell [10] and colorectal cancer cell [11,12]. These results suggest that the activation of PPAR γ is a potential cancer therapeutic method in various cancers. In contrast to that rosiglitazone promotes bladder cancer cell migration and invasion through the activation of PPAR γ [13]. Endogenous PPAR γ ligands include unsaturated fatty acids and several prostanoids [14]. Synthetic ligands comprise the insulin-sensitizing thiazolindinedione (TZD) class (troglitazone, pioglitazone and rosiglitazone) [12,15]. Although extrinsic PPAR γ ligands mediating cancer cell apoptosis have been studied extensively, the detailed mechanisms of rosiglitazone in human bladder cancer 5637 and T24 cell apoptosis remains to be clarified.
In the present study, we investigated the effect of the PPAR γ ligand, rosiglitazone, on the proliferation and apoptosis of the 5637 and T24 human bladder cancer cell lines, and the underlying molecular mechanisms were involved.
Materials and methods
Cell culture
5637 and T24 cell lines were obtained from the Cell Resource Center, Shanghai Institutes for Biological Sciences (SIBS, China) and were maintained in RPMI-1640 (Invitrogen, USA) supplemented with 10% FBS (Invitrogen, USA) at 37°C in a humidified incubator (Thermo, USA), 5% CO2, 95% air atmosphere.
Cell viability detection by MTT
5637 and T24 cells proliferation were monitored by a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) Cell Proliferation/Viability Assay kit (R&D SYSTEMS) in according to the guidelines. The concentration of original rosiglitazone was 10 mM, which was diluted 200, 500, 1000, 2000 and 5000 times with final concentrations of rosiglitazone of 50 μM, 20 μM, 10 μM, 5 μM and 2 μM, respectively.
Transwell Migration assay
The cells were pretreated, trypsinized, and resuspended in 5% FBS medium to achieve a density of 1 × 105 cells/mL. Transwell inserts (8-mm pore size, Corning, Corning, NY) were placed in wells containing media with 10% FBS. A volume of 200 mL of cell suspension medium with 5% FBS was added to the upper chamber and incubated at 37°C with 5% carbon dioxide for indicated times. After incubation for an appropriate period, the upper side of the membrane was washed and wiped off using cotton swabs, and the cells on the lower membrane surface were fixed with methanol for 10 minutes and stained with 1% toluidine blue (wt/vol, prepared in phosphate-buffered saline), for 5 minutes, and then washed with phosphate-buffered saline twice. After the dye had dried, 100 mL 10% acetic acid was added to the upper chamber and vortexed for 10 minutes, then transferred to 96-well plates and the OD570 values were measured with BioTex SynergeMX.
Wound healing assay
5637 and T24 cells were trypsinized and counted. 1 × 105 cells were reseeded in each well of a new 6-well plate. With incubation overnight, the confluent cells monolayers were scratched with a 10 μL sterile pipette tip. Then the non-adherent cells were washed off with sterilized PBS and serum-free medium was added into the wells. The gap area caused by the scratch was monitored by the inverted microscope (Olympus, Japan). Three random non-overlapping areas in each well were pictured at 0 h, 4 h and 8 h post-scratch. Scratch width between the two linear regions was quantitated for assessing capacity of cells migration.
Flow cytometry for the detection of cell cycle progression
5637 and T24 cells were collected after digestion and were washed twice with PBS and centrifuged at 1,000 rpm for 5 min. The supernatant was discarded, and the cells were then resuspended and fixed in ice-cold 75% ethanol and stored at 4°C. After two washes in PBS, the cells were stained with propidium iodide (PI) and subjected to flow cytometric analysis of the percentage of cells in G0/G1, S and G2/M phases (BD Biosciences, USA). Treatment with each drug concentration was conducted in triplicate. The data were processed by Cell Quest Software (BD Biosciences, USA).
Western blotting
5637 and T24 cells were harvested and washed with cold phosphate-buffered saline and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.6; 150 mM NaCl; 1% Triton-X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with complete miniprotease inhibitor cocktail tablets. The protein concentration was estimated using the Bio-Rad protein assay (Bio-Rad, Marnes-la-Coquette, France). Samples containing 30 μg of protein were separated on 10% SDS-PAGE gel, transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). After saturation with 5% (w/v) non-fat dry milk in TBS and 0.1% (w/v) Tween 20 (TBST), the membranes were incubated with the following antibodies: Bcl-2, caspase-3 and PPAR γ (1:1000, Santa Cruz Biotechnoogy, CA, USA). After three washes with TBST, The membranes were next incubated with the appropriate HRP (horseradish peroxidase)-conjugated antibody visualized with chemiluminescence (Thermo, USA).
Statistical analysis
The data from these experiments were reported as mean ± standard deviation (SD) for each group. All statistical analyses were performed by using PRISM version 4.0 (Graph Pad). Inter-group differences were analyzed by one-way ANOVA, and followed by Tukey’s multiple comparison test as a post test to compare the group means if overall P < 0.05. Differences with P value of < 0.05 were considered statistically significant.
Results
Rosiglitazone inhibits 5637 and T24 cells proliferation
We used the MTT assay to monitor the cell growth inhibition rate, and the results demonstrated that growth inhibition of 5637 (Figure 1A) and T24 (Figure 1B) cells were verified in the present of rosiglitazone. The level of inhibition rate was gradually increased by treating the 5637 (Figure 1A) and T24 (Figure 1B) cells with higher doses of rosiglitazone and longer incubation time.
Figure 1.

5637 (A) and T24 (B) cells were incubated with rosiglitazone in different concentration for 24 h, 48 h and 72 h, and the cell viability was examined by MTT assay. Values were expressed as mean ± SD, n = 3 in each group. *P < 0.05 versus control group.
Rosiglitazone inhibits bladder cancer 5637 and T24 cells migration
We examined the potential roles of rosiglitazone on migration capacities of bladder cancer cell lines. After 24 h incubation, the transwell assay was conducted to assess cells migration capability in vitro. Compared with control, rosiglitazone exerted a potent inhibiting effect on migration of the 5637 (Figure 2A) and T24 (Figure 2B) cell lines under the condition of higher concentration. Subsequently, the scratch closed assay was performed to measure the cell migration inhibition of rosiglitazone, and cells were reseeded and then scratches were made 24 h later. Incubation of rosiglitazone led to retarded wound closing compared with control group in T24 cell lines (Figures 3B and 4B). However, the wound closing had no obvious different between the rosiglitazone-treated groups and control group in 5637 cell line (Figures 3A and 4A).
Figure 2.
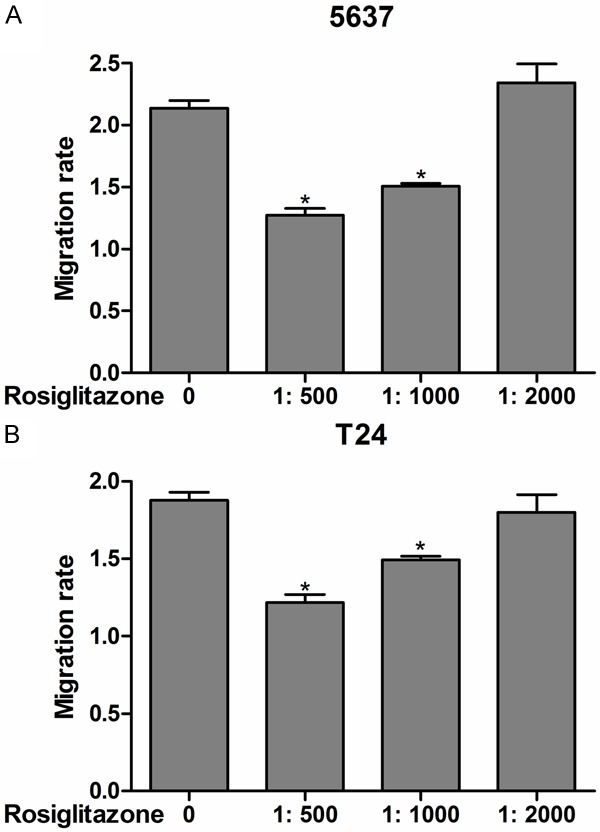
Migration activity of 5637 (A) and T24 (B) cells exposure to rosiglitazone in different concentration for 24 h was measured by transwell assay. Values were expressed as mean ± SD, n = 3 in each group. *P < 0.05 versus control group.
Figure 3.

Cell scratch assay was used to detect the migration of 5637 (A) and T24 (B) cells exposure to rosiglitazone in different concentration for 0 h, 4 h and 8 h.
Figure 4.

The migration inhibition of scratch assay was transformed to the percentage of the initial distance between the two edges for 5637 (A) and T24 (B) cells. Values were expressed as mean ± SD, n = 3 in each group. *P < 0.05 versus control group.
Rosiglitazone regulates bladder cancer 5637 and T24 cells cycle progression and apoptosis by regulating apoptosis-related protein and PPAR γ
Next, we performed flow cytometry to measure cell cycle distribution. In bladder cancer 5637 cell lines, incubation with rosiglitazone resulted in a significant decrease in the G0/G1 population and a corresponding increase in the S phase population compared with control group (Figure 5A and 5B). In T24 cell lines, rosiglitazone administration could significantly increase the population in G0/G1 phase and decrease the population in S phase (Figure 5C and 5D). Furthermore, we examined rosiglitazone-induced apoptosis in human bladder cancer 5637 and T24 cell lines through an apoptotic mechanism. Triphosphate nick-end labeling (TUNEL) staining were measured when 5637 and T24 cells were exposed to rosiglitazone for 24 h. As shown in Figure 6A and 6B, rosiglitazone administration showed a significant cell apoptosis in a concentration-dependent manner. The apoptosis-related markers protein expression were measured by western bltting. Treatment with rosiglitazone led to decrease the anti-apoptotic protein Bcl-2 level and increase the pro-apoptotic protein caspase 3 level in 5637 (Figure 6C) and T24 (Figure 6D) cells. Importantly, the protein expression of PPAR γ was significantly increased in the present of rosiglitazone in 5637 (Figure 6E) and T24 (Figure 6F) cells as compared to control group.
Figure 5.
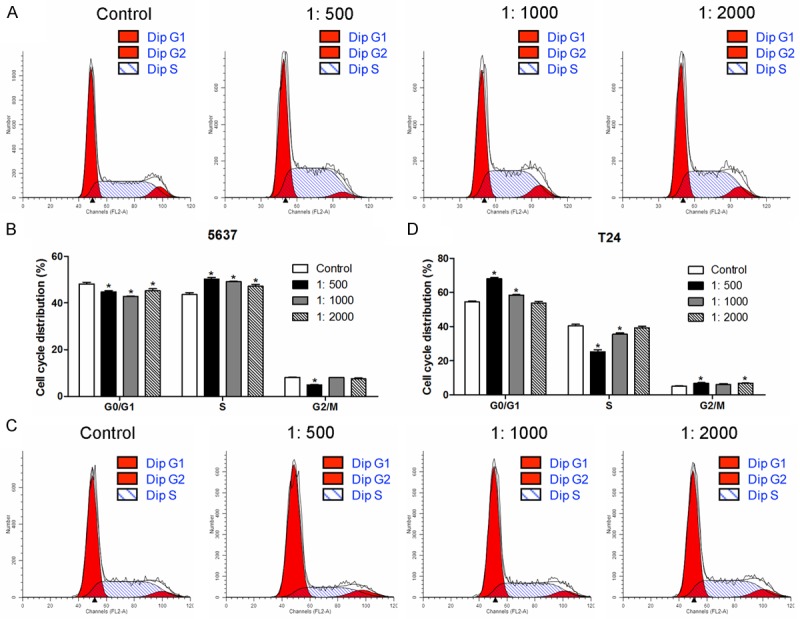
Representative photographs of cell cycle analysis for 5637 (A and B) and T24 (C and D) cells were exposed to rosiglitazone in different concentration for 24 h. Values were expressed as mean ± SD, n = 3 in each group. *P < 0.05 versus control group.
Figure 6.

5637 (A) and T24 (B) cells exposure to rosiglitazone in different concentration for 24 h, triphosphate nick-end labeling (TUNEL) staining was measured by flow cytometry. The protein expression of Bcl-2 and caspase 3 was measured by western blotting in 5637 (C) and T24 (D) cells. The protein expression of PPAR γ was measured by western blotting in 5637 (E) and T24 (F) cells. Values were expressed as mean ± SD, n = 3 in each group. *P < 0.05 versus control group.
Discussion
Our data suggested that rosiglitazone could inhibit human bladder cancer 5637 and T24 cells proliferation and migration and induce cell apoptosis in vitro. Simultaneously, rosiglitazone administration resulted in a significant decrease in anti-apoptotic protein expression and increase in pro-apoptotic protein expression. Moreover, rosiglitazone administration can induce cell cycle arrest in human bladder cancer 5637 and T24 cell lines. These results suggest that rosiglitazone may be a potential therapeutic drug for human bladder cancer.
In this paper we demonstrated that rosiglitazone strongly inhibited human bladder cancer cell proliferation when the concentration of rosiglitazone was more than 10 μM. In HCT-15 human colorectal cancer cell line [11], the IC50 values at 24 and 48 h were 48.84 μmol/L and 33.33 μmol/L, respectively. Interestingly, the similar results in CaCo-2, HT29 and SW480 human colon cancer cells show that the cells viability is markedly suppressed when the concentration of rosiglitazone is more than 10 μM [12]. These results obtained about the inhibition of cell proliferation by rosiglitazone support the hypothesis that PPAR γ ligands can suppress cancer cell proliferation.
PPAR γ, a ligand-activated intracellular transcription factor, belongs to the nuclear receptor superfamily, and PPAR γ agonists can induce antineoplastic signalling pathways in different cancer cell lines, animal models and clinical trials [16]. In HT-29 human colon cancer cells, rosiglitazone induces caveolin-1 by PPAR γ-dependent signaling to improve cancer cell drug resistance [17]. PPAR γ agonists, troglitazone and 15-deoxy-Delta (12, 14)-prostaglandin J2 (15d-PGJ2), show dose-dependent inhibitory effects on the proliferation of the gastric cancer cells [18]. PPAR γ agonists have potent in vitro cytotoxicity, the possible mechanism of which is through induction of apoptosis and cell cycle arrest [19]. Increasing evidence indicates that PPAR γ agonists in combination with other drugs may be the more successful strategies for cancer therapy [11,20]. Notably, there is evidence to show the potential linkage between the development of bladder cancer and long-term, high doses of oral pioglitazone for the treatment of type 2 diabetes [21]. The combination of 15d-PGJ2 and survivin inhibition play a potentially role in the therapeutical manipulation of bladder cancer by inducing the production of reactive oxygen species (ROS) [22]. Moreover, PPARγ agonist DIM-C can be an excellent alternative to bladder tumors resistant to EGFR inhibition, which constitutes rational for combination therapy with EGFR inhibitor in bladder cancer [23].
Previous studies indicate that PPAR γ is widely expressed in bladder cancer cell lines and tumor tissues, and its expression has been correlated with tumor grade and stage [24-26]. Bladder cancer cells with different PPAR γ expression have different capacity of migration and invasion, and there was a positive correlation between the increase in PPAR γ expression and migration and invasion capacity [13]. These results demonstrate that high expression of PPAR γ and PPAR γ agonist play an important role in bladder cancer cell migration and invasion [13]. In contrast, our study indicated that rosiglitazone could induce apoptosis and inhibit migration in human bladder cancer with a time- and dose-dependent manner. The PPAR γ agonist troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells [27].
In conclusion, the present study demonstrates that rosiglitazone is able to exert a antineoplastic activity in human bladder cancer 5637 and T24 cell lines through inhibition of proliferation, induction of apoptosis and cell cycle arrest.
Acknowledgements
Our research was supported by the Science and Technology Development Fund of Macao (Grant No. 064/2012/A).
Disclosure of conflict of interest
None.
References
- 1.Wang C, Ge Q, Zhang Q, Chen Z, Hu J, Li F, Ye Z. Targeted p53 activation by saRNA suppresses human bladder cancer cells growth and metastasis. J Exp Clin Cancer Res. 2016;35:53. doi: 10.1186/s13046-016-0329-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 4.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, Chen DS, Vogelzang NJ. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Zhuang C, Liu Y, Chen M, Zhou Q, Chen Z, He A, Zhao G, Guo Y, Wu H, Cai Z, Huang W. shRNA targeting long non-coding RNA CCAT2 controlled by tetracycline-inducible system inhibits progression of bladder cancer cells. Oncotarget. 2016;7:28989–97. doi: 10.18632/oncotarget.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerstein HC, Yusuf S, Bosch J, Pogue J, Sheridan P, Dinccag N, Hanefeld M, Hoogwerf B, Laakso M, Mohan V, Shaw J, Zinman B, Holman RR. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- 7.Cerquetti L, Sampaoli C, Amendola D, Bucci B, Masuelli L, Marchese R, Misiti S, De Venanzi A, Poggi M, Toscano V, Stigliano A. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp Cell Res. 2011;317:1397–1410. doi: 10.1016/j.yexcr.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Taub M. Cancer drug troglitazone stimulates the growth and response of renal cells to hypoxia inducible factors. Biochem Biophys Res Commun. 2016;471:342–347. doi: 10.1016/j.bbrc.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 9.Cantini G, Lombardi A, Piscitelli E, Poli G, Ceni E, Marchiani S, Ercolino T, Galli A, Serio M, Mannelli M, Luconi M. Rosiglitazone inhibits adrenocortical cancer cell proliferation by interfering with the IGF-IR intracellular signaling. PPAR Res. 2008;2008:904041. doi: 10.1155/2008/904041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hazra S, Batra RK, Tai HH, Sharma S, Cui X, Dubinett SM. Pioglitazone and rosiglitazone decrease prostaglandin E2 in non-small-cell lung cancer cells by up-regulating 15-hydroxyprostaglandin dehydrogenase. Mol Pharmacol. 2007;71:1715–1720. doi: 10.1124/mol.106.033357. [DOI] [PubMed] [Google Scholar]
- 11.Miao R, Xu T, Liu L, Wang M, Jiang Y, Li J, Guo R. Rosiglitazone and retinoic acid inhibit proliferation and induce apoptosis in the HCT-15 human colorectal cancer cell line. Exp Ther Med. 2011;2:413–417. doi: 10.3892/etm.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cerbone A, Toaldo C, Minelli R, Ciamporcero E, Pizzimenti S, Pettazzoni P, Roma G, Dianzani MU, Ullio C, Ferretti C, Dianzani C, Barrera G. Rosiglitazone and AS601245 decrease cell adhesion and migration through modulation of specific gene expression in human colon cancer cells. PLoS One. 2012;7:e40149. doi: 10.1371/journal.pone.0040149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang DR, Lin SJ, Ding XF, Miyamoto H, Messing E, Li LQ, Wang N, Chang C. Higher expression of peroxisome proliferator-activated receptor gamma or its activation by agonist thiazolidinedione-rosiglitazone promotes bladder cancer cell migration and invasion. Urology. 2013;81:1109, e1101–1106. doi: 10.1016/j.urology.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 16.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 17.Tencer L, Burgermeister E, Ebert MP, Liscovitch M. Rosiglitazone induces caveolin-1 by PPARgamma-dependent and PPRE-independent mechanisms: the role of EGF receptor signaling and its effect on cancer cell drug resistance. Anticancer Res. 2008;28:895–906. [PubMed] [Google Scholar]
- 18.Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N, Kinoshita Y. Expression of peroxisome proliferator-activated receptor (PPAR) gamma in gastric cancer and inhibitory effects of PPAR gamma agonists. Br J Cancer. 2000;83:1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiong X, Ye Y, Fu L, Dai B, Liu J, Jia J, Tang J, Li L, Wang L, Shen J, Mei C. Antitumor activity of a novel series of alpha-aryloxy-alpha-methylhydrocinnamic acid derivatives as PPAR gamma agonists against a panel of human cancer cell lines. Invest New Drugs. 2009;27:223–232. doi: 10.1007/s10637-008-9161-0. [DOI] [PubMed] [Google Scholar]
- 20.Dicitore A, Caraglia M, Colao A, Zappavigna S, Mari D, Hofland LJ, Persani L, Vitale G. Combined treatment with PPAR-gamma agonists in pancreatic cancer: a glimmer of hope for cancer therapy? Curr Cancer Drug Targets. 2013;13:460–471. doi: 10.2174/1568009611313040008. [DOI] [PubMed] [Google Scholar]
- 21.Hillaire-Buys D, Faillie JL, Montastruc JL. Pioglitazone and bladder cancer. Lancet. 2011;378:1543–1544. doi: 10.1016/S0140-6736(11)61662-0. author reply 1544-1545. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Tan H, Xu D, Ma A, Zhang L, Sun J, Yang Z, Liu Y, Shi G. The combinatory effects of PPAR-gamma agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int J Mol Med. 2014;34:262–268. doi: 10.3892/ijmm.2014.1774. [DOI] [PubMed] [Google Scholar]
- 23.Mansure JJ, Nassim R, Chevalier S, Szymanski K, Rocha J, Aldousari S, Kassouf W. A novel mechanism of PPAR gamma induction via EGFR signalling constitutes rational for combination therapy in bladder cancer. PLoS One. 2013;8:e55997. doi: 10.1371/journal.pone.0055997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshimura R, Matsuyama M, Segawa Y, Hase T, Mitsuhashi M, Tsuchida K, Wada S, Kawahito Y, Sano H, Nakatani T. Expression of peroxisome proliferator-activated receptors (PPARs) in human urinary bladder carcinoma and growth inhibition by its agonists. Int J Cancer. 2003;104:597–602. doi: 10.1002/ijc.10980. [DOI] [PubMed] [Google Scholar]
- 25.Choi W, Czerniak B, Ochoa A, Su X, Siefker-Radtke A, Dinney C, McConkey DJ. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat Rev Urol. 2014;11:400–410. doi: 10.1038/nrurol.2014.129. [DOI] [PubMed] [Google Scholar]
- 26.Sandes EO, Lodillinsky C, Langle Y, Belgorosky D, Marino L, Gimenez L, Casabe AR, Eijan AM. Inducible nitric oxide synthase and PPAR gamma are involved in bladder cancer progression. J Urol. 2012;188:967–973. doi: 10.1016/j.juro.2012.04.099. [DOI] [PubMed] [Google Scholar]
- 27.Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014;21:188–193. doi: 10.1038/cgt.2014.16. [DOI] [PubMed] [Google Scholar]