

Abstract
IgG4-related lymphadenopathy (IgG4-LAD) is newly described entity, which may occur before, during or after diagnosis of extranodal IgG4-related disease. It is important to recognize IgG4-LAD, especially in cases with lymphadenopathy as the initial presentation, so that the patients can receive prompt treatment. However, it can be challenging to formulate a final diagnosis since IgG4-LAD displays a broad morphologic spectrum. Moreover, morphologic changes alone are not sufficient for diagnosis of IgG4-LAD, and an accurate diagnosis has to take into account of the overall clinical presentations and laboratory studies. Currently, it is not very clear when pathologists should consider workup for potential IgG4-LAD based on the histologic features. Particularly, for some pathologists, it is not certain how to render the diagnosis in reactive lymph nodes with markedly increased IgG4 + cells. In this review, we will attempt to summarize the major clinicopathologic features of IgG4-LAD and its variants, differential diagnoses, and algorithms to establish an accurate diagnosis.
Keywords: IgG4, IgG4-related lymphadenopathy, IgG4-related disease
Introduction
IgG4-related disease (IgG4-RD) is a recently recognized fibroinflammatory condition, which was initially described in the pancreas as “sclerosing pancreatitis”, now referred to as “type I IgG4-related autoimmune pancreatitis” [1]. Nowadays, IgG4-RD has been diagnosed in nearly all organ systems, including the pancreas, lacrimal glands, salivary glands, kidneys, lungs, prostate, lymph nodes, aorta, and skin [2-4]. IgG4-RD mostly occurs in middle-aged to elderly men. The patients typically present with an indolent clinical course, and the signs and symptoms may become evident over months or years. Constitutional symptoms are generally absent, including weight loss, fevers, fatigue, malaise, and night sweats. Up to 30-40% of patients may have a longstanding history of allergies at diagnosis. Diagnosis of IgG4-RD is based upon the clinical presentations, serologic studies, and histomorphologic features [4-10]. Serum IgG4 levels (normal 135 mg/dL) are elevated in the majority of patients, especially those with multi-organ involvement [7,11]. However, elevation in serum IgG4 levels can be detected in conditions other than IgG4-RD, including systemic vasculitis, connective tissue disorders, infections, and malignancies [12]. Glucocorticoids are typically the first line of therapy, which are effective in most patients initially. Rituximab and methotrexate are useful for recurrent or refractory diseases [3,13].
In the extranodal sites, IgG4-RD typically presents as mass or pseudotumour-like lesions in a single or multiple organs with very similar histopathological findings. Three major morphologic characteristics have been described [6] (Figure 1A-E): (1) Dense lymphoplasmacytic infiltrate with increased IgG4+ cells; (2) Fibrosis, typically showing prominent fibroblastic proliferation with a characteristic storiform growth pattern; (3) Obliterative phlebitis, although this feature is difficult to find in many cases of IgG4-RD. Other minor histopathological features associated with IgG4-RD include non-obliterative phlebitis and increased eosinophils in the tissue.
Figure 1.
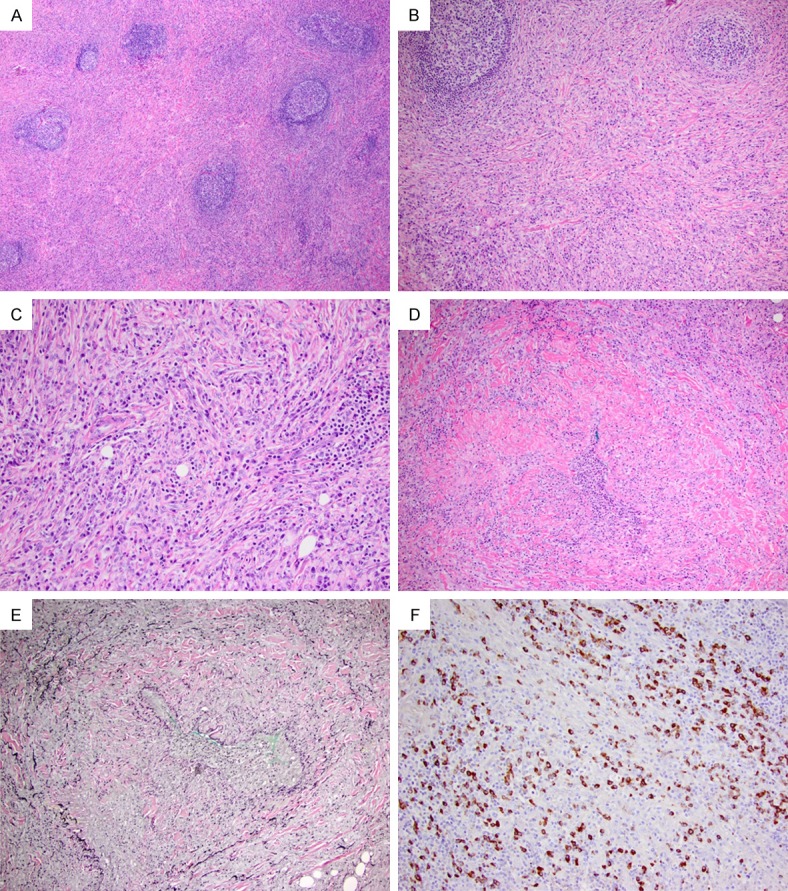
A case of IgG4-RD involving submandibular gland. (A, B) Extensive fibrosis and prominent lymphoid infiltration with scattered reactive lymphoid follicles (H&E, 40 × and 100 ×, respectively). (C) Storiform arrangement of fibroblastic proliferation and prominent lymphoplasmacytic infiltration (H&E, 200 ×). (D) An obliterated vein (H&E, 100 ×) with elastin stain (E) (100 ×). (F) Abundant IgG4+ cells in the tissue (200 ×).
IgG4-related lymphadenopathy (IgG4-LAD) is noted in approximately 80% of patients with extranodal IgG4-RD [14]. It can appear before, concurrent with, or after the diagnosis of the extranodal disease. In the cases with lymphadenopathy as the initial presentation, it is critical to recognize IgG4-LAD so that the patients can receive timely treatment to avoid irreversible vital organ damages, destructive surgeries or life-threatening events in other systems. However, diagnosis of IgG4-LAD can be challenging for several reasons: (1) IgG4-LAD has not been widely recognized by pathologists, and therefore it can be under-diagnosed due to lack of awareness of such entity; (2) The pathologic changes in lymph nodes display a broad morphologic spectrum and greatly overlap with those in non-IgG4-related lymphadenopathy, which could result in potential over-diagnosis; and (3) IgG4-LAD is not purely a morphologic diagnosis, and a definitive diagnosis has to rely on the overall clinical presentations, histology changes, and laboratory studies.
General features of IgG4-LAD
Patients with IgG4-LAD have similar clinical presentations to those with extranodal IgG4-RD, and they typically present with enlarged lymph nodes without constitutional symptoms. The lymph nodes are non-tender and measure 1 to 3 cm in greatest dimension. Lymphadenopathy can be localized or generalized and often associated with extranodal lesions. For example, approximately 80% of patients with IgG4-related pancreatitis have concurrent involvement of lymph nodes, particularly the mediastinal and intraabdominal lymph nodes [14]. However, lymphadenopathy can sometimes be the first manifestation of IgG4-RD, which places great challenge to pathologists in diagnosis.
IgG4-LAD displays a broad morphologic spectrum, and five histologic subtypes have been described, including (1) Multicentric Castleman disease-like; (2) Reactive follicular hyperplasia-like; (3) Interfollicular expansion and immunoblastosis; (4) Progressive transformation of germinal center (PTGC) type; and (5) Inflammatory pseudotumor-like. Morphologic overlaps exist among different subtypes, and therefore sometimes it may not be feasible to accurately subclassify IgG4-LAD. Compared to the extranodal IgG4-RD, the histologic changes in IgG4-LAD are different: obliterative phlebitis is usually absent in all subtype of IgG4-LAD and the characteristic storiform fibrosis is usually absent except for the inflammatory pseudotumor-like subtype.
Studies have suggested that in lymph nodes an IgG4+ cell count of ≥ 100 cells per high power field (HFP) and an IgG4+/IgG+ cell ratio of ≥ 40% are the necessary cutoff values in diagnosis of IgG4-LAD [15]. IgG4 and IgG immunostainings are strongly recommended in any case suspicious for IgG4-LAD, and they have been proved to be very simple and powerful. Distribution of IgG4+ cells in lymph nodes can be patchy, and counting areas with intense IgG4+ cells might be more appropriate. It is recommended that at least three 40 × fields (0.196 mm2) with the highest number of IgG4+ cells should be counted to calculate the average number of IgG4+ cells within these fields [15]. The same three fields should be counted for IgG+ cells to calculate the IgG4+/IgG+ cell ratio. We must emphasize that the morphology alone is essential but not sufficient for a diagnosis of IgG4-LAD, even in cases with significantly increased IgG4+ cells and elevated IgG4+/IgG+ cell ratio. Several non-IgG4-LAD disease entities may also show increased IgG4+ cells and elevated IgG4+/IgG + cell ratios in lymph nodes, including conditions associated with elevated serum interleukin-6 (IL-6) concentrations (such as multicentric Castleman disease and rheumatoid arthritis), infections, Rosai-Dorfman disease, and lymphomas [15-17]. Correlation with clinical findings and laboratory studies is crucial for an accurate diagnosis. The detailed differential diagnoses will be discussed in each section below.
Morphologic subtypes of IgG4-LAD
Type I IgG4-LAD: multicentric Castleman disease-like
The patient with type I IgG4-LAD often presents with systemic lymphadenopathy. Histologically, the nodal architecture is preserved with follicular hyperplasia, variable degrees of interfollicular expansion, and patent sinusoids (Figure 2A). Frequent reactive lymphoid follicles have regressive germinal centers, and occasional follicles may contain penetrating blood vessels, reminiscent of those seen in conventional Castleman disease. Normal to hyperplastic germinal centers are also present. The mantle zones are often expanded. The interfollicular areas are expanded with abundant plasma cells, scattered eosinophils, and prominent proliferation of high endothelial venules. The IgG4+ cells are mostly in the interfollicular areas with an IgG4+ cell count ≥ 100 per HPF and an IgG4/IgG ratio ≥ 40% (Figure 2B).
Figure 2.
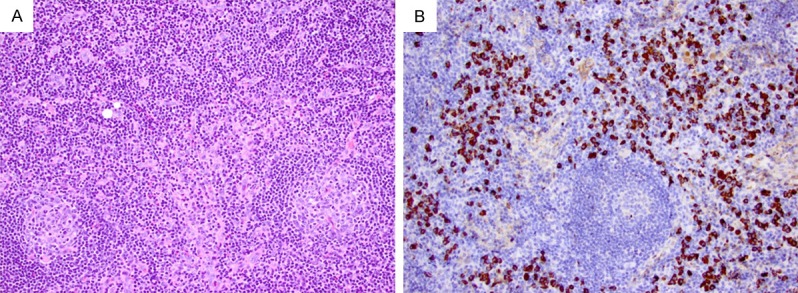
A case of type I IgG4-LAD with multicentric Castleman disease-like features. A. The lymph node shows interfollicular expansion and frequent regressive lymphoid follicles, one of which has a penetrating vessel (right side) (H&E, 200 ×). B. Abundant IgG4+ cells are noted in the interfollicular areas (200 ×).
Type I IgG4-LAD morphologically overlaps significantly with multicentric Castleman disease (MCD) and these two entities have to be separated (Table 1). Clinically, type I IgG4-LAD typically occurs in patients with no history of HIV infection, and the patients mostly lack constitutional symptoms despite systemic lymphadenopathy. Patients usually have no anemia, hypoalbuminemia, or hypocholesterolemia [18]. Serology studies often detect elevated IgG (mostly due to elected IgG4) and IgE, whereas IgA, IgM, IL6 and C-reactive protein (CRP) levels are mostly normal. On the other hand, MCD is often associated with constitutional symptoms in HIV+ patients, and serum immunoglobulins (IgA, IgG and IgM), IL6 and CRP are elevated. The morphologic features are similar in type I IgG4-LAD and MCD; however, in MCD the reactive germinal centers are mostly small and regressed, in contrast to a morphologic spectrum of germinal centers in type I IgG4-LAD. Interfollicular eosinophils are rare or absent in MCD. Although IgG4+ cells are typically not increased in MCD, some cases of MCD may have a marked increase in IgG4+ cell (≥ 100 per HPF), an IgG4/IgG ratio ≥ 40%, and elevated serum IgG4 levels, which would fulfill the morphologic diagnostic criteria for IgG4-RD. Therefore, these two entities may not be fully differentiated just based on the histological findings, IgG4+ cells in the tissue, and serum IgG4 levels; clinical presentations and additional laboratory analyses are essential for a definitive diagnosis.
Table 1.
Comparison between type I IgG4-LAD and multicentric Castleman disease (Modified from [4])
| Type I IgG4-LAD | Multicentric Castleman Disease | |
|---|---|---|
| Presentations | Typically asymptomatic with or without extranodal IgG4-RD. No constitutional symptoms | Abrupt or gradual onset of symptoms, with multiple lymphadenopathy and constitutional symptoms |
| Age | Middle-aged to elderly, median age 69 years | Middle-aged to elderly, median age 64 years |
| Gender | Marked male predilection, M:F=8:1 | M:F=2:1 |
| HIV Status | Negative | Often positive |
| Serum immunoglobulin | Elevated IgG, IgG4 and IgE levels; normal IgM and IgA levels | Globally elevated, including IgG, IgM and IgA levels |
| Serum IL6 | Normal or mildly increased | Increased |
| C-reactive protein | Normal or mildly increased | Increased |
| Anemia | No or mild | Common, often moderate to severe |
| Histology | Preserved architecture. Regressive, normal to hyperplastic follicles. Interfollicular zones often rich in plasma cells and eosinophils | Preserved architecture. Usually small and regressed lymphoid follicles. Interfollicular areas with florid plasmacytosis but no eosinophilia |
| Immunostains | Increased IgG4+ cells and IgG4/IgG ratio. No light chain restriction | Usually no increase in IgG4+ cells. May have lambda light chain restriction. HHV8 often positive |
| Prognosis | Benign clinical course; good response to steroid therapy | Unpredictable clinical course, but often with high mortality |
Non-specific reactive lymphadenopathy with Castleman-like features can be encountered occasionally with no identifiable underlying etiology. The lymphadenopathy is usually localized and lymph node may reach a substantially large size. The reactive lymphoid follicles usually contain regressed to hyperplastic germinal centers and expanded mantle zones. The interfollicular regions often demonstrate a mixed lymphoid proliferation with no significant increase in plasma cells or eosinophils. Most cases have no elevated IgG4+ cells or IgG4/IgG ratio, although some may have a significant increase in IgG4+ cells with no association with IgG4-RD. The lymphocyte-rich variant of classic Hodgkin lymphoma (CHL) may show a nodular growth pattern with an expanded mantle zone and a regressed and eccentrically placed germinal center, sometimes reminiscent of Castleman disease or type I IgG4-LAD. Rare or scattered tumor cells are found in the expanded mantle zones, which may require meticulous morphologic examination at high power fields and the assistance of immunohistochemical stains.
Type II IgG4-LAD: reactive follicular hyperplasia-like
In type II IgG4-LAD, patient usually presents with localized lymphadenopathy, which is frequently found in the regional lymph nodes of extranodal IgG4-RD. Microscopically, the architecture of lymph nodes is well-preserved with reactive follicular hyperplasia and patent sinuses (Figure 3A-D). The lymphoid follicles comprise a reactive germinal center surrounded by a discrete mantle zone. The interfollicular areas contain mostly small reactive lymphocytes, mixed larger transformed lymphocytes, and often scattered eosinophils. The number of interfollicular plasma cells is variable, which is more evident with CD138 or IgG4 immunostaining. The lymphoid follicles may also have increased plasma cells.
Figure 3.
Type II IgG4-LAD with reactive follicular hyperplasia. (A) The lymph node shows preserved architecture with reactive follicular hyperplasia (H&E, 40 ×). (B) The interfollicular regions reveal a mixed lymphoid population with frequent plasma cells and scattered eosinophils (H&E, 400 ×). (C) Abundant IgG+ cells are present (40 ×), most of which are also IgG4+ (D) (40 ×).
The distinction between type II IgG4-LAD and usual non-specific reactive follicular hyperplasia is very difficult without sufficient clinical and laboratory information. There are some minor clinical and morphologic differences which may be more commonly noted in IgG4-LAD, including persistent lymphadenopathy, reactive interfollicular plasmacytosis, and increased interfollicular eosinophils. However, none of the described features is specific enough to separate these two diseases. Moreover, occasional cases of usual reactive follicular hyperplasia may have a significant increase in IgG4+ plasma cells. In patients with lymphadenopathy as an initial presentation and no documented history of extranodal IgG4-RD, it is still uncertain when we should consider the possibility of IgG4-LAD in lymph nodes with reactive follicular hyperplasia. We recommend IgG4 and IgG immunostains in cases with persistent and/or systemic lymphadenopathy and no underlying etiology.
Patients with rheumatoid arthritis (RA) often have persistent lymphadenopathy, and the differential diagnosis from type II IgG4-LAD can be very tricky. Lymphadenopathy associated with RA typically displays hyperplastic follicles, which are relatively uniform in size. Intra- and inter-follicular reactive plasmacytosis is commonly present. Notably, some cases of RA associated lymphadenopathy may have markedly increased IgG4+ cells and IgG4/IgG ratios, which would fit the morphologic criteria for IgG4-LAD. Therefore, it is important to check the clinical information (documented history of rheumatoid arthritis) and essential serum autoantibodies, especially rheumatoid factor.
Type III IgG4-LAD: interfollicular expansion and immunoblastosis
Type III IgG4-LAD usually presents with systemic lymphadenopathy. The architecture of lymph nodes is marked distorted with prominent interfollicular expansion (Figure 4A-D). Sparse residual lymphoid follicles are dispersed with normal or atrophic germinal centers (Figure 4A). The interfollicular regions are composed of a spectrum of lymphocytes, including small lymphocytes, larger transformed cells, plasmacytoid cells, and plasma cells (Figure 4B and 4C). Reactive immunoblasts are often abundant, which may form loose clusters in some cases. Scattered eosinophils and vascular proliferation are observed.
Figure 4.
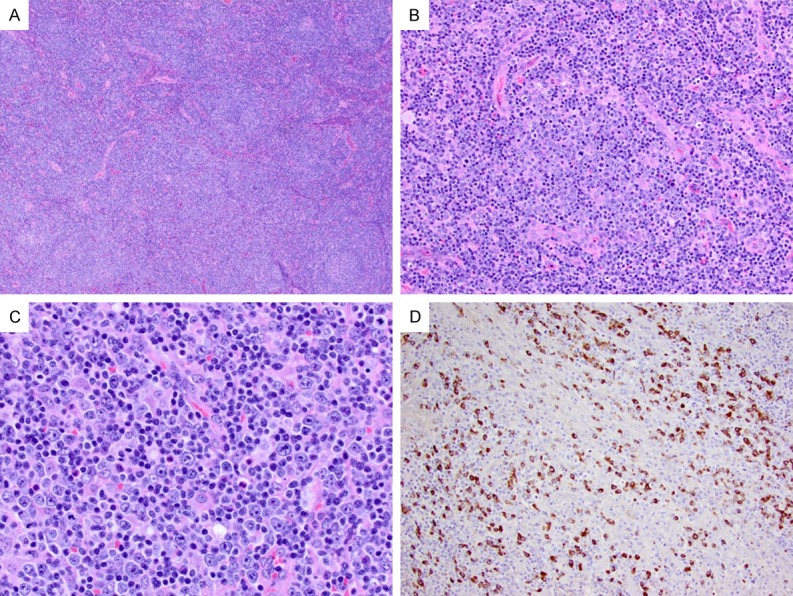
Type III IgG4-LAD with interfollicular expansion and immunoblastosis. A. The lymph node shows architectural distortion with marked interfollicular expansion (H&E, 40 ×). B, C. The expanded interfollicular regions have frequent immunoblasts, many plasma cells, and scattered eosinophils (H&E, 200 × and 400 ×, respectively). D. Abundant IgG4+ cells are present in the interfollicular areas (200 ×).
Diagnosis of type III IgG4-LAD can be very difficult since it mimics several benign and malignant conditions, and it is particularly difficult in cases with needle core biopsies. Acute viral infections, especially EBV and less commonly CMV, typically demonstrate marked interfollicular expansion with reactive immunoblastosis. Patients usually present with fever, laryngitis, splenomegaly, and atypical circulating cells in the peripheral blood. Identification of viral infection in the tissue sections (EBER in situ hybridization for EBV and immunohistochemical stain for CMV) is crucial to reach an accurate diagnosis, in conjunction with viral serology assays. Certain medications (i.e. Dilantin) may induce similar morphologic changes in lymph nodes.
The morphological features of type III IgG4-LAD overlap with those of atypical lymphoplasmacytic and immunoblastic proliferation (ALPIBP) in lymph nodes, which are associated with some autoimmune diseases, including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) [19]. Type III IgG4-LAD has to be distinguished from angioimmunoblastic T-cell lymphoma (AITL) (Table 2), particularly in cases with prominent interfollicular proliferation of high endothelial venules and abundant reactive immunoblasts. Patients with AITL typically present with systemic lymphadenopathy, prominent constitutional symptoms, and markedly increased serum LDH levels. The lymph nodes of AITL reveal architectural effacement with proliferation of often arborized vessels. The expanded and distorted follicular dendritic cell meshwork contains clusters of atypical clear cells, which are CD10+ T-cells. Scattered and loose clusters of EBV-positive cells are mostly present in AITL.
Table 2.
Comparison between type III IgG4-LAD and angioimmunoblastic T-cell lymphoma (AITL)
| Type III IgG4-LAD | AITL | |
|---|---|---|
| Presentation | Often systemic lymphadenopathy, but no constitutional symptoms | Systemic lymphadenopathy and marked constitutional symptoms |
| Serum LDH | Normal or mildly increased | Markedly increased |
| Architecture | Distorted, but at least partially preserved | Markedly distorted or effaced |
| Vascularity | Increased | Markedly increased with arborization |
| Clear cells | Usually inconspicuous | Often abundant |
| FDC meshwork | Normal size and morphology | Markedly expanded and distorted |
| PD1+/CD10+ cells | Limited to the germinal centers | Present in the atypical clear cells |
| EBV | Negative | Scattered, positive in immunoblasts |
| TRG@PCR | Polyclonal | Monoclonal |
| Prognosis | Benign clinical course; good response to steroid therapy | Poor |
Type IV IgG4-LAD: progressive transformation of germinal center (PTGC)-type
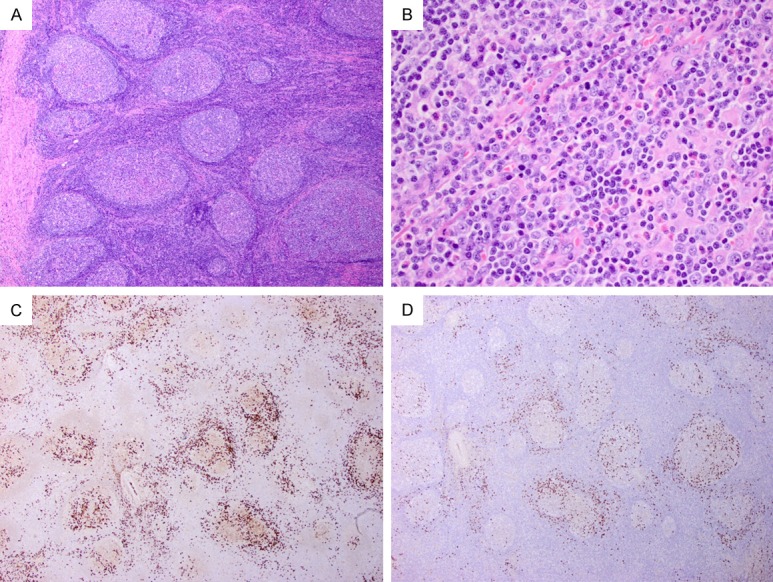
Patients with type IV IgG4-LAD usually present with an asymptomatic localized lymphadenopathy in the cervical region. The lymph node shows preserved architecture with prominent follicular hyperplasia and variable degrees of interfollicular expansion (Figure 5A). The PTGC follicles are scattered in a background of hyperplastic follicles, and they are round to oval with diameters ≥ 2 times of other normal secondary follicles (Figure 5A). The PTGC follicles are composed mostly of mantle cells, which intrude into the germinal centers; scattered and small clusters of residual germinal center B-cells are often noted with the PTGC follicles. Interestingly, frequent reactive plasma cells are noted in the hyperplastic germinal centers but not in the PTGC follicles (Figure 5B). The interfollicular areas contain a mixed population of lymphocytes with frequent eosinophils. Characteristically, in this type of IgG4-LAD, the IgG4+ cells are mostly localized in the hyperplastic germinal centers (Figure 5C and 5D), with only scattered ones in the interfollicular areas.
Figure 5.
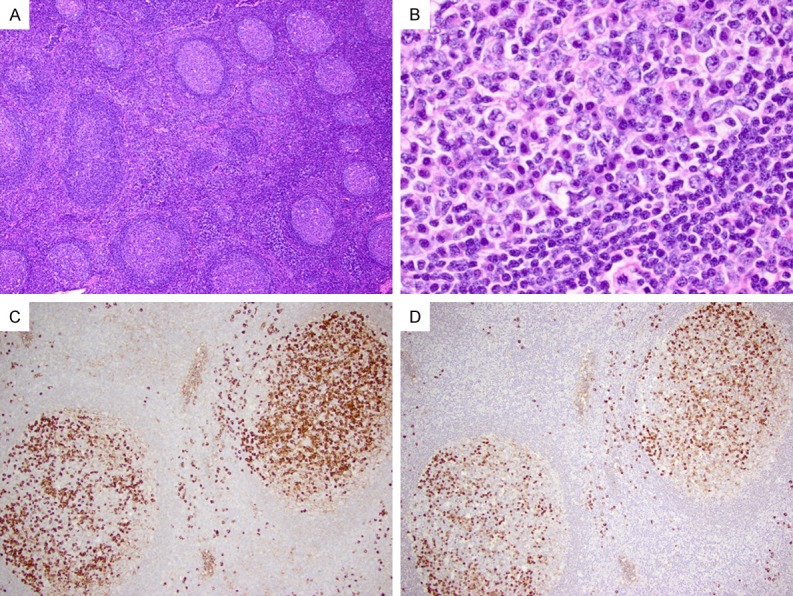
Type IV IgG4-LAD with PTGCs. (A) A PTGC is noted (left middle) in a background of reactive follicular hyperplasia (H&E, 40 ×). (B) A reactive germinal center contains frequent plasma cells (H&E, 600 ×). Abundant IgG+ (C) and IgG4+ (D) cells are noted mostly in the reactive follicles (100 ×).
In general, PTGCs occur in approximately 3.5% of cases of reactive lymphadenopathy [20,21]. PTGCs are associated with nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), and they may arise preceding or following NLPHL or in separate lymph nodes at the same time. Therefore, for lymph nodes with PTGCs, it is essential to search the LP cells (lymphocyte-predominant cells) at high magnifications in the PTGC follicles to rule out NLPHL.
Recent studies have proven that approximately one-third of cases with PTGCs eventually progressed to extranodal IgG4-RD, systemic disease, or both during the follow-up period [22]. In a study from Sato, et al, 40 of 62 PTGC cases (65%) fulfilled the histological criteria of IgG4-LAD, with IgG4+ cells >100 per HPF and an IgG4/IgG ratio > 40% (IgG4+ PTGC) [22]. Thirty-four patients with IgG4+ PTGCs were followed by regular imaging, laboratory findings and clinical evaluation during a median period of 26 months; totally 18 patients (18/34, 53%) were discovered to have extranodal IgG4-RD. Therefore, PTGC alone has a high association with development of extranodal IgG4-RD, with an overall risk of approximately 34% (65% × 53%). The distinction of type IV IgG4-LAD (IgG4+ PTGC) from usual IgG4-negative PTGC mainly relies on the clinical examination and follow-up, although some clinicopathologic differences exist (Table 3). Patients with type IV IgG4-LAD are usually older and have a higher incidence of submandibular lymph node involvement than those with IgG4-negative PTGCs. In contrast to cases with IgG4-negative PTGCs, patients with type IV IgG4-LAD have persistent lymphadenopathy if no further treatment, and the hyperplastic follicles contain many plasma cells, which are more obvious with CD138 or IgG4 immunostaining. The interfollicular regions often have increased eosinophils.
Table 3.
Comparison between type IV IgG4-LAD and IgG4-negative PTGC
| Type IV IgG4-LAD | Usual IgG4 Negative PTGC | |
|---|---|---|
| Age/gender | Mostly middle-aged or elderly male | Younger age, slight male predominance |
| Locations | Localized or systemic, axillary, mediastinal, intraabdominal | Often isolated, mostly cervical, axillary or inguinal |
| Duration | Persistent | Recurs in 20% cases |
| Low-power features | Similar with PTGCs | Similar with PTGCs |
| Plasma cells in germinal center | Markedly increased | Usually not increased |
| Interfollicular eosinophils | Often increased | May or may not be increased |
| IgG4, IgG4/IgG | Markedly increased | Not increased |
| Serum IgG4 | Mostly raised | Not raised |
| Extranodal IgG4-RD | Often present | No |
| Risk | Systemic IgG4-related disease | NLPHL |
Taken together, we recommend performing IgG and IgG4 immunostains in all lymph node cases with PTGCs after excluding NLPHL since PTGCs have a high association with nodal and extranodal IgG4-RD. So far, there are no data on the pediatric PTGCs and more studies are necessary to evaluate the association of pediatric PTGCs with IgG4-RD.
Type V IgG4-LAD: inflammatory pseudotumor (IPT)-like
This type is the least common one and very rare. Patients usually present with asymptomatic localized lymphadenopathy. Histologically, the involved lymph nodes show focal infiltration by poorly demarcated fibrous tissue with hyalinization, which is sprinkled with lymphocytes, plasma cells and scattered eosinophils. The fibrosis may be arranged in a storiform pattern and contain fibroblasts. Focal residual normal lymphoid tissue is present with hyperplastic follicles. The nodal infiltration is similar to that in extranodal IgG4-RD except that there is lack of obliterative phlebitis.
The histological findings often resemble nodal IPT (Table 4). However, patients with nodal IPT typically present with constitutional symptoms, including fever, fatigue, weight loss, and night sweats, whereas patients with type V IgG4-LAD typically lack these symptoms. Morphologically, nodal IPT forms a well-circumscribed mass lesion, which is composed of abundant spindle tumor cells with evenly distributed lymphocytes and plasma cells. The spindle cells express smooth muscle actin in most cases and ALK in a significant subset of cases.
Table 4.
Comparison between type V IgG4-LAD and nodal inflammatory myofibroblastic tumor (IMT)
| Type V IgG4-LAD | Nodal IMT | |
|---|---|---|
| Nature of lesion | An reactive inflammatory fibrosclerosing process | A neoplastic process in many cases |
| Symptoms | Usually no symptoms of fever, fatigue, weight loss, and night sweats | Usually with symptoms of fever, fatigue, weight loss, and night sweats |
| Clinical findings | Mass lesion, variable symptoms, often associated with systemic IgG4-RD | Mass lesion, not often with extranodal involvement |
| Gross features | Usually a poorly demarcated mass lesion | A discrete well-defined mass lesion |
| Histologic features | Fibrosclerotic lesion with poorly defined borders; patchy lymphoplasmacytic infiltration; mixed spindle fibroblasts and myofibroblasts | Well-demarcated lesion; evenly distributed lymphoplasmacytic cells; presence of abundant spindle tumor cells |
| IHC | No significant actin+ myofibroblasts; increased IgG4+ cells and IgG4/IgG ratios | Abundant actin+ cells; ALK+/-; no increased IgG4+ cells or IgG4/IgG ratio |
| Serum IgG4 | Often elevated | Normal |
Further differential diagnoses of IgG4-LAD
In addition to the differential diagnoses listed above in each subtype of IgG4-LAD, other reactive and neoplastic processes should also be considered. In general, lymphadenopathy in IgG4-LAD is localized or generalized but usually asymptomatic except mass effects in some cases. The cases with systemic lymphadenopathy may clinically resemble lymphoma, multicentric Castleman disease, and metastatic malignant tumor. However, the lymph nodes in IgG4-LAD are generally not very large and typically less than 2 cm in greatest dimension. The patients usually do not present with constitutional symptoms, including fever, night sweats, and weight loss. Serum lactate dehydrogenase level is normal or only minimally elevated. It is important to examine the tissue sections to rule out lymphoma, infection, autoimmune process, and medication effect, with the assistance of clinical findings and ancillary studies. IgG4-LAD may display the five major morphologic patterns with mixed features in some cases, but coagulative necrosis, epithelioid granulomas, and prominent neutrophil infiltration are usually absent [6].
Usual reactive lymphadenopathy
The morphologic findings and immunohistochemistry (IgG4+ cells ≥ 100/HFP and IgG4/IgG ≥ 40%) in lymph nodes are essential but not sufficient for diagnosis of IgG4-LAD. Non-IgG4 associated lymphadenopathy may show a significant increase in IgG4+ cells and IgG4/IgG ratio, including rheumatoid arthritis, Rosai-Dorfman disease, and multicentric Castleman disease [15,17,23-25]. Recently, it has been noticed that even nonspecific reactive lymphadenopathies many have increased IgG4+ cells and IgG4/IgG ratio. In an unpublished study, we collected 89 consecutive cases of reactive lymph nodes, which were stained for IgG4. Up to 36% of cases showed a significant increase in IgG4+ cells and IgG4/IgG ratios; during retrospective studies and a short period of follow-up, only a small proportion of patients were found to have extranodal IgG4-RD. The significance of the IgG4+ cells in many non-specific cases is unclear. Our data, in conjunction with others [17], indicate that a substantial number of non-specific or isolated lymphadenopathy with abundant IgG4+ cells may be irrelevant to IgG4-RD. Therefore, an accurate diagnosis of IgG4-LAD requires correlation with further clinical findings and laboratory studies. Moreover, an elevated serum IgG4 level (> 135 mg/dL) is helpful but not essential for diagnosis since serum IgG4 levels may be elevated in non-IgG4 associated conditions as well.
Hyper-interleukin-6 (IL-6) syndromes
Hyper-IL-6 syndromes, including MCD and RA as discussed above, frequently involve lymph nodes, which sometimes fulfill the histological diagnostic criteria for IgG4-LAD with significantly increased IgG4+ cells and IgG4/IgG ratios. In hyper-IL-6 syndromes, with the effects of IL-6, the serum levels of IgA, IgG and subclasses, IgE, IgM, and C-reactive protein (CRP) are globally elevated. In addition, patients present with anemia, thrombocytosis, hypoalbuminemia, and hypocholesterolemia, which are also related to high IL-6 levels. In IgG4-LAD, the major serologic abnormalities are elevated IgG (mostly due to IgG4) and IgE, whereas other parameters are typically within normal limits or only mildly elevated (Table 5). Therefore, histologic findings and immunohistochemical stains for IgG4 are not sufficient to separate IgG4-LAD from hyper-IL-6 syndrome related lymphadenopathy; clinical presentations and further laboratory analyses are crucial to separate the two entities. C-reactive protein and serum IL-6 levels are significantly elevated in MCD, and they are useful markers for differentiating between IgG4-LAD and MCD.
Table 5.
Comparison between IgG4-related disease and hyper-IL-6 syndrome
| IgG4-Related Disease | Hyper-IL-6 Syndrome | |
|---|---|---|
| Serum immunoglobulin | IgG↑ (mainly IgG4↑), IgE↑ | IgG↑ (IgG1~IgG4↑), IgA↑, IgM-/↑, IgE↑ |
| Serum IgG4/IgG ratio | Elevated | Normal or slightly elevated |
| Serum IL-6 | Normal or slightly elevated | Elevated |
| Serum CRP | Normal or slightly elevated | Elevated |
| Thrombocytosis | No | Yes |
| Anemia | No | Yes |
| Hypoalbuminemia | No | Yes |
| Hypocholesterolemia | No | Yes |
↑: increased.
Rosai-Dorfman diseases
A subset (up to 30%) of nodal and extranodal Rosai-Dorfman diseases (RDD) shows features of IgG4-RD with significantly increased IgG4+ cells and IgG4/IgG ratio [26,27], indicating that these two diseases may overlap in certain aspects, although the studies from Liu, et al. suggested that RDD does not belong to the spectrum of IgG4-RD [28]. We also encountered two nodal cases of RDD with a prominent increase in IgG4+ cells and IgG4/IgG ratio, and interestingly both patients also had markedly elevated serum IgG4 levels. One patient presented with systemic lymphadenopathy, and the lymph nodes were significantly reduced in size after treatment with steroids and low-dose rituximab. Extranodal RDD may also closely resemble IgG4-RD with prominent fibrosis and lymphoplasmacytic infiltrate, and the characteristic histiocytes with emperipolesis may not be evident compared to the nodal cases; it can be challenging to make an accurate diagnosis of these extranodal RDD, particularly in cases with elevated IgG4+ cells in the tissue.
Lymphoma
T-cell and B-cell lymphomas must be excluded incases suspicious for IgG4-LAD. For instances, lymphocyte-rich CHL needs to be ruled out in lymph nodes with Castleman-like features, NLPHL has to be excluded in cases with PTGC, and AITL is also in the differential diagnoses in cases with marked interfollicular expansion, prominent vascular proliferation and immunoblastosis. Detailed clinical information, meticulous morphologic examination, and necessary ancillary studies are critical to reach an accurate diagnosis. The lymph node cases with florid lymphoplasmacytic infiltrates may mimic primary nodal lymphoma, especially nodal marginal zone lymphoma and lymphoplasmacytic lymphoma with plasmacytic differentiation. In lymphoma cases, the lymphoid and plasmacytic populations often exhibit atypical morphologic features, and they may express aberrant markers with light chain restriction by flow cytometric assays, immunohistochemical stains, and in situ hybridization studies.
We have to point out that certain B-cell lymphomas and plasma cell neoplasms may express IgG4, which can potentially resemble IgG4-RD particularly in the extranodal sites [29-31]. In a recent study, 19 cases of orbital MALT lymphoma were investigated for IgG4-related disease, and in 10 out of 19 cases the lymphoma cells had a significant expression of IgG4, which would meet the criteria of IgG4-RD immunohistochemi-cally [31].
Diagnostic algorithm for IgG4-LAD
When should we consider IgG4 immunostain in reactive lymph nodes?
IgG4 immunostain is certainly not recommended for every single reactive lymph node during our routine practice, and the decision may be made based upon the overall clinical presentations and morphologic changes. We feel that the IgG4 immunostain may not be necessary under the following circumstances, to list a few examples: (1) Lymphadenopathy is limited to a single node or a localized region in patients with no clinical suspicion for IgG4-RD; (2) Lymphadenopathy in patients with known clinical history of infections, autoimmune disorders (i.e. RA and SLE) or certain medications (i.e. Dilantin and methotrexate); (3) Lymphadenopathy associated with malignancies or procedures in the vicinity. It is not uncommon to identify very large reactive lymph nodes in regional lymph node dissection sample for carcinoma or melanoma, despite no metastatic tumor in the lymph nodes; (4) Rather small quiescent lymph node with no significant follicular/interfollicular hyperplasia and no marked increase in reactive plasma cells and eosinophils; and (5) Reactive lymphadenopathy with known etiology, i.e. dermatopathic lymphadenopathy and Kikuchi-Fujimoto lymphadenitis.
In general, we recommend IgG and IgG4 immunostains in the following settings: (1) Lymphadenopathy in patients with a documented clinical history of extranodal IgG4-RD; (2) Persistent and/or systemic lymphadenopathy with no underlying causes, including lymphoma, autoimmune diseases, medications, and infections; (3) Lymphadenopathy with increased plasma cells in germinal centers and/or interfollicular regions; and (4) Reactive lymph nodes with PTGCs after excluding NLPHL.
How should we render the diagnosis of reactive lymph nodes with increased IgG4+ cells?
As we emphasized above, the presence of compatible histology and immunohistochemistry (abundant IgG4+ cells and high IgG4/IgG ratio) is essential but not sufficient for a diagnosis of IgG4-LAD, and appropriate clinical findings and laboratory tests are still required for an accurate diagnosis. Many other non-IgG4-related diseases may meet these criteria, including rheumatoid arthritis, multicentric Castleman disease, and even some non-specific reactive conditions. Therefore, great caution should be taken in diagnosis of IgG4-LAD. We recommend the following algorithms in working up reactive lymph nodes with markedly increased IgG4+ cells and IgG4/IgG ratio:
1. Specific entities have to be excluded first based on the clinical features, morphologic changes and laboratory studies, including multicentric Castleman disease, rheumatoid arthritis, Rosai-Dorfman disease, and other inflammatory/infectious process. For example, if the morphology and immunohistochemistry fit well with Rosai-Dorfman disease, the nodal disease may be signed out as “Rosai-Dorfman disease with increased IgG4+ cells”; 2. If the patient has documented or newly confirmed extranodal IgG4-RD, the final diagnosis in the lymph node may be rendered as “consistent with IgG4-related lymphadenopathy”; 3. It is not uncommon that the lymphadenopathy occurs as the initial presentation in patients with no documented history of IgG4-RD, which often places great challenge to the pathologists for a definitive diagnosis. Under these circumstances, if the lymphadenopathy is systemic and/or persistent with no apparent underlying etiology, a tentative diagnosis can be made descriptively as “reactive lymphoid hyperplasia with increased IgG4+ cells; possible IgG4-related lymphadenopathy if clinical presentations and laboratory results fit”. A comment is necessary to recommend further clinical workup and correlation, including serology studies (particularly immunoglobulin fractions and levels) and possibly biopsies of extranodal lesions if present. The patient may be managed with steroids, methotrexate or rituximab if clinical treatment is required to relieve the symptoms; 4. Some patients may have localized lymphadenopathy and no documented history of extranodal IgG4-RD, and morphologically the suspicion for IgG4-LAD is not high. Therefore, it is appropriate to make a descriptive diagnosis of “reactive lymphoid hyperplasia with increased IgG4+cells; significance unclear”. In the comment, further clinical examinations, laboratory studies, and close follow-up should be advised.
Conclusion
IgG4-LAD is a clinicopathologic entity, and an accurate diagnosis has to be established based on the overall clinical presentations, laboratory studies and histologic findings. The morphologic changes and immunohistochemistry (> 100 IgG4+ cells/HPF and IgG4/IgG ratio > 40%) are essential but not sufficient for diagnosis since other non-IgG4-related conditions may fulfill these criteria and have to be excluded, including autoimmune diseases, infections, neoplastic processes, and non-specific reactive changes.
IgG4 and IgG immunostains are recommended in systemic and/or persistent reactive lymphadenopathy with no obvious underlying causes, particularly in cases with reactive plasmacytosis and PTGCs. For reactive lymph nodes with markedly increased IgG4+ cells and IgG4/IgG ratios, the interpretation has to be formulated based upon the clinical history (documented extranodal IgG4-RD or not), clinical examinations (possible presence of IgG4-RD or not), and laboratory studies (particularly levels of immunoglobulin subtypes and IgG fractions, autoimmune antibodies, and IL-6). Effective communication between the pathologists and the treating physicians is also critical in establishing a final diagnosis and for further management of patients.
Disclosure of conflict of interest
None.
References
- 1.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 2.Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, Azumi A, Bloch DB, Brugge WR, Carruthers MN, Cheuk W, Cornell L, Castillo CF, Ferry JA, Forcione D, Kloppel G, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Masaki Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani D, Sato Y, Smyrk T, Stone JR, Takahira M, Umehara H, Webster G, Yamamoto M, Yi E, Yoshino T, Zamboni G, Zen Y, Chari S. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–3067. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E, Frulloni L, Goto H, Hart PA, Kamisawa T, Kawa S, Kawano M, Kim MH, Kodama Y, Kubota K, Lerch MM, Lohr M, Masaki Y, Matsui S, Mimori T, Nakamura S, Nakazawa T, Ohara H, Okazaki K, Ryu JH, Saeki T, Schleinitz N, Shimatsu A, Shimosegawa T, Takahashi H, Takahira M, Tanaka A, Topazian M, Umehara H, Webster GJ, Witzig TE, Yamamoto M, Zhang W, Chiba T, Stone JH Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67:1688–1699. doi: 10.1002/art.39132. [DOI] [PubMed] [Google Scholar]
- 4.Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol. 2010;17:303–332. doi: 10.1097/PAP.0b013e3181ee63ce. [DOI] [PubMed] [Google Scholar]
- 5.Deshpande V. IgG4-related disease. Introduction. Semin Diagn Pathol. 2012;29:175–176. doi: 10.1053/j.semdp.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Kloppel G, Heathcote JG, Khosroshahi A, Ferry JA, Aalberse RC, Bloch DB, Brugge WR, Bateman AC, Carruthers MN, Chari ST, Cheuk W, Cornell LD, Fernandez-Del Castillo C, Forcione DG, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Lauwers GY, Masaki Y, Nakanuma Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani DV, Smyrk TC, Stone JR, Takahira M, Webster GJ, Yamamoto M, Zamboni G, Umehara H, Stone JH. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 7.Inoue D, Yoshida K, Yoneda N, Ozaki K, Matsubara T, Nagai K, Okumura K, Toshima F, Toyama J, Minami T, Matsui O, Gabata T, Zen Y. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore) 2015;94:e680. doi: 10.1097/MD.0000000000000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol. 2014;9:315–347. doi: 10.1146/annurev-pathol-012513-104708. [DOI] [PubMed] [Google Scholar]
- 9.Stone JH, Chan JK, Deshpande V, Okazaki K, Umehara H, Zen Y. IgG4-Related Disease. Int J Rheumatol. 2013;2013:532612. doi: 10.1155/2013/532612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 11.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, Hamano H, Kamisawa T, Shimosegawa T, Shimatsu A, Nakamura S, Ito T, Notohara K, Sumida T, Tanaka Y, Mimori T, Chiba T, Mishima M, Hibi T, Tsubouchi H, Inui K, Ohara H. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 12.Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74:14–18. doi: 10.1136/annrheumdis-2013-204907. [DOI] [PubMed] [Google Scholar]
- 13.Della-Torre E, Campochiaro C, Bozzolo EP, Dagna L, Scotti R, Nicoletti R, Stone JH, Sabbadini MG. Methotrexate for maintenance of remission in IgG4-related disease. Rheumatology (Oxford) 2015;54:1934–1936. doi: 10.1093/rheumatology/kev244. [DOI] [PubMed] [Google Scholar]
- 14.Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol. 2006;41:1197–1205. doi: 10.1007/s00535-006-1908-9. [DOI] [PubMed] [Google Scholar]
- 15.Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol. 2012;29:226–234. doi: 10.1053/j.semdp.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Cheuk W, Yuen HK, Chu SY, Chiu EK, Lam LK, Chan JK. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:671–681. doi: 10.1097/PAS.0b013e318157c068. [DOI] [PubMed] [Google Scholar]
- 17.Martinez LL, Friedlander E, van der Laak JA, Hebeda KM. Abundance of IgG4+ plasma cells in isolated reactive lymphadenopathy is no indication of IgG4-related disease. Am J Clin Pathol. 2014;142:459–466. doi: 10.1309/AJCPX6VF6BGZVJGE. [DOI] [PubMed] [Google Scholar]
- 18.Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, Mizobuchi K, Fujihara M, Kuraoka K, Nakai T, Ichimura K, Tanaka T, Tamura M, Nishikawa Y, Yoshino T. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol. 2009;22:589–599. doi: 10.1038/modpathol.2009.17. [DOI] [PubMed] [Google Scholar]
- 19.Kojima M, Motoori T, Asano S, Nakamura S. Histological diversity of reactive and atypical proliferative lymph node lesions in systemic lupus erythematosus patients. Pathol Res Pract. 2007;203:423–431. doi: 10.1016/j.prp.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann S, Winkelmann R, Metcalf RA, Treetipsatit J, Warnke RA, Natkunam Y, Hansmann ML. Immunoarchitectural patterns of progressive transformation of germinal centers with and without nodular lymphocyte-predominant Hodgkin lymphoma. Hum Pathol. 2015;46:1655–1661. doi: 10.1016/j.humpath.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Hansmann ML, Fellbaum C, Hui PK, Moubayed P. Progressive transformation of germinal centers with and without association to Hodgkin's disease. Am J Clin Pathol. 1990;93:219–226. doi: 10.1093/ajcp/93.2.219. [DOI] [PubMed] [Google Scholar]
- 22.Sato Y, Inoue D, Asano N, Takata K, Asaoku H, Maeda Y, Morito T, Okumura H, Ishizawa S, Matsui S, Miyazono T, Takeuchi T, Kuroda N, Orita Y, Takagawa K, Kojima M, Yoshino T. Association between IgG4-related disease and progressively transformed germinal centers of lymph nodes. Mod Pathol. 2012;25:956–967. doi: 10.1038/modpathol.2012.54. [DOI] [PubMed] [Google Scholar]
- 23.Asano N, Sato Y. Rheumatoid lymphadenopathy with abundant IgG4 (+) plasma cells : a case mimicking IgG4-related disease. J Clin Exp Hematop. 2012;52:57–61. doi: 10.3960/jslrt.52.57. [DOI] [PubMed] [Google Scholar]
- 24.Grimm KE, Barry TS, Chizhevsky V, Hii A, Weiss LM, Siddiqi IN, Brynes RK, O’Malley DP. Histopathological findings in 29 lymph node biopsies with increased IgG4 plasma cells. Mod Pathol. 2012;25:480–491. doi: 10.1038/modpathol.2011.177. [DOI] [PubMed] [Google Scholar]
- 25.Sato Y, Kojima M, Takata K, Morito T, Mizobuchi K, Tanaka T, Inoue D, Shiomi H, Iwao H, Yoshino T. Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol. 2010;63:1084–1089. doi: 10.1136/jcp.2010.082958. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Hyjek E, Vardiman J. A subset of Rosai-Dorfman disease exhibits features of IgG4-related disease. Am J Clin Pathol. 2013;139:622–632. doi: 10.1309/AJCPARC3YQ0KLIOA. [DOI] [PubMed] [Google Scholar]
- 27.Menon MP, Evbuomwan MO, Rosai J, Jaffe ES, Pittaluga SA. Subset of Rosai-Dorfman disease cases show increased IgG4-positive plasma cells: another red herring or a true association with IgG4-related disease? Histopathology. 2014;64:455–459. doi: 10.1111/his.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Perry AM, Cao W, Smith LM, Hsi ED, Liu X, Mo JQ, Dotlic S, Mosunjac M, Talmon G, Weisenburger DD, Fu K. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013;140:395–402. doi: 10.1309/AJCPFH0SJ6YILXJU. [DOI] [PubMed] [Google Scholar]
- 29.Geyer JT, Niesvizky R, Jayabalan DS, Mathew S, Subramaniyam S, Geyer AI, Orazi A, Ely SA. IgG4 plasma cell myeloma: new insights into the pathogenesis of IgG4-related disease. Mod Pathol. 2014;27:375–381. doi: 10.1038/modpathol.2013.159. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama R, Matsumoto Y, Horiike S, Kobayashi S, Nakao R, Nagoshi H, Tsutsumi Y, Nishimura A, Shimura K, Kobayashi T, Uchiyama H, Kuroda J, Taki T, Inaba T, Nishida K, Yokota S, Yanagisawa A, Taniwaki M. Close pathogenetic relationship between ocular immunoglobulin G4-related disease (IgG4-RD) and ocular adnexal mucosa-associated lymphoid tissue (MALT) lymphoma. Leuk Lymphoma. 2014;55:1198–1202. doi: 10.3109/10428194.2013.823494. [DOI] [PubMed] [Google Scholar]
- 31.Oles K, Skladzien J, Szczepanski W, Okon K, Leszczynska J, Bojanowska E, Bartus K, Mika J. Immunoglobulin G4-related disease (IgG4-RD) in the orbit: mucosa-associated lymphoid tissue (MALT)-type lymphomas. Med Sci Monit. 2015;21:1043–1050. doi: 10.12659/MSM.893043. [DOI] [PMC free article] [PubMed] [Google Scholar]