

Abstract
Background: Distal arthrogryposis (DA) is the most common congenital limb malformation secondary to the functional defects of joints and muscles. DA1 is one of the most commonly described forms of DA. The characteristics of DA1 include bilateral and symmetric clenched fist, overlapping fingers, camptodactyly, ulnar deviation of fingers, and positional foot deformities such as talipes equinovarus. Previous studies demonstrate that mutations of TPM2, TNNI2, TNNT3, MYH3 and MYBPC1 may contribute to DA1. Materials and methods: The present study investigated 8 DA1 families/patients and 1 DA2B patient, determined sequences of TPM2, TNNI2, TNNT3, MYH3 and MYBPC1 and detected the mutation by multiple sequence alignments and bioinformatic prediction of mutation. Results: We identified a novel missense mutation of TPM2 (c.463G>A; p.A155T) in a DA1 family without genetic mutant of TNNI2, TNNT3, MYH3 and MYBPC1. Conclusion: The mutation of TPM2 (c.463G>A; p.A155T) led to DA1 of the family. The identification of the mutation expands the spectrum of known TPM2 mutations, and it may contribute to novel approaches to genetic diagnosis and counseling of families with DA1.
Keywords: TPM2, mutation, DA1, DA2
Introduction
Distal arthrogryposis (DA) occurring in 1/20000 human live births, is classified as arthrogryposis multiplex congenita (AMC), and is the most common congenital limb malformation secondary to the functional defects of joints and muscles [1-3]. The typical phenotype of DA mainly is congenital joint contractures of the hands and feet. Hall et al. (1982) described DA as arthrogryposis with mainly hand and foot involvement, which was classified into 5 subtypes (type I and type IIA-IID) [4]. Then, Banshad et al. (1996) revised and extended the classification [5]. And now, DAs have been recognized and classified into 10 different forms (DA1-DA10) (Table 1) [6-8].
Table 1.
Classification of distal arthrogryposis
| Classification | OMIM number | Key features | Causative genes |
|---|---|---|---|
| DA1 | 108120 | Joint contractures of the hands and feet | TPM2, TNNI2, TNNT3, MYH3, MYBPC1 |
| DA2A | 193700 | DA with facial contractures, small pursed mouth | MYH3 |
| DA2B | 601680 | Intermediate between DA1 and DA2A | TPM2, TNNI2, TNNT3, MYH3 |
| DA3 | 114300 | DA with cleft palate, short stature | PIEZO2 |
| DA4 | 609128 | DA with scoliosis | Unknown |
| DA5 | 108145 | DA with ptosis, limited ocular mobility | ECEL1, PIEZO2 |
| DA6 | 108200 | DA with sensorineural hearing loss | Unknown |
| DA7 | 158300 | DA with trismus, facultative finger contractures | MYH8 |
| DA8 | 178110 | DA with multiple pterygium | Unknown |
| DA9 | 121050 | DA with ear deformity, long fingers | FBN2 |
| DA10 | 187370 | DA with plantar flexion contractures | Unknown |
DA, distal arthrogryposis; unknown, causative genes have been recognized.
The most commonly described forms of DA are DA1 (OMIM 108120) and DA2 [9]. The characteristics of DA1 are bilateral and symmetric clenched fist, overlapping fingers, camptodactyly, ulnar deviation of fingers, and positional foot deformities such as talipes equinovarus [10-12]. Besides aforementioned features, DA2A (OMIM 193700) also has facial phenotypes including a very small orifice, H-shaped dimpling of the chin, prominent nasolabial folds, increased philtrum length, small nose, blepharophimosis, deep-sunken eyes with hypertelorism [5,13,14]. And DA2B (OMIM 601680) has features intermediate between DA1 and DA2A [12,15].
9 genes encoding muscle proteins, ion channels and converting enzymes are recognized as causative genes of DA [7]. Mutations of TPM2, TNNI2, TNNT3 and MYH3 were identified in DA1 and DA2B, and MYBPC1 in DA1 [6,7,10,16-18]. Of these genes, TPM2 (9p13.2-p13.1) is a significant disease-causing gene of DA1. TPM2 encodes bate-tropomyosin, which is an isoform of tropomyosins [19]. The tropomyosins exist as coiled-coil homo- or heterodimers forming head-to-tail polymers, running along the length of the actin filament [20]. There are 7 alternating actin bonding sites in every tropomyosin [19]. It stabilizes the actin filament of the sarcomere and mediates the interactions between the troponin complex and actin to regulate muscle contraction in the striated muscle [21].
In this study, we collected 9 cases including 8 DA1 families/patients and 1 DA2B patient and investigated the potential causative gene of them by sequence analysis of TPM2, TNNI2, TNNT3, MYH3 and MYBPC1.
Materials and methods
Patients
We collected 8 DA1 families/patients and 1 DA2B patient who went to Xiangya Hospital in 2016. The DA1 family of 8 living members, which was indentified mutations of diseasing-cause gene, is from Hunan province, China. Except 3 adults absent for working in other cities, 5 of these members across three generations participated in this study (Figure 1A). The proband, family member 3 from the 3rd generation (III: 3), I: 2 and III: 1 were diagnosed with DA1. The remaining 2 members (II: 4 and III: 2) were phenotypically normal. The proband (3-year-old) had congenital ulnar bilateral and symmetric arthrogryposes of 2-5 fingers without other malformation, I: 2 had arthrogryposes of 5th fingers, and III: 1 had arthrogryposes of 4/5 fingers (Figure 1C). In addition, it is said that II: 2 and II: 3 had DA, but I: 1 and II: 1 was normal. The Review Board of Xiangya Hospital of the Central South University (Hunan, China) approved this research and all family members involved gave written informed consent.
Figure 1.
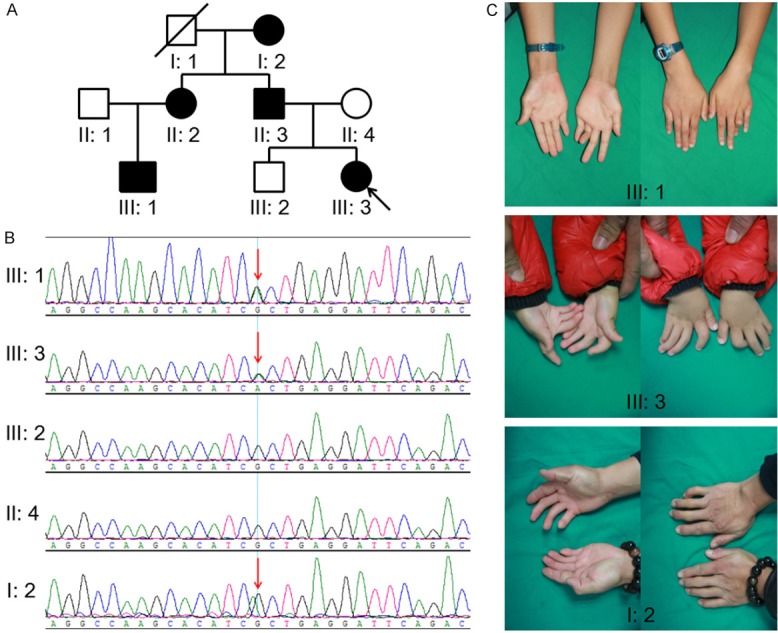
A. Pedigree of the family affected with AD1. Family members are identified by generations and number. Squares indicate male family members; circles, female members; a closed symbol, the affected member; open symbols, unaffected members; arrow, proband. B. Sequencing result of the TPM2 mutation. Sequence chromatogram indicates a heterozygous mutation (c.463G>A; p.A155T). C. Phenotypes of the part patients. III: 1 had arthrogryposes of 4/5 fingers, III: 3 had arthrogryposes of 2-5 fingers, I: 2 had arthrogryposes of 5th fingers.
DNA extraction
Genomic DNA was extracted from the peripheral blood of the patient and the other family members using a DNeasy Blood & Tissue kit (Qiagen, Inc., Valencia, CA, USA) on the QIAcube automated DNA extraction robot (Qiagen, Inc.).
Mutation sequencing
The entire coding regions, including the flanking intronic sequences of TPM2 [Refseq (https://www.ncbi.nlm.nih.gov/refseq/), NM_213674], TNNI2 (NM_001145829), TNNT3 (NM_001297646), MYH3 (NM_002470) and MYBPC1 (NM_002465) were amplified by polymerase chain reaction (PCR; primer sequences will be provided upon request). PCR product sequences were determined using the ABI 3100 Genetic Analyzer (Thermo Fisher Scientific, Inc., Waltham, MA, USA) as previously described [22].
Multiple sequence alignments and bioinformatic prediction of mutation
The multiple TPM2 protein sequences across mammals were aligned using the multiple sequence comparison by log-expectation program (version 3.6; an online program at http://www.ncbi.nlm.nih.gov) [23].
Results
The present study investigated 8 DA1 families/patients and 1 DA2B patient and confirmed a novel mutation in 1 DA1 family from Hunan province, China. There are 8 living members in this family, and 5 of them across three generations participated in this study. The proband (III: 4), I: 2 and III: 1 were diagnosed with DA1 characterized by congenital ulnar bilateral and symmetric arthrogryposes of fingers. These arthrogryposes involved 2-5 fingers, 5th fingers and 4/5 fingers in III: 4, I: 2 and III: 1, respectively. Furthermore, II: 2 and II: 3, absent from this research, were narrated having ulnar arthrogryposes of fingers. All potential causative genes among all families/patients including TPM2, TNNI2, TNNT3, MYH3 and MTBPC1, are investigated and only a mutation is identified in the 8-member family emphasized above. It is a novel heterozygous missense mutation (c.463G>A; p.A155T) in exon 4 of TPM2, and co-segregated with the affected family members (Figure 1B). No further relevant mutations were identified by direct sequencing of the genes for TNNI2, TNNT3, MYH3 and MYBPC1. This newly discovered mutation of TPM2 (c.463G>A; p.A155T) was not identified in the 200 control cohorts that our group studied previously. In addition, this mutation was not present in the dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) or Exome Variant Server databases (http://evs.gs.washington.edu/EVS).
Discussion
Minttu et al. (2014) summarized the genotype-phenotype correlations of mutations in TPM2 [20]. He identified the mutation (A155T, our identified mutation) in 2 “congenital myopathy patients lacking clinical details” from different families in a supplemental table, but he didn’t provide information of these families and sequence chromatograms. The present study identified the mutation in a DA1 family and co-segregated with the affected family members. Therefore, our study at least confirmed the existence and pathogenicity of the mutation.
TPM2 belongs to the family of tropomyosin genes (including TPM1-4), and encodes the skeletal muscle isoforms Tm2 [20,24-27]. The isoform has 284 amino acid residues and a highly conserved N-terminal region [28]. TPM2 is a major causative gene of congenital myopathies and had been reported at least 35 mutations (Figure 2). The phenotypes resulting from these mutations are observably heterogeneous, mainly including DA1, DA2B, nemaline myopathy (NM), congenital fibre type disproportion (CFTD), cap disease and so on [12,29,30]. For example, the mutation (A3G) was reported causing NM, G97K causing DA1, and R105P causing DA2B [12,20]. Moreover, the change of the same amino acid could lead to different diseases: E41K could cause NM, CFTD or cap disease [20,31]. R133W could cause DA2B or NM, but R133P cause CFTD [9,32]. A155V was reported to lead to CFTD by Nigel et al., but our identified mutation (A155T) resulted in symptoms of DA1 [33]. Although, phenotypes resulting from the change of 155th AA are different, the result of Nigel aspect demonstrated the pathogenicity of the mutation (A155T). It indicates that the genotype-phenotype correlations of mutations in TPM2 need more clinical date and research to support.
Figure 2.
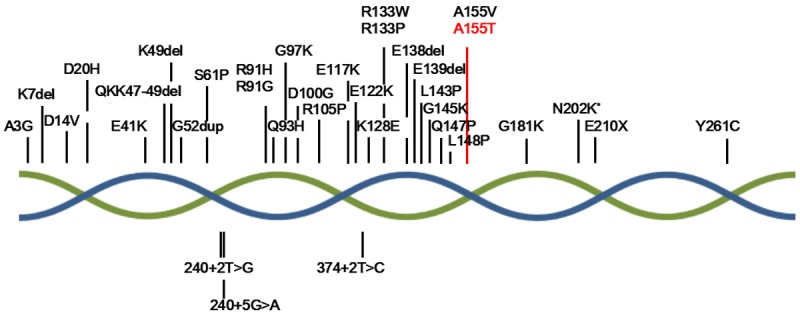
TPM2 disease mutations in congenital myopathy patients. The double helix represents the tropomyosin consisted by 2 bate-tropomyosin. The red word represents our mutation. *indicates the mutation caused by change of two different bases.
TPM2, TNNI2, TNNT3 and MYH3 had been recognized as disease-causing genes of DA1 and DA2B, and mutations of MYBPC1 were identified in DA1. In this study, of 8 DA1 families/patients and 1 DA2B patient, we only identified a mutation of TPM2 in a DA1 family by sequence analysis of aforementioned genes. All forms of arthrogryposis are associated with decreased fetal akinesia [34]. Possible etiology and potential cause of fetal akinesia including: 1) myopathic processes, 2) neuropathic processes, 3) neuromuscular endplate abnormalities, 4) abnormalities of connective tissue, 5) the restriction of movement in utero, 6) maternal illness or exposures, 7) intrauterine vascular compromise, and 8) metabolic disturbances [35,36]. The pathopoiesis of 8 remaining cases need to be proved by further study.
In conclusion, the present study identified a novel heterozygous missense mutation (c.463G>A; p.A155T) of TPM2 in a Chinese family with DA1. The present identification of the mutation expands the spectrum of known TPM2 mutations, and it may contribute to novel approaches to genetic diagnosis and counseling of families with DA1.
Acknowledgements
The National Natural Science Foundation of China (81370394) and the Fundamental Research Funds for Central Universities of Central South University (2016zzts581). We thank the patients and their families for participating in this study.
Disclosure of conflict of interest
None.
References
- 1.Darin N, Kimber E, Kroksmark AK, Tulinius M. Multiple congenital contractures: birth prevalence, etiology, and outcome. J Pediatr. 2002;140:61–67. doi: 10.1067/mpd.2002.121148. [DOI] [PubMed] [Google Scholar]
- 2.Hall JG. Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach, and general aspects. J Pediatr Orthop B. 1997;6:159–166. [PubMed] [Google Scholar]
- 3.Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M. A mutation in the fast skeletal muscle troponin I gene causes myopathy and distal arthrogryposis. Neurology. 2006;67:597–601. doi: 10.1212/01.wnl.0000230168.05328.f4. [DOI] [PubMed] [Google Scholar]
- 4.Hall JG, Reed SD, Greene G. The distal arthrogryposes: delineation of new entities--review and nosologic discussion. Am J Med Genet. 1982;11:185–239. doi: 10.1002/ajmg.1320110208. [DOI] [PubMed] [Google Scholar]
- 5.Bamshad M, Jorde LB, Carey JC. A revised and extended classification of the distal arthrogryposes. Am J Med Genet. 1996;65:277–281. doi: 10.1002/(SICI)1096-8628(19961111)65:4<277::AID-AJMG6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 6.Culic V, Miyake N, Jankovic S, Petrovic D, Simunovic M, Dapic T, Shiina M, Ogata K, Matsumoto N. Distal arthrogryposis with variable clinical expression caused by TNNI2 mutation. Hum Genome Var. 2016;3:16035. doi: 10.1038/hgv.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang B, Zheng Z, Wang Z, Zhang X, Yang H, Cai H, Fu Q. A novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1. Am J Med Genet A. 2016;170A:135–141. doi: 10.1002/ajmg.a.37391. [DOI] [PubMed] [Google Scholar]
- 8.Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009;91(Suppl 4):40–46. doi: 10.2106/JBJS.I.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M. Distal arthrogryposis: clinical and genetic findings. Acta Paediatr. 2012;101:877–887. doi: 10.1111/j.1651-2227.2012.02708.x. [DOI] [PubMed] [Google Scholar]
- 10.Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI, Talerico M, Miller SE, Ju JS, Pestronk A, Connolly AM, Druley TE, Weihl CC, Dobbs MB. Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Hum Mol Genet. 2010;19:1165–1173. doi: 10.1093/hmg/ddp587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bamshad M, Bohnsack JF, Jorde LB, Carey JC. Distal arthrogryposis type 1: clinical analysis of a large kindred. Am J Med Genet. 1996;65:282–285. doi: 10.1002/(SICI)1096-8628(19961111)65:4<282::AID-AJMG7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 12.Beck AE, McMillin MJ, Gildersleeve HI, Kezele PR, Shively KM, Carey JC, Regnier M, Bamshad MJ. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet A. 2013;161A:550–555. doi: 10.1002/ajmg.a.35809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, Ashley CC, Redwood CS. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21:896–905. doi: 10.1096/fj.06-6899com. [DOI] [PubMed] [Google Scholar]
- 14.Stevenson DA, Carey JC, Palumbos J, Rutherford A, Dolcourt J, Bamshad MJ. Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics. 2006;117:754–762. doi: 10.1542/peds.2005-1219. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen S, Siu R, Dewey S, Cui Z, Gomes AV. Amino acid changes at arginine 204 of troponin i result in increased calcium sensitivity of force development. Front Physiol. 2016;7:520. doi: 10.3389/fphys.2016.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mroczek M, Kabzinska D, Chrzanowska KH, Pronicki M, Kochanski A. A novel TPM2 gene splice-site mutation causes severe congenital myopathy with arthrogryposis and dysmorphic features. J Appl Genet. 2017;58:199–203. doi: 10.1007/s13353-016-0368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daly SB, Shah H, O’Sullivan J, Anderson B, Bhaskar S, Williams S, Al-Sheqaih N, Mueed Bidchol A, Banka S, Newman WG, Girisha KM. Exome sequencing identifies a dominant TNNT3 mutation in a large family with distal arthrogryposis. Mol Syndromol. 2014;5:218–228. doi: 10.1159/000365057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Racca AW, Beck AE, McMillin MJ, Korte FS, Bamshad MJ, Regnier M. The embryonic myosin R672C mutation that underlies Freeman-Sheldon syndrome impairs cross-bridge detachment and cycling in adult skeletal muscle. Hum Mol Genet. 2015;24:3348–3358. doi: 10.1093/hmg/ddv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis WG, Smillie LB. The amino acid sequence of rabbit cardiac tropomyosin. J Biol Chem. 1980;255:6854–6859. [PubMed] [Google Scholar]
- 20.Marttila M, Lehtokari VL, Marston S, Nyman TA, Barnerias C, Beggs AH, Bertini E, Ceyhan-Birsoy O, Cintas P, Gerard M, Gilbert-Dussardier B, Hogue JS, Longman C, Eymard B, Frydman M, Kang PB, Klinge L, Kolski H, Lochmuller H, Magy L, Manel V, Mayer M, Mercuri E, North KN, Peudenier-Robert S, Pihko H, Probst FJ, Reisin R, Stewart W, Taratuto AL, de Visser M, Wilichowski E, Winer J, Nowak K, Laing NG, Winder TL, Monnier N, Clarke NF, Pelin K, Gronholm M, Wallgren-Pettersson C. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum Mutat. 2014;35:779–790. doi: 10.1002/humu.22554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolska BM, Wieczorek DM. The role of tropomyosin in the regulation of myocardial contraction and relaxation. Pflugers Arch. 2003;446:1–8. doi: 10.1007/s00424-002-0900-3. [DOI] [PubMed] [Google Scholar]
- 22.Tan ZP, Huang C, Xu ZB, Yang JF, Yang YF. Novel ZFPM2/FOG2 variants in patients with double outlet right ventricle. Clin Genet. 2012;82:466–471. doi: 10.1111/j.1399-0004.2011.01787.x. [DOI] [PubMed] [Google Scholar]
- 23.Xiang R, Fan LL, Huang H, Zhao SP, Chen YQ. Whole-exome sequencing identifies a novel mutation of DSG2 (Y198C) in a Chinese arrhythmogenic right ventricular cardiomyopathy patient. Int J Cardiol. 2016;214:1–3. doi: 10.1016/j.ijcard.2016.03.136. [DOI] [PubMed] [Google Scholar]
- 24.Gateva G, Kremneva E, Reindl T, Kotila T, Kogan K, Gressin L, Gunning PW, Manstein DJ, Michelot A, Lappalainen P. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr Biol. 2017;27:705–713. doi: 10.1016/j.cub.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pleines I, Woods J, Chappaz S, Kew V, Foad N, Ballester-Beltran J, Aurbach K, Lincetto C, Lane RM, Schevzov G, Alexander WS, Hilton DJ, Astle WJ, Downes K, Nurden P, Westbury SK, Mumford AD, Obaji SG, Collins PW, Delerue F, Ittner LM, Bryce NS, Holliday M, Lucas CA, Hardeman EC, Ouwehand WH, Gunning PW, Turro E, Tijssen MR, Kile BT. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest. 2017;127:814–829. doi: 10.1172/JCI86154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weymouth KS, Blanton SH, Powell T, Patel CV, Savill SA, Hecht JT. Functional assessment of clubfoot associated HOXA9, TPM1, and TPM2 variants suggests a potential gene regulation mechanism. Clin Orthop Relat Res. 2016;474:1726–1735. doi: 10.1007/s11999-016-4788-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan H, Gu L, Liu B, Li Y, Wang Y, Bai X, Li L, Wang B, Peng Q, Yao Z, Tang Z. Tropomyosin-1 acts as a potential tumor suppressor in human oral squamous cell carcinoma. PLoS One. 2017;12:e0168900. doi: 10.1371/journal.pone.0168900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tajsharghi H, Ohlsson M, Lindberg C, Oldfors A. Congenital myopathy with nemaline rods and cap structures caused by a mutation in the beta-tropomyosin gene (TPM2) Arch Neurol. 2007;64:1334–1338. doi: 10.1001/archneur.64.9.1334. [DOI] [PubMed] [Google Scholar]
- 29.Jarraya M, Quijano-Roy S, Monnier N, Behin A, Avila-Smirnov D, Romero NB, Allamand V, Richard P, Barois A, May A, Estournet B, Mercuri E, Carlier PG, Carlier RY. Whole-Body muscle MRI in a series of patients with congenital myopathy related to TPM2 gene mutations. Neuromuscul Disord. 2012;22(Suppl 2):S137–147. doi: 10.1016/j.nmd.2012.06.347. [DOI] [PubMed] [Google Scholar]
- 30.Sung SS, Brassington AM, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA, Jorde LB, Carey JC, Bamshad M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet. 2003;72:681–690. doi: 10.1086/368294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ochala J, Li M, Ohlsson M, Oldfors A, Larsson L. Defective regulation of contractile function in muscle fibres carrying an E41K betatropomyosin mutation. J Physiol. 2008;586:2993–3004. doi: 10.1113/jphysiol.2008.153650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karpicheva OE, Simonyan AO, Kuleva NV, Redwood CS, Borovikov YS. Myopathy-causing Q147P TPM2 mutation shifts tropomyosin strands further towards the open position and increases the proportion of strong-binding cross-bridges during the ATPase cycle. Biochim Biophys Acta. 2016;1864:260–267. doi: 10.1016/j.bbapap.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Clarke NF, Waddell LB, Sie LT, van Bon BW, McLean C, Clark D, Kornberg A, Lammens M, North KN. Mutations in TPM2 and congenital fibre type disproportion. Neuromuscul Disord. 2012;22:955–958. doi: 10.1016/j.nmd.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Riemersma S, Vincent A, Beeson D, Newland C, Hawke S, Vernet-der Garabedian B, Eymard B, Newsom-Davis J. Association of arthrogryposis multiplex congenita with maternal antibodies inhibiting fetal acetylcholine receptor function. J Clin Invest. 1996;98:2358–2363. doi: 10.1172/JCI119048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall JG. Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur J Med Genet. 2014;57:464–472. doi: 10.1016/j.ejmg.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Xiao Y, Zuo X, You Y, Luo H, Duan L, Zhang W, Li Y, Xie Y, Zhou Y, Ning W, Li T, Liu S, Zhu H, Jiang Y, Wu S, Zhao H. Investigation into the cause of mortality in 49 cases of idiopathic inflammatory myopathy: a single center study. Exp Ther Med. 2016;11:885–889. doi: 10.3892/etm.2016.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]