

Abstract
Oral squamous cell carcinoma (OSCC) is a common and aggressively malignant tumor of the head and neck region. Long non-coding RNAs (lncRNAs) are important regulatory molecules in many types of cancer. However, there are limited studies on the role of lncRNAs in OSCC. In this study, we identified that lncRNA P4713 was one of the most up-regulated lncRNAs in OSCC by exploring the expression profile of lncRNAs/mRNAs in four pairs of OSCC samples and adjacent non-cancer tissues. In addition, silencing of P4713 inhibited OSCC proliferation, migration, and invasion in vitro. Furthermore, through bioinformatics analysis and functional experiments, we found that decreased P4713 expression affected the expression and phosphorylation of Janus Kinase (JAK) 2 and signal transducer and activator of transcription (STAT) 3. Taken together, the results suggest that P4713 contributes to OSCC cell proliferation, migration, and invasion by activating the JAK/STAT3 pathway. Accordingly, this molecule could be a potential biomarker and therapeutic target in the treatment of OSCC.
Keywords: OSCC, lncRNA-P4713, JAK/STAT3 pathway, proliferation, migration, invasion
Introduction
Oral squamous cell carcinoma (OSCC) is one of the most common malignancies of the head and neck region, accounting for approximately 90% of oral cancers [1]. Approximately 274,000 new cases are reported worldwide each year. Although valid advancements have been made in the study of OSCC, its mortality rate has not significantly decreased [2-5]. Mounting evidence reveals that oral carcinogenesis is a complicated process that involves multiple epigenetic and genetic alterations in the expression of both coding and non-coding RNAs [6]. However, most studies have focused on protein-coding genes, with only limited studies on non-coding RNA, particularly long non-coding RNA (lncRNA).
Long non-coding RNAs (lncRNAs) are defined as transcripts more than 200 bp without evident protein coding capacity [7]. With the widespread use of high throughput sequencing technology and bioinformatics, a growing number of lncRNAs have been discovered in different types of human cancer [8,9]. These molecules can localize to the nucleus, cytoplasm, or both [10]. In addition, they can function as either oncogenes or tumor suppressors [11]. Moreover, recent studies have reported that many lncRNAs are dysregulated in OSCC. These differentially expressed lncRNAs could play important roles in OSCC progression, but their precise mechanisms remain unclear [12-15].
Janus kinase (JAK) is a tyrosine kinase that meditates signal transducer and activator of transcription (STAT) signaling [16]. STAT3 is one of the seven members of the STAT protein family. It is primarily phosphorylated by activated JAK1 and 2 at tyrosine 705 [17,18] in the cytoplasm [19]. Phosphorylated STAT3 subsequently dimerizes and translocates to the nucleus, where it acts as a transcription factor. JAK/STAT3 signaling plays critical roles in tumor cell proliferation, survival, angiogenesis, and invasion [19-21].
In this study, we discovered a novel long non-coding RNA termed P4713 in OSCC based on analysis of lncRNA microarray data. Through RNA interference (RNAi) experiments, P4713 was found to contribute to OSCC cell proliferation, migration, and invasion. Furthermore, we demonstrated that P4713 might contribute to OSCC progression by targeting JAK/STAT3 signaling. Our results provide novel insight into the underlying molecular mechanisms of OSCC. These results indicate that P4713 may serve as a promising target for its diagnosis and treatment.
Materials and methods
Patients and samples
Twenty-six paired OSCC and adjacent non-tumor samples were collected from the Department of Craniofacial Surgery, Guanghua School of Stomatology, Sun Yat-sen University (Guangzhou, Guangdong, China). All samples were immediately frozen in liquid nitrogen and stored at -80°C. Informed consent was obtained from all patients. The study was approved by the Ethics Committee of Guanghua School of Stomatology, Sun Yat-sen University. Tumor and non-tumor samples were confirmed by pathological examination.
Microarray analysis
The OSCC lncRNA/mRNA microarray analysis was performed by CapitalBio Corporation (Beijing, China). Differentially expressed lncRNAs/mRNAs with statistical significance (P < 0.05; T/N fold change [FC] > 1.5) were identified by performing paired t-tests after comparing normalized expression levels in tumor and non-tumor samples.
Bioinformatics, coding potential of lncRNA-P4173, and plasmid construction
The evolutionary conservation of P4713 was analyzed using the University of California Santa Cruz (UCSC) Genome Browser (genome.ucsc.edu). The coding potential of P4713 was analyzed using LNCipedia.org. A P4713 expression vector was constructed through PCR-based amplification of cDNA from HSC-3 cells and subsequent subcloning into the ORF of the eukaryotic expression vector pReceiver-M29, which incorporates an EGFP tag such that P4713 and EGFP shared the same CMV promoter. pReceiver-M02R with only the EGFP tag and pReceiver-M02R with P4713 but without the EGFP tag served as controls. All constructs were confirmed by DNA sequencing.
Cell culture
The human OSCC cell line UM1 was provided by Dr. Xiaofeng Zhou (University of Illinois at Chicago, IL, USA), and HSC-3 (OSCC) and normal oral keratinocyte (NOK) cell lines were kindly provided by J. Silvio Gutkind (National Institutes of Health, Bethesda, MD, USA). The human OSCC cell lines SCC25 and CAL27 were obtained from ATCC (Rockville, MD, USA). The HEK 293 cell line was purchased from the National Centre for Cell Science (NCCS; Pune, India). HSC-3, CAL27, and HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium/high glucose (DMEM; Gibco, Rockville, MD, USA) with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA). SCC15 and UM1 cells were cultivated in DMEM/F12(1:1) (Gibco) supplemented with 10% FBS. NOK cells were maintained in keratinocyte serum-free medium (KSFM; Gibco). All cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C.
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted from tissues or cells using the miRNeasy Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. Total RNA was reverse-transcribed using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). SYBR GREEN I Master Mix (Roche, Mannheim, Germany) and a Light Cycler 480 system (Roche) were used for qRT-PCR, which was performed in triplicate. The sequences of primers were shown in Table 1. Relative expression levels were calculated using the 2-ΔΔCt method after normalization to GAPDH.
Table 1.
Primer sets used in the qRT-PCR
| Gene name | Primer (5’→3’) |
|---|---|
| GAPDH sense | GCACCGTCAAGGCTGAGAAC |
| GAPDH antisense | TGGTGAAGACGCCAGTGGA |
| lncRNA P4713 sense | GGTGACGGTGTCGTGGAA |
| lncRNA P4713 antisense | GCAGGTGTAGGTCTGGGTG |
| JAK2 sense | CAGGCAACAGGAACAAGATG |
| JAK2 antisense | CCATTCCCATGCAGAGTCTT |
| STAT3 sense | CCTCTGCCGGAGAAACAG |
| STAT3 antisense | CTGCTCCAGGTACCGTGTGT |
Small interfering RNA (siRNA) transfection
Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, CA, USA) was used to transfect 50 nM P4713 or negative control (NC) siRNA (RiboBio, Guangzhou, China) into OSCC cells, according to the manufacturer’s instructions. Target sequences for P4713 siRNAs and negative control were shown in Table 2. Cells were harvested after 48 h for qRT-PCR and western blot analyses.
Table 2.
Target sequences of P4713 siRNAs and negative control
| Name | Target sequence |
|---|---|
| P4713 siRNA-1 | CGCCGGTTTCAATCCTGTT |
| P4713 siRNA-2 | GGAAGACAAATAGCAGCTG |
| Negative Control | GCTGACCCTGAAGTTCATCTG |
Proliferation assays
Cell proliferation was assayed using the Cell Counting Kit-8 (CCK-8; Dojindo Lab, Kumamoto, Japan). Cells were seeded into 96-well plates at 2 × 103 cells/well in triplicate. Cell growth was assessed at 12, 24, 48, 72, and 96 h post-transfection. Absorbance was measured at 450 nm using a microplate reader (Genios TECAN, Männedorf, Switzerland).
Colony-formation assays
Single-cell suspensions were prepared 48 h post-transfection by trypsinization, and 1 × 103 cells were plated in 6-well plates. After incubation for 14 d, visible colonies were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet. Colonies with more than 50 cells were counted using Image J software (NIH, Bethesda, MD).
Cell cycle analysis
Cell cycle progression was measured using a Cell Cycle Staining Kit (MultiSciences Biotech Co, Ltd, China). Transiently transfected cells (48 h post-transfection) were collected, trypsinized, and resuspended in cold phosphate-buffered saline (PBS) in triplicate. The cells were stained with DNA staining solution and permeabilization solution. Cell cycle distribution was analyzed using the FC500 flow cytometer and MXP software (Beckman Coulter, Brea, CA, USA).
Migration and invasion assays
For transwell migration and invasion assays, we used BD 24-well transwell units (BD Bioscience, Miami, FL, USA) coated with or without Matrigel according to the manufacturer’s protocol. Cells (6-8 × 104) were resuspended in 200 µl serum-free medium and added to the upper compartment of inserts 48 h post- transfection; 700 µl of medium containing 10% FBS was added to the lower chamber as a chemoattractant. After incubation for 24 h, non-migrating/invading cells in the upper inserts were gently removed with a cotton swab and migrating cells were fixed with 4% formaldehyde and stained with 0.2% crystal violet (Sigma, Santa Clara, CA, USA). Stained cells were imaged using a Zeiss microscope, and cells in five random fields per chamber were quantified. Experiments were conducted in triplicate.
Wound healing assays
UM1 and HSC-3 cells transfected with si-P4713-1, si-P4713-2, or si-NC were seeded into six-well plates. When cell growth reached approximately 90% confluence, the cell monolayer was scratched using a 200 µl pipette tip and dislodged cells were washed away with PBS. The gap area was measured at 0 and 24 h using a Zeiss microscope.
Western blotting
Cells were lysed in RIPA buffer (Sigma-Aldrich) containing protease inhibitors (Cell Signaling Technology, Danvers, MA, USA). Protein concentrations were measured using the BCA Protein Assay kit (Sigma-Aldrich). Equal amounts (40 µg) of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, MA, USA). After blocking for 1 h at room temperature in 5% non-fat milk, the membranes were incubated with specific primary antibodies overnight at 4°C. The membrane was washed with Tris-buffered saline containing Tween 20 and incubated with HRP-conjugated secondary antibody for 1 h. Bound antibodies were visualized using the enhanced chemiluminescence (ECL) detection system (Millipore, MA, USA). Three individual experiments were conducted. The following primary antibodies were used: JAK2 (1:1000), p-JAK2 (Tyr1007/1008; 1:1000), STAT3 (1:1000), p-STAT3 (Tyr705; 1:1000), E-cadherin (1:1000), N-cadherin (1:1000), vimentin (1:1000), cyclin D1 (1:1000), cyclin-dependent kinase 4 (CDK4; 1:1000), cyclin-dependent kinase 6 (CDK6; 1:1000); all were purchased from Cell Signaling Technology (Beverly, MA, USA).
Fluorescent in situ hybridization (FISH) analysis
To observe the location of lncRNA-P4713, HSC-3 cells were rinsed briefly with PBS and fixed in 4% formaldehyde for 15 min. Subsequently, cells were permeabilized with 0.1% Triton X-100 and washed with PBS. Hybridization was performed using digoxin-labeled (Roche) lncRNA-P4713 cDNA probes in a moist chamber at 37°C overnight. After hybridization, cells were incubated with anti-digoxin-rhodamine antibody (Roche) and subjected to confocal microscopy. For colocalization after RNA-FISH, cells were again fixed for 5 min in 2% formaldehyde and subjected to immunofluorescence staining using 4’, 6-diamidino-2-phenylindole (DAPI). Cells were then observed with a Zeiss (LSM 780) confocal laser scanning microscope.
Nuclear and cytoplasmic RNA extraction
Nuclear and cytoplasmic RNA fractions were isolated using the Ambion PARIS™ kit (Carlsbad, CA, USA). Briefly, 3 × 105 cultured cells were collected and washed once in ~1 ml PBS and placed on ice. PBS was removed and 500 μl of ice-cold cell disruption buffer was added to the cells. Cells were vortexed vigorously for complete lysis and incubated on ice for 5-10 min. Samples were centrifuged for 5 min at 4°C and 500 × g. The supernatant, containing the cytoplasmic fraction, was carefully collected, avoiding the pellet. Next, 500 μl of ice-cold Cell Disruption Buffer was added to the nuclear pellet and 700 μl QIAzol Lysis Reagent was added to the cytoplasmic and nuclear fractions. RNA isolation and RT-PCR were performed as described previously.
Immunofluorescence
UM1 and HSC-3 cells were maintained on 15-mm confocal dishes (Nest, Jiangsu, China) in an incubator with complete culture medium for 48 h after transfection with siRNA. Cells were washed with PBS three times and fixed in 4% paraformaldehyde for 15 min at 37°C. Subsequently, cell membranes were permeabilized using 0.1% Triton X-100 for 15 min. Cells were then blocked with PBS containing 5% BSA for 30 min, and incubated with an anti-STAT3 antibody (1:400 dilution) in 1% BSA overnight. Cells were washed three times with PBS containing Tween 20 at room temperature, followed by incubation with secondary antibody conjugated with AlexaFluor 488. The samples were washed three times with PBS, and the cell nuclei were stained with 1 mg/ml DAPI (Cell Signaling Technology) for 5 min. Finally, the confocal dishes were washed and imaged using a Zeiss (LSM 780) microscope.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (La Jolla, CA, USA). Data are presented as the means ± SD. Student’s t-tests were used to compare two groups. A P-value < 0.05 was considered statistically significant.
Results
P4713 expression is upregulated in OSCC tissues and cell lines
A human lncRNA microarray and hierarchical clustering analysis were used to compare lncRNA expression between four OSCC tissues and paired adjacent non-tumor tissues (Figure 1A). Among the differentially expressed lncRNAs, we selected one significantly up-regulated lncRNA, namely P4713 (upregulated > 30-fold). To verify the microarray data, the relative expression of P4713 was confirmed by qRT-PCR in 22 OSCC tissues and paired adjacent non-cancerous tissues (Figure 1B). Furthermore, P4713 expression was higher in SCC-25, Cal-27, and particularly UM1 and HSC-3 cells compared to NOK cells (Figure 1C). Moreover, qRT-PCR and FISH assays demonstrated that P4713 was mainly enriched in nuclear fractions (Figure 1D). P4713 is located on chromosome 14 (Figure 1E), and its coding potential was suggested to be weak. The absence of protein expression was confirmed experimentally by confocal microscopy and western blot analysis (Figure 1F, 1G). Taken together, the results demonstrate that P4713 is a lncRNA that is upregulated in OSCC.
Figure 1.
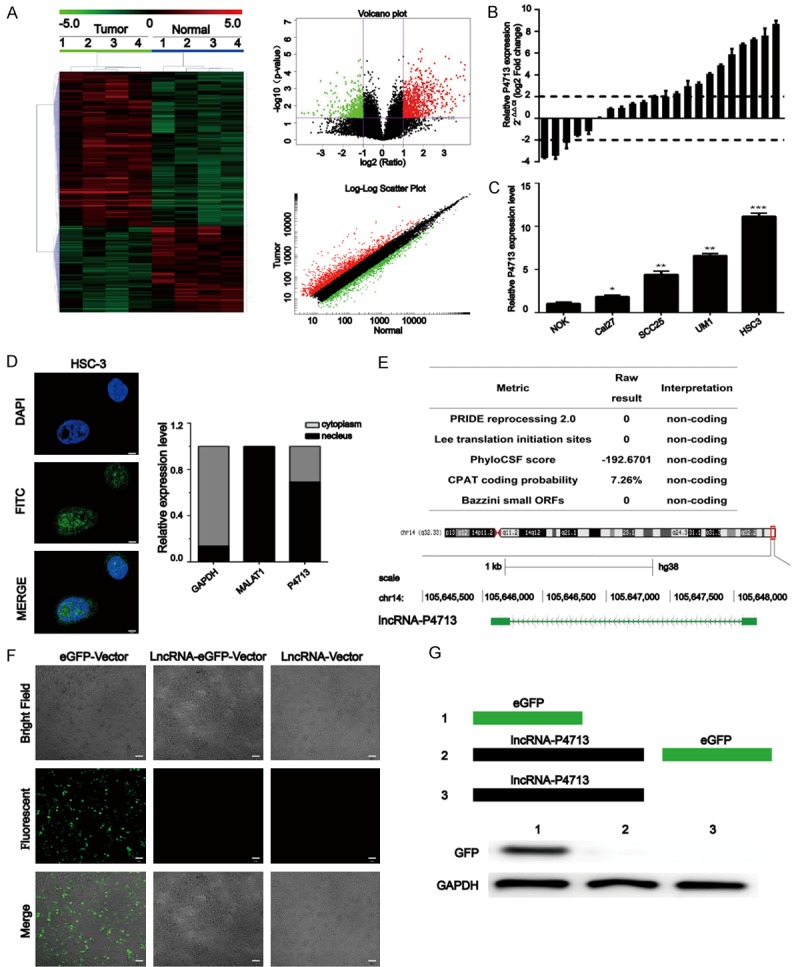
Analysis of long non-coding RNAs (lncRNAs) in oral squamous cell carcinoma (OSCC). A: Heat map, volcano plot, and log-log scatter plot showing differentially expressed lncRNAs between four OSCC samples and paired adjacent normal tissues (fold change > 2.0, P < 0.05). B: The expression of lncRNA P4713 was detected by quantitative real-time PCR (qRT-PCR) in 22 OSCC tissues and adjacent non-cancerous tissues. The results are expressed as log10 (2-ΔΔCt). A log2 fold change ≥ +2 or ≤ -2 was considered significant upregulation or downregulation (dotted lines). C: Relative P4713 expression in OSCC cell lines was measured by qRT-PCR. Columns represent the mean of three independent experiments; bars, the s.d; *P < 0.05; **P < 0.01. D: Confocal microscopic fluorescent in situ hybridization images and qRT-PCR results. Scale bar = 10 µm. E: Genome location analysis of human P4713 by the UCSC Genome Browser. F: Representative fluorescent images of at least three independent experiments. Scale bar = 10 µm. G: Relative levels of GFP expression by Western blot.
Silencing of P4713 suppresses OSCC cell proliferation in vitro
To investigate whether P4713 contributes to OSCC malignant phenotypes, we performed P4713 RNAi in vitro and observed the effects on cell proliferation. First, qRT-PCR was used to confirm the efficiency of the siRNAs in UM1 and HSC-3 cells (Figure 2A). CCK-8 assays showed that depletion of P4713 significantly decreased the proliferation rates of UM1 and HSC-3 cells at 72 and 96 h (P < 0.05; Figure 2B). In addition, plate colony-formation assays indicated markedly decreased UM1 and HSC-3 colonies after transfection with P4713 siRNA (Figure 2C). Knockdown of P4713 induced G0/G1 arrest (P < 0.05; Figure 2D). Consistently, cyclin D1, CDK4, and, CDK6 were significantly downregulated in UM1 and HSC-3 cells transfected with si-P4713 compared to cells transfected with si-NC (Figure 2E). These results suggest that P4713 might facilitate OSCC proliferation.
Figure 2.

The effects of P4713 on oral squamous cell carcinoma cell proliferation in vitro. A: The relative expression of P4713 was examined by qRT-PCR in HSC-3 and UM1 cells. B: Cell proliferation was measured by CCK-8 assay. C: Detection for colony-formation assays after knockdown of P4713. D: Cell cycle analysis using propidium iodide staining. E: Western blot analysis of cyclin D1, CDK4, and CDK6 after P4713 knockdown.
Knockdown of P4713 inhibits the migration and invasion of OSCC cells in vitro
To assess the role of P4713 in OSCC migration and invasion, wound healing experiments and transwell assays were performed. Knockdown of P4713 caused a significant reduction in the number of migrating cells (P < 0.05; Figure 3A). In Matrigel invasion experiments, the number of invading cells decreased after treatment with P4713 siRNA (Figure 3B). Wound healing assays revealed that P4713 knockdown inhibited cell motility (Figure 3C). In addition, E-cadherin, N-cadherin, and vimentin protein levels were significantly decreased after P4713 depletion (Figure 3D). These results indicate that P4713 could regulate the migration and invasion of OSCC cells.
Figure 3.
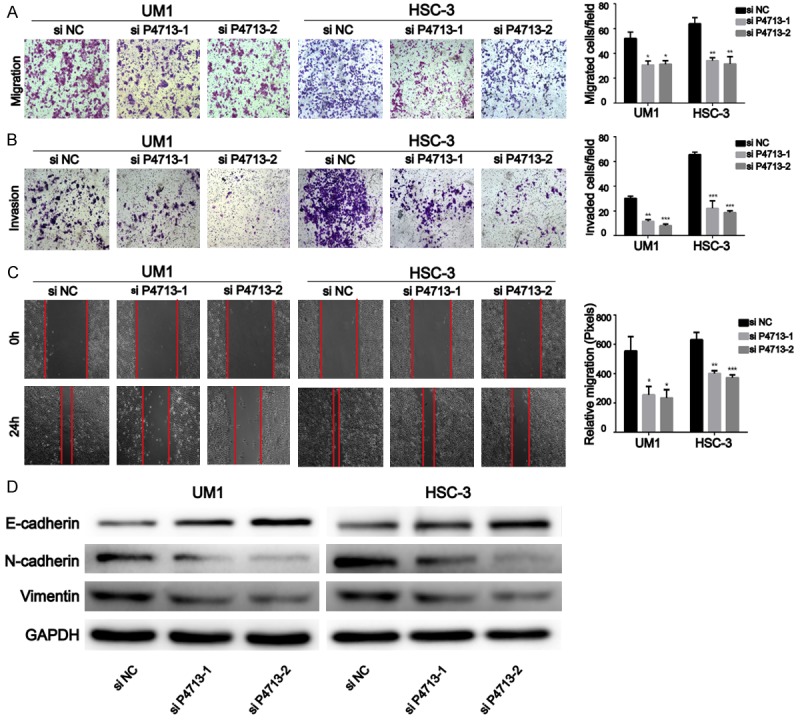
Silencing of P4713 suppressed the migration and invasion of oral squamous cell carcinoma (OSCC) cells. A: Inhibition of migration in HSC-3 and UM1 cells after P4713 knockdown. B: A Matrigel invasion assay was performed using an invasion chamber after treatment with si-P4713. C: In vitro migration was assessed by wound healing experiments. D: E-cadherin, N-cadherin, and vimentin were analyzed by western blotting.
P4713 might contribute to the malignant phenotype of OSCC by activating the JAK/STAT3 signaling
By mRNA microarray, a distinguishable mRNA expression profile was revealed in the OSCC tissues (Figure 4A). A P4713/mRNA co-expression network was constructed based on strong interactions with other genes (Figure 4B). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the P4713-associated mRNAs were mainly involved in adhesion, motility, JAK/STAT signaling, and pathways in cancer (Figure 4C, 4D). In UM1 and HSC-3 cells, qRT-PCR revealed that JAK2 and STAT3 were downregulated after P4713 knockdown (Figure 4E). The expression and phosphorylation of JAK2 and STAT3 were assayed by western blotting (Figure 4F). Consistent with the qRT-PCR results, the expression of both proteins decreased after P4713 depletion, and their phosphorylation levels decreased as well. To determine if P4713 affects STAT3 activation and translocation, STAT3 localization was examined by immunofluorescence. While STAT3 localized mainly in the nucleus in cells treated with control siRNA, depletion of P4713 caused its accumulation in the cytoplasm (Figure 4G). Collectively, the results indicate that P4713 might modulate the activation of JAK/STAT3 pathway, which is involved in OSCC.
Figure 4.

The relationship between P4713 and the JAK/STAT3 pathway. (A) Hierarchically clustered heatmaps of mRNAs altered in oral squamous cell carcinoma (OSCC; fold change > 2, P < 0.05). (B) The lncRNA-P4713 subnetwork in the OSCC co-expression network. (C) The top twenty GO terms of upregulated and downregulated mRNAs in OSCC cases (P < 0.05). (D) Significantly enriched pathways of the indicated gene sets. (E) qRT-PCR and (F) western blotting were used to measure the JAK/STAT3 pathway affected by P4713. (G) Representative fluorescent images of the location of STAT3. Scale bar = 10 µm.
Discussion
LncRNAs play a critical role in the development of human cancer and is suggested to act as a probable biomarker for cancer diagnose and prognosis [22,23]. Recently, aberrant expression of lncRNAs has been reported in OSCC [6,24,25]. However, the exact role and mechanism of these lncRNA in OSCC initiation and progression is still not clear. In this study, we identified a novel lncRNA, P4713, and demonstrated that it could promote the malignant progression of OSCC. P4713 was found to be significantly upregulated in both OSCC tissues and cell lines. Functional assays showed that P4713 contributed to the OSCC cell growth, proliferation, migration, and invasion. In addition, knockdown of P4713 inhibited the activity of JAK/STAT3 signaling in OSCC. These results indicated the potential role of P4713 in OSCC malignancy via JAK/STAT3 signaling pathway.
It is well recognized that lncRNAs play important regulatory role in the epigenetic modification of genomic DNA, gene transcription and protein translation [26,27]. LncRNAs could protect the expression of miRNA-targeted mRNA by binding to specific miRNA as a sponge. On the other hand, lncRNAs could interact with certain mRNA to regulate their function directly or indirectly [28]. Altered co-expression between lncRNAs and mRNAs has been involved in several diseases, including human cancer. Therefore, an mRNA expression microarray was performed in four OSCC tissues and adjacent non-cancerous tissues to explore the regulatory role of P4713. The result of GO and KEGG pathway analysis revealed that mRNAs with cell proliferation-, invasion-, and migration-related functions changed significantly, particularly components of the JAK/STAT pathway. In addition, knockdown of P4713 inhibited the activity of JAK/STAT3 signaling in OSCC cell lines. These results suggested that P4713 might contribute to regulating the activation of JAK/STAT3 signaling pathway in OSCC.
Several studies have reported the importance of JAK/STAT pathway in progression of many tumors, such as breast cancer, ovarian cancer, and hepatocellular carcinoma [16,29,30]. Moreover, emerging evidence indicates that aberrant activation of the JAK/STAT3 signaling may be involved in OSCC development and progression [31]. Likewise, we noticed that inhibition of P4713 could decrease the expression and phosphorylation level of JAK2/STAT3. Therefore, we suggest that P4713 may promote the expansion of OSCC via activating JAK/STAT3 signaling. In addition, lncRNA DC has been suggested to regulate STAT3 posttranslational modification by directly interacting with STAT3 in the cytoplasm [14]. Similarly, STAT3 was found to translocate from nucleus to cytoplasm while down regulating the expression level of P4713 in this study. Moreover, it has been reported that STAT3 attenuates tumor cell proliferation and migration capacity through downregulating cyclin D1 and vimentin expression [32,33]. Here, depletion of P4713 also downregulated the expression of cyclin D1 and vimentin. Thereby, we considered that P4713 regulates cyclin D1 and vimentin by influencing STAT3 in OSCC. These data suggested that P4713 promotes OSCC cell growth, proliferation, migration, and invasion via the JAK/STAT3 pathway. Whereas, the exact mechanisms involved require further study.
In summary, the results above demonstrated that lncRNA-P4713 contribute to OSCC malignant phenotypes via JAK/STAT3 signaling pathway. And it might provide a potential biomarker for OSCC. Meanwhile, further efforts are needed to fully elucidate the possible molecular mechanisms and biological role of P4713 in future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81502356&81500864) and the Postdoctoral Science Foundation of China Grant (No. 2015M570748&2016T90819).
Disclosure of conflict of interest
None.
References
- 1.Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma--an update. CA Cancer J Clin. 2015;65:401–421. doi: 10.3322/caac.21293. [DOI] [PubMed] [Google Scholar]
- 2.Liang S, Zhang S, Wang P, Yang C, Shang C, Yang J, Wang J. LncRNA, TUG1 regulates the oral squamous cell carcinoma progression possibly via interacting with Wnt/beta-catenin signaling. Gene. 2017;608:49–57. doi: 10.1016/j.gene.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 3.Tang H, Wu Z, Zhang J, Su B. Salivary lncRNA as a potential marker for oral squamous cell carcinoma diagnosis. Mol Med Rep. 2013;7:761–766. doi: 10.3892/mmr.2012.1254. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Ramirez I, Soto-Reyes E, Sanchez-Perez Y, Herrera LA, Garcia-Cuellar C. Histones and long non-coding RNAs: the new insights of epigenetic deregulation involved in oral cancer. Oral Oncol. 2014;50:691–695. doi: 10.1016/j.oraloncology.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Bag S, Banerjee DR, Basak A, Das AK, Pal M, Banerjee R, Paul RR, Chatterjee J. NMR ((1)H and (13)C) based signatures of abnormal choline metabolism in oral squamous cell carcinoma with no prominent Warburg effect. Biochem Biophys Res Commun. 2015;459:574–578. doi: 10.1016/j.bbrc.2015.02.149. [DOI] [PubMed] [Google Scholar]
- 6.Arunkumar G, Murugan AK, Prasanna SRH, Subbiah S, Rajaraman R, Munirajan AK. Long non-coding RNA CCAT1 is overexpressed in oral squamous cell carcinomas and predicts poor prognosis. Biomed Rep. 2017;6:455–462. doi: 10.3892/br.2017.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D, Ji Y, Li W, Yu Y, Liu C. Long noncoding RNA GACAT1 promotes proliferation and invasion of gastric cancer cells by targeting miR-378. Int J Clin Exp Pathol. 2017;10:6364–6374. [Google Scholar]
- 9.Ma Y, Sun N, Sheng F, Ji Y, Ding H, Zhang Q, Sun S, Li W. Long noncoding RNA H19 inhibits the growth and invasion of trophoblasts by inactivating Wnt/beta-catenin signaling via downregulation of DDX3X. Int J Clin Exp Pathol. 2017;10:6560. [Google Scholar]
- 10.Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145:178–181. doi: 10.1016/j.cell.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Zhu H, Zhang L, Yan S, Li W, Cui J, Zhu M, Xia N, Yang Y, Yuan J, Chen X, Luo J, Chen R, Xing R, Lu Y, Wu N. LncRNA16 is a potential biomarker for diagnosis of early-stage lung cancer that promotes cell proliferation by regulating the cell cycle. Oncotarget. 2017;8:7867–7877. doi: 10.18632/oncotarget.13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J, Liu Y, Gong Z, Zhang S, Guo C, Li X, Tang Y, Yang L, He Y, Wei F, Wang Y, Liao Q, Zhang W, Li X, Li Y, Li G, Xiong W, Zeng Z. Overexpression long non-coding RNA LINC00673 is associated with poor prognosis and promotes invasion and metastasis in tongue squamous cell carcinoma. Oncotarget. 2017;8:16621–16632. doi: 10.18632/oncotarget.14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 14.Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, Cao X. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–313. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 15.Zheng J, Zhao S, He X, Zheng Z, Bai W, Duan Y, Cheng S, Wang J, Liu X, Zhang G. The upregulation of long non-coding RNA CCAT2 indicates a poor prognosis for prostate cancer and promotes metastasis by affecting epithelialmesenchymal transition. Biochem Biophys Res Commun. 2016;480:508–514. doi: 10.1016/j.bbrc.2016.08.120. [DOI] [PubMed] [Google Scholar]
- 16.Shang AQ, Wu J, Bi F, Zhang YJ, Xu LR, Li LL, Chen FF, Wang WW, Zhu JJ, Liu YY. Relationship between HER2 and JAK/STAT-SOCS3 signaling pathway and clinicopathological features and prognosis of ovarian cancer. Cancer Biol Ther. 2017;18:314–322. doi: 10.1080/15384047.2017.1310343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halachmi S, Aitken KJ, Szybowska M, Sabha N, Dessouki S, Lorenzo A, Tse D, Bagli DJ. Role of signal transducer and activator of transcription 3 (STAT3) in stretch injury to bladder smooth muscle cells. Cell Tissue Res. 2006;326:149–158. doi: 10.1007/s00441-006-0204-6. [DOI] [PubMed] [Google Scholar]
- 18.Joung YH, Na YM, Yoo YB, Darvin P, Sp N, Kang DY, Kim SY, Kim HS, Choi YH, Lee HK, Park KD, Cho BW, Kim HS, Park JH, Yang YM. Combination of AG490, a Jak2 inhibitor, and methylsulfonylmethane synergistically suppresses bladder tumor growth via the Jak2/STAT3 pathway. Int J Oncol. 2014;44:883–895. doi: 10.3892/ijo.2014.2250. [DOI] [PubMed] [Google Scholar]
- 19.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 20.Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 2011;19:429–431. doi: 10.1016/j.ccr.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen ZZ, Huang L, Wu YH, Zhai WJ, Zhu PP, Gao YF. LncSox4 promotes the self-renewal of liver tumour-initiating cells through Stat3-mediated Sox4 expression. Nat Commun. 2016;7:12598. doi: 10.1038/ncomms12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Wu Z, Yuan J, Sun L, Lin L, Huang N, Bin J, Liao Y, Liao W. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017;395:31–44. doi: 10.1016/j.canlet.2017.02.035. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Yu S, Jiang L, Wang X, Song X. HOTAIR is a promising novel biomarker in patients with thyroid cancer. Exp Ther Med. 2017;13:2274–2278. doi: 10.3892/etm.2017.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C. Long non-coding RNA FTH1P3 facilitates oral squamous cell carcinoma progression by acting as a molecular sponge of miR-224-5p to modulate fizzled 5 expression. Gene. 2017;607:47–55. doi: 10.1016/j.gene.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Yang YT, Wang YF, Lai JY, Shen SY, Wang F, Kong J, Zhang W, Yang HY. Long non-coding RNA UCA1 contributes to the progression of oral squamous cell carcinoma by regulating the WNT/beta-catenin signaling pathway. Cancer Sci. 2016;107:1581–1589. doi: 10.1111/cas.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Liang T, Wang Y, Huang Y, Li Y. Long non-coding RNA AK093407 promotes proliferation and inhibits apoptosis of human osteosarcoma cells via STAT3 activation. Am J Cancer Res. 2017;7:892–902. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Xu X, Ji S, Li W, Yi B, Li H, Zhang H, Ma W. LncRNA H19 promotes the differentiation of bovine skeletal muscle satellite cells by suppressing Sirt1/FoxO1. Cell Mol Biol Lett. 2017;22:10. doi: 10.1186/s11658-017-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz Esparza-Garrido R, Rodríguez-Corona JM, López-Aguilar JE, Rodríguez-Florido MA, Velázquez-Wong AC, Viedma-Rodríguez R, Salamanca-Gómez F, Velázquez-Flores MÁ. Differentially expressed long non-coding RNAs were predicted to be involved in the control of signaling pathways in pediatric astrocytoma. Mol Neurobiol. 2017;54:6598–6608. doi: 10.1007/s12035-016-0123-9. [DOI] [PubMed] [Google Scholar]
- 29.Sengupta S, Nagalingam A, Muniraj N, Bonner MY, Mistriotis P, Afthinos A, Kuppusamy P, Lanoue D, Cho S, Korangath P, Shriver M, Begum A, Merino VF, Huang CY, Arbiser JL, Matsui W, Gyorffy B, Konstantopoulos K, Sukumar S, Marignani PA, Saxena NK, Sharma D. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene. 2017;36:5709–5721. doi: 10.1038/onc.2017.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, Zhao YM, Xiao YS, Sun QM, Ding ZB, Fan J. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol. 2015;46:587–596. doi: 10.3892/ijo.2014.2761. [DOI] [PubMed] [Google Scholar]
- 31.Huang JS, Yao CJ, Chuang SE, Yeh CT, Lee LM, Chen RM, Chao WJ, Whang-Peng J, Lai GM. Honokiol inhibits sphere formation and xenograft growth of oral cancer side population cells accompanied with JAK/STAT signaling pathway suppression and apoptosis induction. BMC Cancer. 2016;16:245. doi: 10.1186/s12885-016-2265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Z, Huang W, Chen B, Bai PD, Wang XG, Xing JC. Up-regulation of miR-124 inhibits invasion and proliferation of prostate cancer cells through mediating JAK-STAT3 signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:2338–2345. [PubMed] [Google Scholar]
- 33.Xiang Y, Liao XH, Yu CX, Yao A, Qin H, Li JP, Hu P, Li H, Guo W, Gu CJ, Zhang TC. MiR-93-5p inhibits the EMT of breast cancer cells via targeting MKL-1 and STAT3. Exp Cell Res. 2017;357:135–144. doi: 10.1016/j.yexcr.2017.05.007. [DOI] [PubMed] [Google Scholar]