

Abstract
Objective: Sclerostin (SOST), acting as a Wnt antagonist, has been shown to play a key role in regulating bone homestasis, and has also been linked to osteoarthritis (OA) development. Here, we investigated whether overexpressing SOST could affect OA development after destabilization of the medial meniscus (DMM) using SOST transgenic (Tg) mice. Methods: Bone and cartilage phenotypes of SOST Tg mice at 10 weeks of age were investigated by dual x-ray absorptiometry (DXA) and histology. Subsequently, 10-week-old SOST Tg mice and their wild-type (WT) littermates were subjected to DMM or sham surgery. Knee joints were isolated to evaluate the cartilage damage and the subchondral bone plate thickness at 2 and 8 weeks post-surgery. The changes of chondrocyte anabolic and catabolic responses after IL-1β or TNFα stimulation, β-catenin signaling and apoptosis were also measured. Results: Ten-week-old SOST Tg mice were identical to their WT littermate males except that they displayed digit abnormalities and osteopenic, whereas more severe OA was observed in SOST Tg mice at 2 and 8 weeks post-DMM. In addition, DMM resulted in significantly greater subchondral bone changes compared with sham surgery in SOST Tg mice at 8 weeks post-surgery. The accelerated OA in SOST Tg mice may be associated with reduced β-catenin signaling and increased chondrocyte apoptosis. Conclusion: Overexpressing SOST led to accelerated development of instability-induced OA. Our data further highlight that cartilage homeostasis requires finely tuned Wnt signaling.
Keywords: Sclerostin, osteoarthritis, cartilage, subchondral bone
Introduction
Osteoarthritis (OA) is a degenerative joint disease, characterized by cartilage degradation, osteophyte formation and subchondral bone sclerosis. The manifestations of the disease are pain, decreased mobility and disability [1]. Heart or metabolic diseases in these patients may be exacerbated due to lack of mobility. Unfortunately, there are no effective disease-modifying therapies for OA and great efforts are made on discovering molecular pathway and targets for drug development.
The Wnt/β-catenin signaling pathway is a key regulator in bone and cartilage homeostasis and has been linked to the cartilage degradation in OA [2]. Sclerostin (SOST) is a potent soluble antagonist of Wnt signaling and a regulator of bone homeostasis [3,4]. It has been demonstrated that patients afflicted by van Buchem disease (MIM 239100) [5,6] or sclerosteosis (MIM 269500) [7,8] display a high bone mass phenotype owing to SOST loss-of-function mutations. Moreover, Sost knockout (KO) mice have a high bone mineral density [9], whereas transgenic mice overexpressing human SOST are osteopenic [10,11].
While SOST has been widely viewed as a secreted peptide produced by osteocytes, recent studies have shown that chondrocytes are capable of producing SOST [10,12,13]. The number of SOST-positive chondrocytes increases in surgery-induced OA in mice [13,14] and sheep [14], as well as late stage human OA [13-16]. What is more, interleukin-1α (IL-1α) upregulates the SOST mRNA [14]. Recently, studies have documented that increased SOST could protect against cartilage damage. The initial study by Chan et al. demonstrated that SOST dose-dependently inhibited IL-1α-stimulated aggrecanolysis in vitro [14]. A separate study confirmed the in vitro results and showed that Sost KO mice have accelerated surgery-induced cartilage damage [17].
The number of SOST-positive osteocytes decreased in femoral neck of hip OA, which was associated with increased cortical bone density [18]. Another study further demonstrated that SOST expression was the lowest under full thickness cartilage defects with the thickest subchondral bone in hip OA [19]. In addition, SOST in subchondral bone osteocytes was the lowest with the thickest subchondral bone plate in late stage knee OA [20]. SOST expression was also low in sclerotic subchondral bone in a mice model of knee OA [14]. These studies suggest the inhibition of osteogenesis in the subchondral bone by systematically increasing the SOST level may serve as a therapeutic strategy for OA.
As upregulating the SOST level in OA cartilage and subchondral bone may play a protective role [14,17-20], we performed the present study to assess whether overexpressing SOST could decelerate the progression of OA using the SOST transgenic (SOST Tg) mice. SOST Tg mice were bred and subjected to the destabilization of medial meniscus model (DMM) of OA, a model offering an excellent translational perspective on OA [21].
Materials and methods
Ethics board approval
All animal procedures and protocols in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the Model Animal Research Center of Nanjing University (Animal Care and Use Protocol Permit Number: JQ01).
Animals
SOST Tg mice were kindly provided by Professor Gabriela G Loots, who generated these mice [11,22]. Generally, the limb phenotype is more severe in homozygous mice, whose ulna or fibula may be absent [23]. Missing fibula may affect joints due to loading changes. To obtain a consistent phenotype, hemizygous male mice were breed with WT females to get hemizygous and WT littermate mice for experiments. SOST transgene was genotyped by PCR. SOST transgene primer sequences: upper primer, 5’-ATGTCCACCTTGCTGGACTC-3’ and lower primer, 5’-GTCTGTGGGCTGGTTTGCAT-3’. All mice were caged in groups (n = 3-6 mice per cage), allowed free access to water and a standard rodent chow and maintained under 12-hour light/dark cycle.
X-ray analysis and BMD measurement
X-ray images were obtained using Philips Bucky Diagnosis CS DR System (Philips Medical System, Best, Netherlands) as previously reported [24]. BMD of the whole body and left femur were monitored in mice at 10 weeks of age, using DXA (PIXImus II; GE-Lunar, Madison, WI, USA) as previously described [24]. The region of interest (ROI) for whole body excluded the head while the ROI for the left femur included the whole bone.
DMM model
DMM was performed to introduce joint instability as previously described [21,25]. Briefly, after anesthesia with 100 mg/kg ketamine and 5 mg/kg xylazine, the medial meniscotibial ligament, which anchored the medial meniscus to the tibial plateau of the right knee, was transected. Separate mice were sham operated with medial capsulotomy only. Only male mice were used in the present study because of sex-related differences in this murine DMM-induced OA model [26].
A total of 100 mice male mice at 10 weeks old were operated and these mice were sacrificed at 2 or 8 weeks post-surgery. The right knee joints were harvested for subsequent histologic analyses. Only one mouse in the WT sham group died during the study. The number of each group was as follows: 2 weeks post-surgery (1) WT sham (n = 10), (2) WT DMM (n = 10), (3) SOST Tg sham (n = 13), (4) SOST Tg DMM (n = 13); 8 weeks post-surgery (1) WT sham (n = 10), (2) WT DMM (n = 17), (3) SOST Tg sham (n = 10), (4) SOST Tg DMM (n = 16).
Histology
Mouse knee joints were fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) overnight at 4°C, decalcified with 0.5 M EDTA (pH 7.4) and then embedded in paraffin. For quantification of surgery-induced cartilage lesions, serial frontal sections (5 μm thick) were cut, with 3 sections one slide. Each paraffin block yielded about 60 slides. Every six slide was stained with Safranin O/Fast Green. Cartilage damage in the medial femoral condyle and tibial plateau was graded using the Osteoarthritis Research Society International (OARSI) recommended 0 to 6 scoring system [27]. All the slides stained were evaluated by two independent investigators (SZ and YXG) in a blinded manner. The highest OARSI score among all the slides stained was selected for the severity of cartilage damage. The OARSI scores of the two independent observers were averaged for each mouse prior to statistical analysis.
Subchondral bone plate histomorphometry
Histomorphometry was performed using a published method [28]. One slide per animal was analyzed and sections were chosen from the weight-bearing area of the knee joint. Digital image was analyzed using Image J software. For each Safranin O/Fast Green stained section, represented field with a fixed width (600 μm) was chosen based on the following: the upper limit was the transition of cartilage to subchondral bone while the lower limit was the transition of subchondral bone to growth plate (referred to as subchondral bone area, which was defined by black line in Figure 3A). The upper limit of the subchondral bone plate area was just the one of subchondral bone area and the lower limit was the transition of the subchondral bone plate to trbecular bone (yellow line in Figure 3A).
Figure 3.
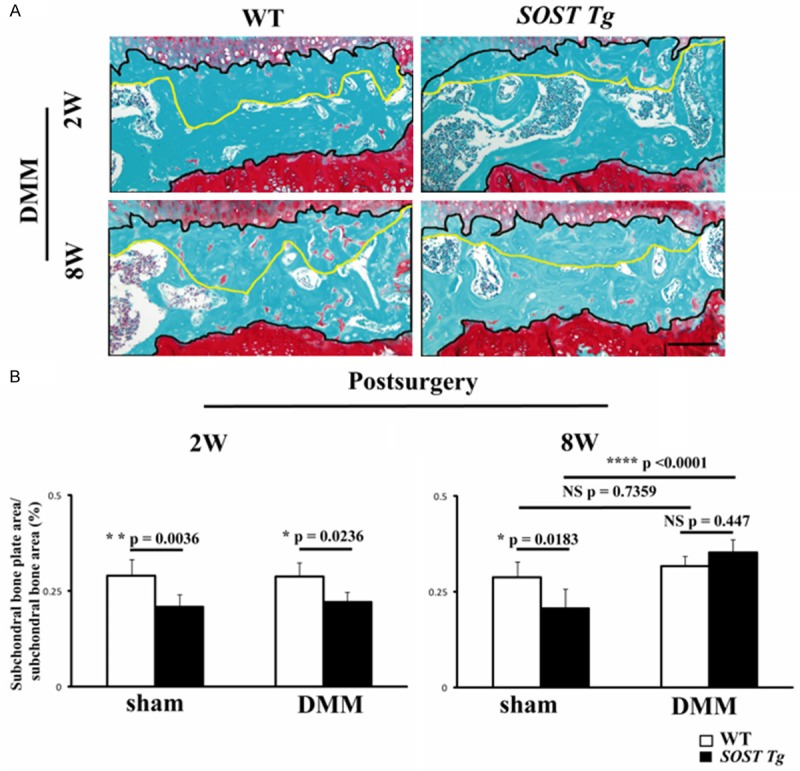
Histomorphometric analyses of subchondral bone in SOST Tg mice and their WT littermates. A. Representative photographs of subchondral bone in SOST Tg mice and their WT littermates at 2 and 8 weeks post-DMM. Black line delineates the subchondral bone area and the transition of the subchondral bone plate to trbecular bone was defined as yellow line. Sections were stained with Safranin O/Fast Green. Scale bars = 100 μm. B. Quantification of the subchondral bone plate. *P < 0.05. ****P < 0.0001. NS = not significant. Data are shown as means + 95% CI.
Chondrocyte primary cell culture
Mouse knee articular cartilage (femoral chondyle and tibial plateau) isolated from 5-day-old SOST Tg mice and their WT littermates was digested with 1% collagenase II (Gibco), as previously described [29]. After reaching 80% confluence, chondrocytes were detached and plated in 6-well plates (5×105/well). Cells were then stimulated with IL-1β (10 ng/ml; R & D Systems) or TNFα (10 ng/ml; R & D Systems) or vehicle for 24 h followed by RNA extraction. In the present study, only the first passage chondrocytes were used for assays.
Real-time reverse transcriptase-polymerase chain reaction (q-PCR)
Total RNA was extracted from murine chondrocytes using TRIzol reagent (Invitrogen). About 1 μg mRNA was used for reverse transcription using the PrimeScript RT Reagent Kit (TaKaRa). Q-PCR was performed using SYBR Green PCR Master Mix (Thermo) in an ABI Step One Plus instrument (Applied Biosystems). Relative transcript levels were calculated using the 2-ΔΔCt method. Data were normalized to the mouse β-actin (Actb) and all reactions were performed in triplicate. Primers names and sequences are list in Table S1.
Immunohistochemisty (IHC) and TUNEL
Anatomically equivalent sections from SOST Tg and their WT littermates were used for IHC or TUNEL. Sections were deparaffinized by xylene and rehydrated. After antigen retrieval in a sodium citrate (0.01 M, pH 6.0), sections were washed and deprived of endogenous peroxidase activity using 3% hydrogen peroxide. Then slides were blocked with 1% bovine serum albumin (Sigma) at room temperature and immunolabeled with primary antibodies against type II collagen (Col2a1, 1:100, Boster), Sox9 (1:100, Millipore) or β-catenin (1:100, Proteintech) at 4°C overnight. Subsequently, sections were incubated with biotinylated goat anti-rabbit secondary antibodies (Vector). Signals were amplified using a Vectstain ABC kit (Vector). After reaction with DAB (Vector), sections were counterstained with hematoxylin and photographed under a Leica light microscope.
TUNEL was performed with the In Situ Cell Death Detection Kit (Roche). Nuclei were counterstained with DAPI (Invitrogen) and scanned using a confocal microscope (Leica). The numbers of total chondrocytes and TUNEL-positive chondrocytes across the entire tibial plateau were determined with Image J software. Sections from 3 sections per animal were imaged. The final results were averaged and presented as the percentage of TUNEL-positive cells.
Statistical analysis
GraphPad Prism version 6 was used for all analyses. Data did not violate the assumptions of normality based on normality test. To derive the number of mice required in this study, we performed power calculations with reference to our published article [25]. For body weight and in vivo DXA findings, data were analyzed by Student’s two-tailed t-test. For OA severity, qPCR and TUNEL analyses, we performed 2-way analysis of variance (ANOVA). When multiple comparisons were performed, post-hoc Sidak test was used to adjust for multiplicity. For cell culture studies, data were collected from three individual experiments. All data were expressed as means + 95% CI. P < 0.05 was the threshold of statistical significance.
Results
Characterization of SOST Tg mice
SOST Tg mice were breed by the above strategy. SOST transgene was genotyped by PCR (Supplementary Figure 1A) and only hemizygous mice and WT littermates were used in this study. Consistent with previously reported findings [11,22], 10-week-old SOST Tg male mice grew to skeletal maturity with normal weight and body size but develop osteopenia (Supplementary Figure 1B). Total body and femur BMD and BMC were significantly lower in SOST Tg mice relative to their WT littermates, as evaluated in vivo by DXA analyses (Supplementary Figure 1B).
Ten-week-old SOST Tg mice were indistinguishable from their WT littermates, except that they displayed digit abnormalities (Supplementary Figure 2A). Missing digits were only observed in the forelimbs of SOST Tg mice (Supplementary Figure 2A). The joints and articular cartilage of the transgenic mice were then evaluated by X-rays and histology. No obvious abnormalities in knee joints were observed in SOST Tg mice by X-rays (Supplementary Figure 2B). The structure and proteoglycans in the articular cartilage of SOST Tg mice did not exhibited any abnormality (Figure 1A and 1B). SOST Tg mice also exhibited similar levels of Col2a1 and Sox9 to their WT littermate males in the articular cartilage (Figure 1C).
Figure 1.
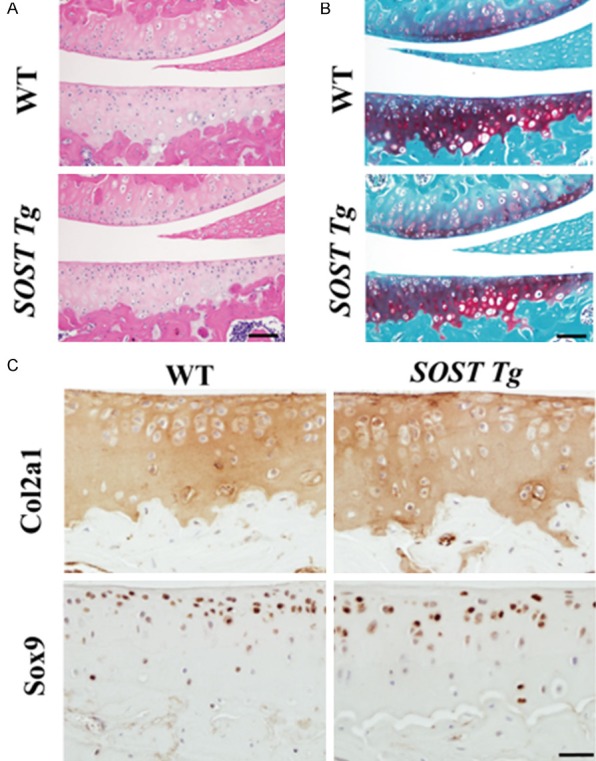
Basal articular cartilage in SOST Tg mice and their WT littermates. (A) Safranin-O/Fast green and (B) H&E staining of the cartilage from 10-week-old SOST Tg mice and their WT littermates (n = 6/group). Scale bars = 50 μm. Representative (C) IHC images of Col2a1 and Sox9 in the cartilage from 10-week-old SOST Tg mice and their WT littermates (n = 6/group). Col2a1 = Type II collagen. Scale bars = 20 μm.
SOST Tg mice exhibit exacerbated OA following surgical destabilization of the knee
The knee joints of 10-week-old SOST Tg mice exhibited no obvious abnormalities compared with WT littermates, indicating that they are suitable for instability-induced OA studies. Subsequently, the DMM OA mouse model was induced in 10-week-old SOST Tg mice and their WT littermates. SOST Tg mice were more responsive to surgery-induced OA than were WT littermate males. Loss of chondrocyte cellularity and proteoglycans were observed at 2 weeks post-DMM, whereas complete loss of the entire non-calcified cartilage was noted at 8 weeks post-surgery in both the medial femoral condyle and tibial plateau of SOST Tg mice (Figure 2A). The OARSI scores for morphological structure changes in the medial tibia were significantly higher in knee joints from SOST Tg mice than in those of their WT littermate males at 2 and 8 weeks post-surgery (Figure 2B; P = 0.0176; P < 0.0001 respectively). For the femur, the OARSI scores were only significantly increased in SOST Tg mice compared with those in the WT littermate males at 8 weeks post-DMM (Figure 2B; P < 0.0001). However, sham-operated knees from SOST Tg mice exhibited no obvious cartilage lesions (Figure 2B).
Figure 2.
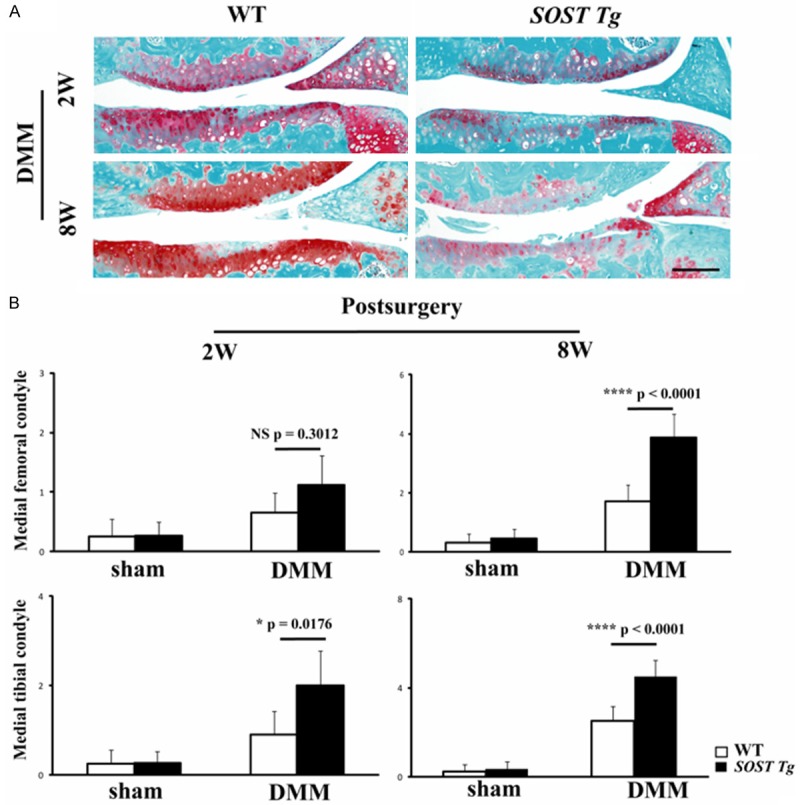
Accelerated OA in SOST Tg mice following destabilization of the medial meniscus (DMM). A. Representative photographs of articular cartilage destruction in SOST Tg mice and their WT littermates at 2 and 8 weeks post-DMM. Sections were stained with Safranin O/Fast Green. Scale bars = 100 μm. B. The OARSI scores for the medial femoral condyle and the medial tibial condyle at 2 and 8 weeks post-DMM in SOST Tg mice and their WT littermates. The OARSI score in the medial tibia were significantly higher in DMM knees of SOST Tg mice compared with their WT littermate mice at 2 (P = 0.0176) and 8 (P < 0.0001) weeks post-surgery. For the femur, the OARSI score was only significantly higher in knees of SOST Tg mice compared to WT littermates at 8 weeks post-DMM (P < 0.0001). *P < 0.05. ****P < 0.0001. NS = not significant. Data are shown as means + 95% CI.
We then measured the subchondral bone plate thickness in the medial tibial plateau of sham-operated and DMM-induced knee joints at 2 and 8 weeks post-DMM. Subchondral bone plate area/subchondral bone area ratio was significantly lower in SOST Tg mice than their WT littermates in the sham-operated knees at 2 and 8 post-surgery and DMM-induced knees at 8 weeks post-surgery (Figure 3A and 3B; P = 0.0036; P = 0.0183; P = 0.0236 respectively). Subchondral bone plate thickness in the SOST Tg mice was significantly greater in the DMM group relative to the sham group at 8 weeks post-surgery (P < 0.0001). However, there was no significant difference between the DMM group of SOST Tg mice and littermates at 8 weeks post-surgery (P = 0.447).
Exacerbated OA in SOST Tg mice was associated with increased chondrocyte apoptosis
To further investigate the mechanism underlying the exacerbation of OA in SOST Tg mice, we performed the primary chondrocyte culture experiments. Isolated primary chondrocytes from 5-day-old SOST Tg mice and their WT littermates were cultured with or without IL-1β or TNFα for analysis. Chondrocytes from SOST Tg mice has a similar basal expression of anabolic genes (Col2a1, Aggrecan and Sox9) and catabolic genes (MMP-3, MMP-13, Adamts4 and Adamts5) to those from their WT littermates. Next, we treated primary chondrocytes with IL-1β or TNFα for 24 hours. Both IL-1β and TNFα led to reduced Col2a1, Aggrecan and Sox9 and increased MMP-3, MMP13, Adamts4 and Adamts5. However, the more MMP-3 and MMP-13 mRNA levels via IL-1β were significantly lessened in chondrocytes from SOST Tg mice (Figure 4; P = 0.0386; P = 0.0328 respectively).
Figure 4.
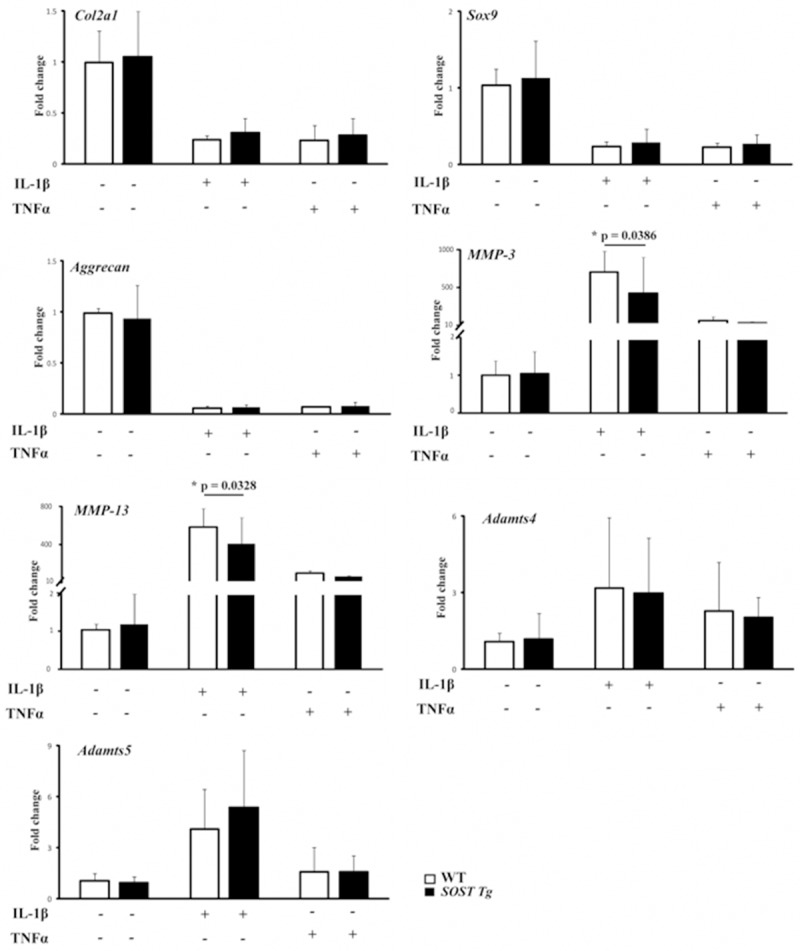
Primary articular chondrocytes were isolated from SOST Tg mice and their WT littermates and treated with 4-hydroxytamoxifen for 48 h as described in the Methods. The expression levels of Col2a1, Aggrecan, Sox9, MMP-3, MMP-13, Adamts4 and Adamts5 messenger RNA (mRNA) in primary murine chondrocytes treated with IL-1β (10 ng/ml; R & D Systems) or TNFα (10 ng/ml; R & D Systems) or vehicle for 24 h were determined by real-time reverse transcriptase-PCR. Data are representative of three individual experiments. Col2a1 = Type II collagen; MMP-3 = matrix metalloproteinase-3; MMP-13 = matrix metalloproteinase-13; *P < 0.05.
Given the fact that imbalance between chondrocyte anabolism and catabolism was not aggravated in SOST Tg mice, the more severe cartilage degeneration in those mice must occur via an alternative mechanism. As SOST is a negative regulator of Wnt signaling, we then analyzed the β-catenin IHC staining of the medial tibial plateau of sham-operated and surgery-induced knee cartilage at 2 weeks post-DMM. The IHC results suggested that more β-catenin was present in WT mice compared with SOST Tg mice upon DMM surgery (Figure 5A). In the sham-operated knee cartilage, β-catenin was relatively low in both SOST Tg mice and their WT littermates (Figure 5A).
Figure 5.
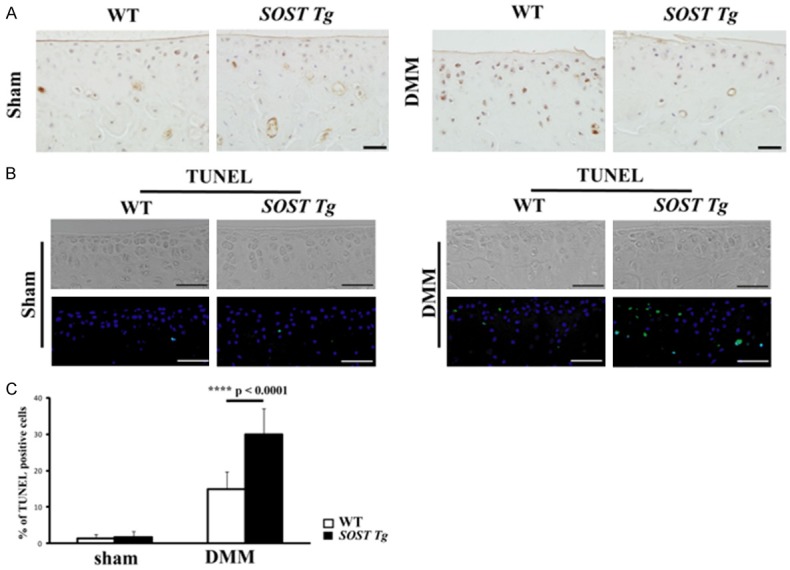
A. Representative IHC images of β-catenin in the medial tibial plateau in SOST Tg mice and their WT littermates 2 weeks post-sham operation and DMM surgery. Scale bars = 20 μm. The cellularity of the section was confirmed with hematoxylin staining. B. Representative TUNEL images in the medial tibial plateau in SOST Tg mice and their WT littermates 2 weeks post-sham operation and DMM surgery. Green represents positive staining. Blue indicates DAPI staining. No intensity or background adjustments were made between sections. Scale bars = 50 μm. C. Quantifications of the percentage of TUNEL-positive cells are presented as percentages relative to cells stained for DAPI. (n = 6/group) TUNEL = terminal deoxynucleotidyl transferase dUTP nick end labelling; ****P < 0.0001. Data areshown as means + 95% CI.
As increased chondrocyte apoptosis may contribute to the articular cartilage lesions observed in chondrocyte-specific knockout of β-catenin mice [30], we then analyzed chondrocyte apoptosis by TUNEL and found that the percent of TUNEL-positive chondrocytes in SOST Tg mice was significantly more than that in WT littermates at 2 weeks post-DMM (Figure 5B and 5C; P < 0.0001).
Discussion
Multiple lines of evidence suggest that a certain level of Wnt/β-catenin signaling activity is critical important for maintaining the health of articular cartilage while the precise mechanism still remains uncovered [2]. Aberrant activation and inhibition of Wnt/β-catenin signaling both led to acceleration of articular cartilage degeneration [30,31]. As a Wnt antagonist, SOST was mainly detected in osteocytes, suggesting that SOST might play a tissue-specific role in bone. However, increased SOST was detected in articular cartilage of OA [13-17] and Sost KO mice resulted in an acceleration of OA development upon DMM [17]. Unexpectedly, we found the exacerbated but not attenuated development of OA in SOST Tg mice compared with WT littermates upon surgery. We speculate that a certain level of SOST is required for regulating Wnt/β-catenin signaling activity in articular cartilage under pathological conditions.
Wnt/β-catenin signaling has been shown to be associated with not only chondrocyte survival but also differentiation [32,33] and Lrp6 mutant mice display chondrocyte apoptosis and cartilage destruction [34]. Consistently, we found that increased chondrocyte apoptosis in SOST Tg mice compared with WT littermates upon DMM.
The reduced subchondral bone plate thickness observed in SOST Tg mice implied that sclerostin could also inhibit bone formation in the subchondral bone as described in the whole skeleton [9-11], whereas the DMM-induced knees exhibited a significant increase in the subchondral bone plate thickness compared with the sham-operated ones in the SOST Tg mice at 8 weeks post-surgery while their WT littermates exhibited a nonsignificant increase. Most probably, the severe cartilage damage upon DMM in SOST Tg mice had an effect on this subchondral sclerosis.
Previous study reported that injection of recombinant SOST (rSOST) did not alter the disease progression in the proteoglycan-induced sondylitis (PGISp) mouse model [35]. The authors attributed the failure to the efficacy of rSOST, low stability of rSOST, insufficient dosage, etc. To avoid these multiple factors, we chose SOST Tg mice, in which human SOST showed effectiveness at blocking Wnt signaling, to perform this study.
In summary, our findings revealed that mice overexpressing SOST have accelerated OA development after DMM, which is associated with reduced Wnt/β-catenin signaling activity, and further highlight the fine-tuning and complexity of Wnt signaling in joint homeostasis and disease.
Acknowledgements
Supported by National Natural Science Foundation of China (81472116), the Projects of International Cooperation and Exchanges NSFC (81420108021), National Key Technology Support Program (2015BAI08B02), Excellent Young Scholars NSFC (81622033), Jiangsu Provincial Key Medical Center Foundation, Jiangsu Provincial Medical Talent Foundation and Jiangsu Provincial Medical Outstanding Talent Foundation.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lories RJ, Corr M, Lane NE. To Wnt or not to Wnt: the bone and joint health dilemma. Nat Rev Rheumatol. 2013;9:328–39. doi: 10.1038/nrrheum.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blom AB, van Lent PL, van der Kraan PM, van den Berg WB. To seek shelter from the WNT in osteoarthritis? WNT-signaling as a target for osteoarthritis therapy. Curr Drug Targets. 2010;11:620–9. doi: 10.2174/138945010791011901. [DOI] [PubMed] [Google Scholar]
- 4.Atkins GJ, Rowe PS, Lim HP, Welldon KJ, Ormsby R, Wijenayaka AR, Zelenchuk L, Evdokiou A, Findlay DM. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J Bone Miner Res. 2011;26:1425–36. doi: 10.1002/jbmr.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–52. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 7.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 8.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–69. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 10.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, Rubin EM. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, Oostenbroek HJ, Van Hul W, Hamersma H, Dikkers FG, Hamdy NA, Papapoulos SE, Lowik CW. Sclerostin in mineralized matrices and van Buchem disease. J Dent Res. 2009;88:569–74. doi: 10.1177/0022034509338340. [DOI] [PubMed] [Google Scholar]
- 13.Roudier M, Li X, Niu QT, Pacheco E, Pretorius JK, Graham K, Yoon BR, Gong J, Warmington K, Ke HZ, Black RA, Hulme J, Babij P. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum. 2013;65:721–31. doi: 10.1002/art.37802. [DOI] [PubMed] [Google Scholar]
- 14.Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN, Little CB. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–85. doi: 10.1016/j.joca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Karlsson C, Dehne T, Lindahl A, Brittberg M, Pruss A, Sittinger M, Ringe J. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010;18:581–92. doi: 10.1016/j.joca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Papathanasiou I, Kostopoulou F, Malizos KN, Tsezou A. DNA methylation regulates sclerostin (SOST) expression in osteoarthritic chondrocytes by bone morphogenetic protein 2 (BMP-2) induced changes in Smads binding affinity to the CpG region of SOST promoter. Arthritis Res Ther. 2015;17:160. doi: 10.1186/s13075-015-0674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouaziz W, Funck-Brentano T, Lin H, Marty C, Ea HK, Hay E, Cohen-Solal M. Loss of sclerostin promotes osteoarthritis in mice via beta-catenin-dependent and -independent Wnt pathways. Arthritis Res Ther. 2015;17:24. doi: 10.1186/s13075-015-0540-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Power J, Poole KE, van Bezooijen R, Doube M, Caballero-Alias AM, Lowik C, Papapoulos S, Reeve J, Loveridge N. Sclerostin and the regulation of bone formation: effects in hip osteoarthritis and femoral neck fracture. J Bone Miner Res. 2010;25:1867–76. doi: 10.1002/jbmr.70. [DOI] [PubMed] [Google Scholar]
- 19.Zarei A, Hulley PA, Sabokbar A, Javaid MK. Co-expression of DKK-1 and sclerostin in subchondral bone of the proximal femoral heads from osteoarthritic hips. Calcif Tissue Int. 2017;100:609–618. doi: 10.1007/s00223-017-0246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Guo H, Sun K, Zhao X, Ma T, Jin Q. Sclerostin expression in the subchondral bone of patients with knee osteoarthritis. Int J Mol Med. 2016;38:1395–402. doi: 10.3892/ijmm.2016.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–9. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25:178–89. doi: 10.1359/jbmr.090730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collette NM, Genetos DC, Murugesh D, Harland RM, Loots GG. Genetic evidence that SOST inhibits WNT signaling in the limb. Dev Biol. 2010;342:169–79. doi: 10.1016/j.ydbio.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 25.Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, Shi D, Dai J, Li J, Ju H, Cao Y, Qin J, Chen S, Teng H, Jiang Q. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. 2017;7:43245. doi: 10.1038/srep43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007;15:695–700. doi: 10.1016/j.joca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Amiable N, Martel-Pelletier J, Lussier B, Kwan Tat S, Pelletier JP, Boileau C. Proteinaseactivated receptor-2 gene disruption limits the effect of osteoarthritis on cartilage in mice: a novel target in joint degradation. J Rheumatol. 2011;38:911–20. doi: 10.3899/jrheum.100710. [DOI] [PubMed] [Google Scholar]
- 29.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3:1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 30.Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O’Keefe RJ, Chen D. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008;58:2053–64. doi: 10.1002/art.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier RN, O’Keefe RJ, Zuscik M, Chen D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009;24:12–21. doi: 10.1359/JBMR.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaguchi H. Regulation of osteoarthritis development by Wnt-beta-catenin signaling through the endochondral ossification process. J Bone Miner Res. 2009;24:8–11. doi: 10.1359/jbmr.081115. [DOI] [PubMed] [Google Scholar]
- 33.Dao DY, Jonason JH, Zhang Y, Hsu W, Chen D, Hilton MJ, O’Keefe RJ. Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res. 2012;27:1680–94. doi: 10.1002/jbmr.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joiner DM, Less KD, Van Wieren EM, Hess D, Williams BO. Heterozygosity for an inactivating mutation in low-density lipoprotein-related receptor 6 (Lrp6) increases osteoarthritis severity in mice after ligament and meniscus injury. Osteoarthritis Cartilage. 2013;21:1576–85. doi: 10.1016/j.joca.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Haynes KR, Tseng HW, Kneissel M, Glant TT, Brown MA, Thomas GP. Treatment of a mouse model of ankylosing spondylitis with exogenous sclerostin has no effect on disease progression. BMC Musculoskelet Disord. 2015;16:368. doi: 10.1186/s12891-015-0823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.