

Abstract
Chemokine (C-C motif) ligand 4 (CCL4) and vascular endothelial growth factor-A (VEGF-A) are involved in the progression and metastasis of some tumors, including ovarian cancer, colon cancer and prostate cancer. However, the roles of CCL4 and VEGF-A in human endometrial cancer (EC) are still unclear. Here, we demonstrated that the production of CCL4 and VEGF-A was significantly higher in EC tissues than in normal tissues, and their expression profiles were associated with the clinical stage of EC. In addition, we found that CCL4 promoted the angiogenesis and invasive ability of EC tumors by increasing the production of VEGF-A. We further confirmed the effect of CCL4 in the growth of EC tumors by silencing the expression of CCL4 in EC cell lines. Finally, we found that CCL4 upregulated VEGF-A expression by activating STAT3, and it enhanced the progression and metastasis of EC. Our study showed that CCL4 promoted tumor growth by upregulating VEGF-A expression, which affected the STAT3 signal pathway in the EC cells.
Keywords: Endometrial cancer, CCL4, VEGF-A, STAT3
Introduction
Endometrial carcinoma (EC) is thought to be the most common gynecologic malignancy in the world, with a mean age at diagnosis of 60 years [1]. Epidemiological data have shown that due in large part to the obvious clinical symptoms, approximately 73% of EC patients have stage I at diagnosis, which has a low recurrence rate, and the five year survival is approximately 85% [2,3]. However, approximately 10% of patients are diagnosed at stage II, which has a very poor prognosis [2,4]. Advanced stage (III-IV) EC is less common, and it is often associated with metastasis. Previous studies have indicated that advanced stage EC can metastasize to the ovaries, lymph nodes and even outside the abdomen, which is fatal [5]. Currently available treatments, such as surgery, radiation and chemotherapy, are not very effective for EC, especially in patients with advanced stage disease [5,6]. It is very important to understand the underlying mechanism of EC so that we can explore more effective strategies for the detection, prevention and treatment of this disease.
Cancer cells need oxygen and nutrients for their survival, and they are hence located near blood vessels; without blood vessel support, tumors could not survive or metastasize to other organs [7,8]. Numerous studies have shown that angiogenesis is regulated by pro- and anti-angiogenic agents, such as transforming growth factor β (TGF-β), matrix metalloproteinases (MMPs) and vascular endothelial growth factor-A (VEGF-A). Among these agents, VEGF-A is thought to be the major inducer of angiogenesis, and evidence has shown that VEGF-A is involved in the growth of various tumors [9,10]. While the roles of VEGF-A in EC remain unclear, it is important to understand the effects of VEGF-A in EC.
Previous studies have demonstrated that many factors can stimulate cancer cells to release VEGF-A, such as chemokines, a kind of chemotactic cytokine that is produced by stimulation like cytokines and growth factors [11,12]. The activation of chemokines results in the transcription regulation of a target gene, which is involved in the cancer cell proliferation, invasion, and metastasis [13]. Chemokine (CC motif) ligand 4 (CCL4) is a member of the chemokine family, and it is associated with the leukocyte traffic, angiogenesis, and metastasis of various tumors, such as ovarian cancer, colon cancer and prostate cancer [14-16]. Recently, several studies have demonstrated that CCL3 promotes the metastasis of some cancers through the VEGF-A signal pathway, although the roles of CCL4 in VEGF-A and angiogenesis in endometrial cancer have not been clarified [10,17,18]. In this article, we showed that CCL4 promoted tumor angiogenesis by upregulating VEGF-A expression via the phosphorylation of STAT3 in EC.
Materials and methods
Acquisition of tissue specimens
We collected 22 EC tissue specimens from patients who were diagnosed with endometrial cancer and treated at Huai’an First People’s Hospital between Jun 2014 and Jun 2016. Informed consent was obtained from all patients.
Cell culture
The human endometrial cancer cell lines AN3CA, HEC-1B, and the endothelial progenitor cells (EPCs) were obtained from ATCC (Rockville, MD, USA). AN3CA and HEC-1B were cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco, Gaithersburg, MD), which was supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, USA). While EPCs were maintained in RPMI-1640 (Gibco), they were also supplemented with 10% FBS. All cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C.
Construction of a stable expression CCL4 shRNA cell line
The CCL4 shRNA and control shRNA were designed and obtained from GenePharma (Shanghai, China). HEC-1B and AN3CA cells (5 × 104 cells/well) were seeded in 24-well plates and incubated overnight, and then, they were transduced with CCL4-shRNA or control shRNA lentiviral supplemented with 8 mg/mL polybrene (Sigma-Aldrich, the Netherlands). Finally, the media was removed, and the resuspended cells were maintained in fresh medium containing 2 μg/ml puromycin to select for stable transfectants.
Detection of cell growth phenotypes
The effect of CCL4-shRNA on the proliferation of EC cells was evaluated by MTT, colony formation and apoptosis assays. The stable expression CCL4 AN3CA and HEC-1B cells were plated in 96-well culture plates (3 × 103 per well) and incubated for 12, 24, 36 and 48 hours. Then, the MTT (0.5 mg/ml; Sigma-Aldrich, USA) was added to each well (20 μl/well). After 4 hours of additional incubation, the MTT solution was discarded, 200 ml of DMSO (Sigma, USA) was added, and the plates were shaken gently. The absorbance was measured on an ELISA reader at a wavelength of 490 nm. For the colony formation assay, cells were counted and seeded in 12-well plates (in triplicate) at 100 cells per well. Fresh culture medium was replaced every 3 days. The number of viable cell colonies was determined after 14 days, and colonies were fixed with methanol, stained with crystal violet, photographed and counted. For the cell apoptosis assay, cell apoptotic rate was determined by using Annexin V-FITC and a PI staining flow cytometry kit (KeyGEN BioTECH, China) according to manufacturer’s instructions. Briefly, the cells in the different groups were harvested and washed twice with PBS. Next, the cells were resuspended in 500 ml of binding buffer included in the kit. Then, 5 ml Annexin V and 5 ml propidium iodide (PI) were added to the cells and incubated at room temperature for 15 minutes in the dark. The cells’ apoptotic rate was then tested by flow cytometry within 1 h. Each experiment was performed in triplicate.
Quantitative real-time PCR
The total RNA of the EC cells was prepared using a TRIzol kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions, and then, it was reverse transcribed into cDNA using an oligo primer using the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher). A quantitative real-time polymerase chain reaction (q-PCR) assay was carried out using SYBR Premix Ex Taq (Takara, Japan). The sequence of all the primers used in the current study were designed and purchased from Sangon, China, and the sequences of these primers are listed in Table 1.
Table 1.
Primer sequences for real-time PCR analysis
| mRNA | Primer sequence |
|---|---|
| CCL4 | Forward: 5’-GCTGTGTTTGTGCTGATGCT-3’ |
| Reverse: 5’-GCTGGCTGGTCTTTTGGTAG-3’ | |
| VEGF-A | Forward: 5’-CCTTGCCTTGCTGCTCTACCTC-3’ |
| Reverse: 5’-TTCTGCCCTCCTCCTTCTGC-3’ | |
| β-actin | Forward: 5’-CTGGGACGACATGGAGAAAA-3’ |
| Reverse: 5’-AAGGAAGGCTGGAAGAGTGC-3’ |
Western blot
Total proteins of AN3CA and HEC-1B cells were extracted using RIPA buffer (0.1% SDS, 1% Triton X-100, 1 mM MgCl2, 10 mM Tris-HCl, pH 7.4) including a protease inhibitor in 4°C for at least 30 mins. The total protein concentration was estimated using the BCA method (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer’s instructions. The proteins (50 μg) were separated by SDS-PAGE, and then transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). Nonspecific binding sites of membranes were blocked by 5% bovine serum albumin (BSA) for 2 h at room temperature, and primary antibodies were incubated overnight at 4°C. Horseradish peroxidase-conjugated secondary antibodies were applied for 2 h. Proteins were then detected by enhanced chemiluminescent reagents. Actin was used as an internal control. The following antibodies were used: anti-pSTAT3 (1:200, Abcam, Cambridge, UK), anti-STAT3 (1:200, Abcam, Cambrige, UK), and anti-actin (1:2000, CST Inc., CST, Danvers, Massachusetts, USA).
Immunohistochemical (IHC) staining
The samples of normal or EC tissues were prepared and analyzed using a Histostain-Plus kit (MRBiotech, Emeryville, USA), as previously described [19]. Briefly, the paraffin-embedded specimens were stained with CCL4, CCR5 and VEGF-A primary antibodies at a dilution of 1:300 and incubated at 4°C for 12 h. Next, biotinylated secondary antibodies (MRBiotech) were applied to the sections for 2 h at room temperature. Then, a horseradish peroxidase-conjugated avidin-biotin complex was added, and the signal was detected by diaminobenzidine according the manufacturer’s instructions. The IHC results were scored by the intensity of the stain, and the percentage of staining was analyzed in the following manner: Intensity: 0, negative; 1, weak; 2, moderate; 3, strong; and percentage: 0, 0-5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; and 4, 75-100%.
Cell invasion assay
The EPCs invasion assay was performed using Transwell chambers (8 μm pore size; BD Biosciences, USA). The treated cells (1 × 104 cells/well) were seeded in the upper chamber of each individual well, and the complete medium was transfused into the bottom chamber. After 24 h of incubation at 37°C in 5% CO2, cells on the upper side were removed with cotton-tipped swabs, and cells that were attached to the other side of the membrane were fixed and stained with 5% crystal violet. At least five random fields were selected for statistical purposes, and the invasion assay was repeated three times.
Tube formation assay
A plate was coated with 150 μl Matrigel per well and incubated at 37°C for approximately 2 h. Next, the EPCs (1 × 104 cells/well) were suspended in 2 ml CM of AN3CA, and HEC-1B cells were seeded in the pre-coated plate for 16 h at 37°C. Finally, tube formation was observed under a microscope, and the total length of the tube was calculated using three randomly selected fields using MacBiophotonics ImageJ software.
Mouse xenograft assay
Ten male 5-week-old BALB/c nude mice were randomly divided into 2 groups. HEC-1B cells transfected with CCL4-shRNA or control shRNA were suspended in serum-free medium and then subcutaneously injected into the flanks of the mice. Three weeks after the injections, the mice were euthanized, and tumor volumes and weights were measured. The volumes were calculated using the following standard formula: tumor volume (cm3) = (the longest diameter) × (the shortest diameter)2 × 0.5.
Statistical analysis
The data were analyzed by the Student’s t-test and variance (ANOVA), and the correlation between CCL4 expression and VEGF-A expression was analyzed by Pearson’s correlation coefficient. The data was calculated using GraphPad (GraphPad Prism Software, La Jolla, CA, USA) and SPSS statistics (IBM SPSS Statistics 20, Chicago, IL, USA). All experiments were completed at least three times. All results were counted and presented as the means ± SD. P < 0.05 was considered statistically significant.
Results
CCL4, CCR5 and VEGF-A were upregulated in EC tissues
To explore the roles of CCL4 and VEGF-A in endometrial cancer, we first examined the expression levels of CCL4, CCR5 and VEGF-A in 22 EC tissues and their corresponding normal tissues from patients. We confirmed this result by detecting the mRNA expression level of CCL4 and VEGF-A using a qRT-PCR assay in the 22 EC tissues and normal tissues, and the results demonstrated that CCL4 and VEGF-A were highly expressed in the EC tissues compared with normal individuals (Figure 1A and 1B). In addition, we found that the mRNA expression level of CCL4 had a positive correlation with VEGF-A (r2 = 0.7089, P < 0.05, Figure 1C). The immunohistochemistry results also showed that the expression levels of CCL4, CCR5 and VEGF-A were higher in the EC tissues than in the normal individuals (Figure 1D), and their expression profiles were associated with the clinical stage of EC (Figure 1E). Therefore, these results demonstrated that CCL4 and VEGF-A may be involved in the pathogenesis of EC.
Figure 1.
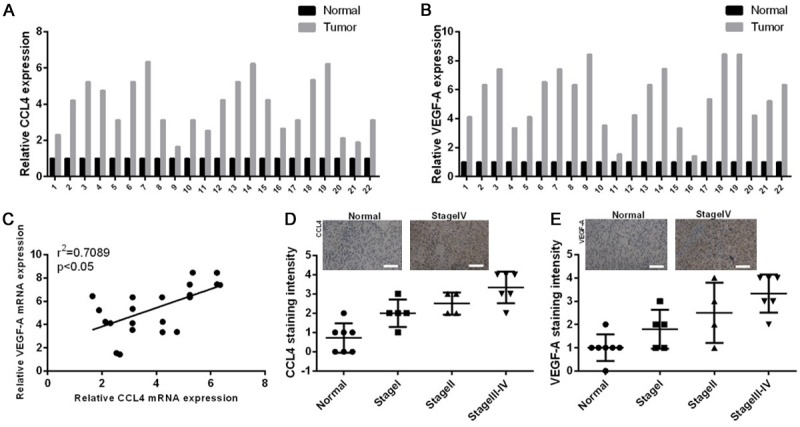
The expression levels of CCL4 and VEGF-A were highly expressed in the EC patients and showed positive correlation. A. The mRNA expression levels of CCL4 were detected by qRT-PCR in twenty-two tumor specimens and their corresponding normal specimens. B. VEGF-A expression was measured by qRT-PCR in twenty-two tumor specimens and their corresponding normal specimens. C. The correlation between CCL4 expression and VEGF-A expression was analyzed (r2 = 0.7089, P < 0.05). D. Quantitative intensity of CCL4 was counted in the normal and tumor specimens of different clinical stages using IHC, the representative images were shown at the top from one stage IV patient. E. VEGF-A intensity was analyzed in the normal and tumor specimens of different clinical stages using IHC, the representative images were shown at the top from one stage IV patient.
CCL4 promoted the migration and invasion abilities of EC cells by increasing VEGF-A expression
As the preliminary results showed that CCL4 and VEGF-A are upregulated in the EC tissues, we next examined the effects of CCL4 and VEGF-A on the migration and invasion of EC cells by Transwell migration and invasion assays in vitro. The invasive abilities of two EC cell lines (AN3CA and HEC-1B) were evaluated following CCL4 treatment for 30, 60, or 100 μg/ml. The results demonstrated that CCL4 promoted the invasion abilities of AN3CA and HEC-1B cells in a dose-dependent manner. Invasion cell numbers were obviously increased following 30, 60, and 100 μg/ml CCL4 treatment compared with the control group (*P< 0.5, **P < 0.01, ***P < 0.001, respectively), but the 10 μg/ml condition did not induce a significant alteration in invasion cell numbers. However, the CCL4 (100 μg/ml) induced enhancement of invasive ability was reversed when the neutralizing antibody of CCR5 or VEGF-A was added (Figure 2A and 2B). In addition, we found that CCL4 promoted the migration abilities of AN3CA and HEC-1B cells in a dose-dependent manner. The neutralizing antibody of CCR5 or VEGF-A then reversed CCL4 (100 μg/ml) induced enhancement of the migration abilities of AN3CA and HEC-1B cells (*P < 0.5, **P < 0.01, ***P < 0.001, Figure 2C and 2D).
Figure 2.
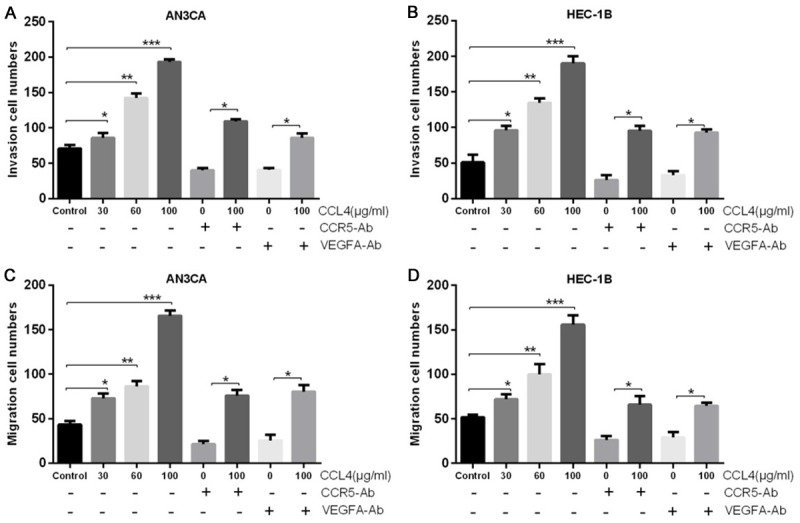
CCL4 promoted the migration and invasion abilities of EC cells by increasing VEGF-A expression. AN3CA and HEC-1B cells were treated with various concentrations of CCL4 (0, 10, 30, 60, and 100 μg/ml), or they were pretreated with CCR5 antibody or VEGF-A antibody and then treated with CCL4 (100 μg/ml). A and B. Transwell assays were performed to detect the invasion abilities of AN3CA and HEC-1B cells (*P < 0.05, **P < 0.01, ***P < 0.001). C and D. The migration abilities of AN3CA and HEC-1B cells were measured by Transwell assays (*P < 0.05, **P < 0.01, ***P < 0.001).
CCL4 promoted angiogenesis in a VEGF-A dependent manner
It is widely accepted that tumor growth needs to be supplied by blood vessels, and some evidence has demonstrated that VEGF-A is a critical regulator of tumor angiogenesis. To determine whether CCL4 affects VEGF-A dependent angiogenesis in EC cells, we applied the tube formation assay to investigate it. The results showed that the CM from CCL4 (30, 60, and 100 μg/ml) treated AN3CA or HEC-1B cells significantly promoted tube formation in a CCL4 dose-dependent manner, and this effect was completely blocked when a VEGF-A antibody was added into the CM (Figure 3A and 3B). These results indicated that CCL4 induced angiogenesis was VEGF-A mediated.
Figure 3.
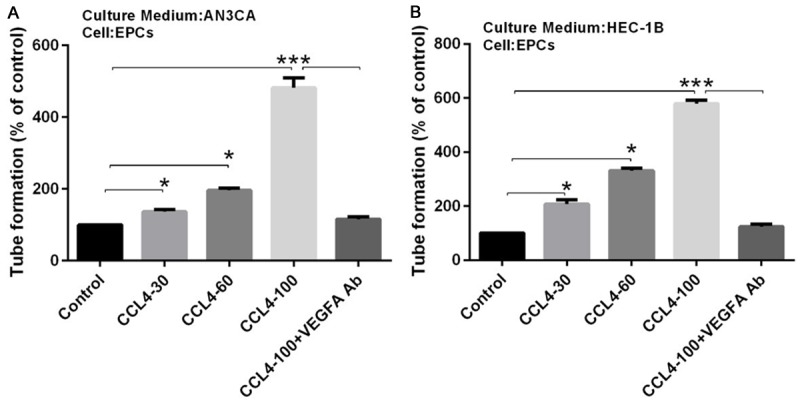
CCL4 promoted angiogenesis in a VEGF-A dependent manner. A and B: AN3CA and HEC-1B cells were pretreated with VEGF-A antibody for 30 mins, followed by stimulation with CCL4 (30, 60, 100 μg/ml) for 24 h, and then the culture medium was collected as conditional medium (CM) and applied to EPCs for 24 h. Tube formation in EPCs was quantified by averaging the length of the tubes in three randomly chosen microscope fields (*P < 0.05, ***P < 0.001).
Knockdown of CCL4 by shRNA inhibited tumor growth in vitro
To explore the role of CCL4 in regulating cell growth, the stable expression sh-CCL4 AN3CA and HEC-1B cells were established. Compared with the control group, the transfection of sh-CCL4 markedly decreased the level of CCL4 in EC cells (Figure 4A and 4B). Next, we tested the effects of sh-CCL4 on cellular growth. The MTT and colony formation assays results showed that the knockdown of CCL4 can inhibit cell growth in EC cells (Figure 4C-F). Furthermore, the Annexin V assay showed that sh-CCL4 caused a significant increase in cell apoptosis compared with the control group (Figure 4G and 4H). Taken together, these results revealed a functional role for CCL4 in regulating EC cell proliferation in vitro.
Figure 4.

CCL4 by shRNA inhibited tumor growth and induced cellular apoptosis in vitro. A and B: The efficiency of CCL4 shRNA (sh-CCL4) was detected with RTPCR in AN3CA and HEC-1B cells. C and D: Cell viability was determined for 12 h, 24 h, 36 h, and 48 h using an MTT assay with stable sh-CCL4 expression and control EC cells. E and F: The long-term cell proliferation capacity was determined by a colony formation assay with stable sh-CCL4 expression and control EC cells. G and H: Cell apoptosis was detected using an Annexin V assay with stable sh-CCL4 expression and control EC cells (*P < 0.05).
Knockdown of CCL4 by shRNA inhibited tumor growth of the mouse xenograft model in vivo
To further understand the role of CCL4 in the progression of EC, we established a tumor xenograft model in nude mice in vivo. We first constructed HEC-1B cell lines that were transfected with either CCL4 shRNA or control shRNA. Then, the transfected cells were injected into the nude mice, and several weeks after the injection, the size and weight of the tumors were calculated; we found that the tumor size and weight was significantly smaller in the CCL4 knockdown group compared with the control group (Figure 5A-C). In addition, we found that the CCL4 expression was positively correlated with VEGF-A (r2 = 0.8605, P < 0.01, Figure 5D).
Figure 5.
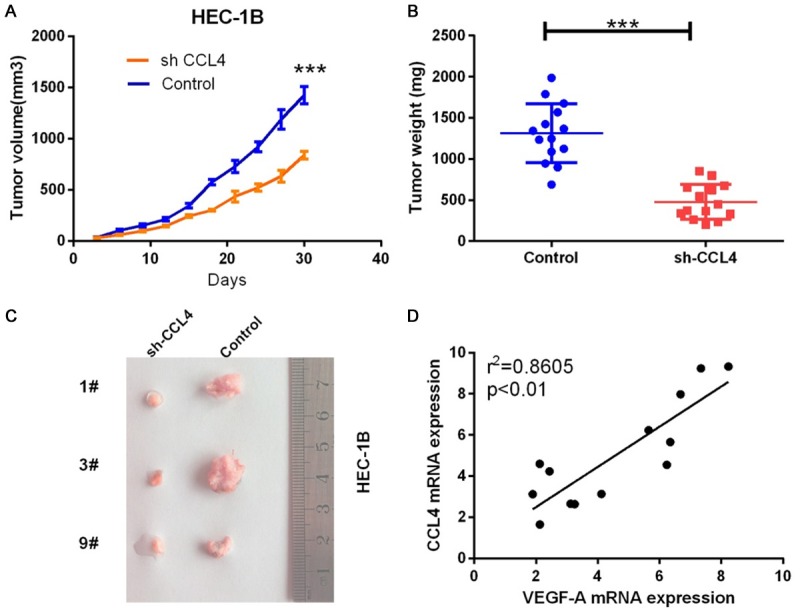
CCL4 by shRNA inhibited tumor growth in a mouse xenograft model in vivo. Control shRNA and CCL4 shRNA were injected into the HEC-1B cells and then inoculated into nude mice for 21 days. Finally, the nude mice were sacrificed, and the tumors were excised. A: The tumor volume was measured (***P < 0.001). B: The tumor weight was measured (***P < 0.001). C: The tumors were photographed with a microscope and shown. D: The correlation between CCL4 expression and VEGF-A expression was analyzed in a mouse xenograft model (r2 = 0.8605, P < 0.01).
CCL4 promoted angiogenesis by upregulating VEGF-A and pSTAT3
Although the phosphorylation of STAT3 occurs in several cancer cells, it is still undetermined whether CCL4 can lead to STAT3 activation in EC cells. Thus, we treated AN3CA and HEC-1B cells with CCL4 (30, 60, and 100 μg/ml), and then, the protein expression levels of pSTAT3 and STAT3 were detected by Western blot assay. The results showed that CCL4 upregulated the production of pSTAT3 in AN3CA and HEC-1B cell lines, and this effect was blocked by the application of VEGF-A antibody (Figure 6A and 6B). Additionally, the expression of STAT3 did not change significantly after treatment with CCL4 or CCL4 and VEGF-A antibody (Figure 6C and 6D). These results indicated that CCL4 may promote the angiogenesis and invasion of EC cells by increasing the production of VEGF-A, which upregulated the expression levels of the STAT3 signal pathway (Figure 6E).
Figure 6.
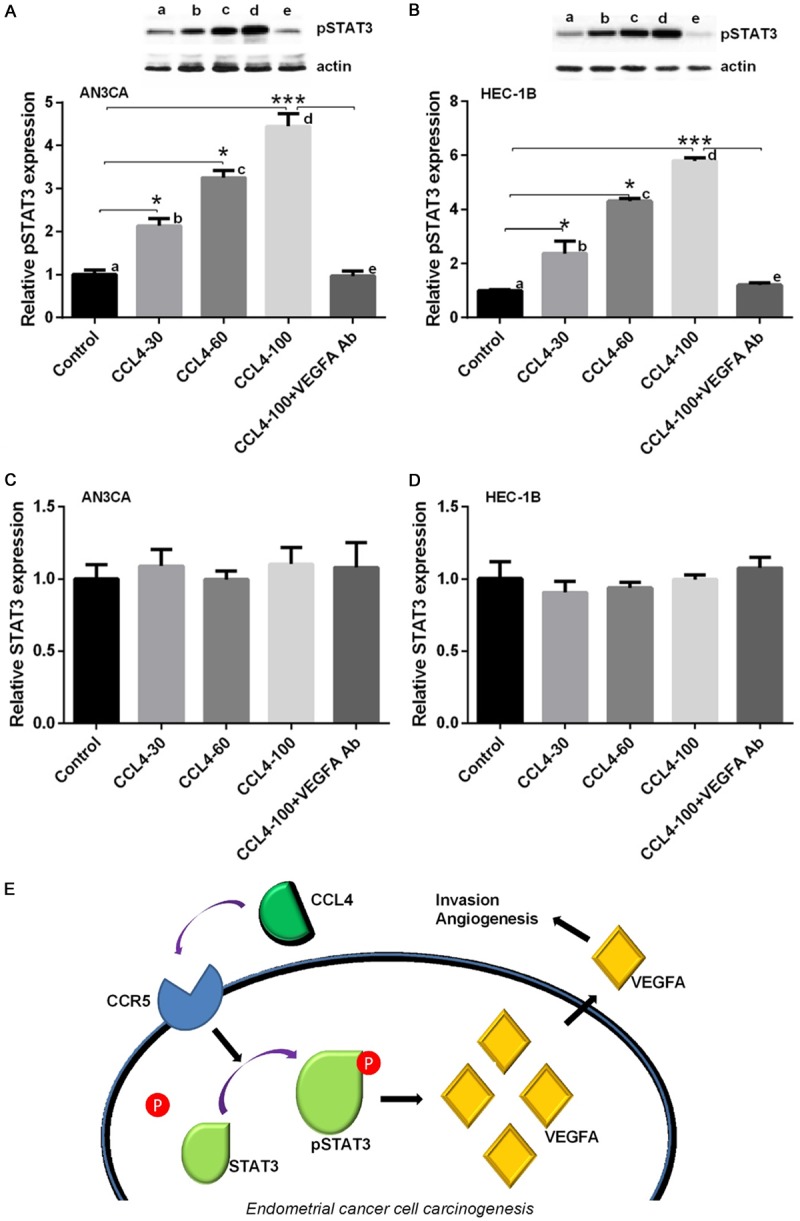
CCL4 promoted angiogenesis by upregulating VEGF-A and pSTAT3. AN3CA and HEC-1B cells were treated with CCL4 (30, 60, 100 μg/ml) or pretreated with the antibody of VEGF-A followed by the application of CCL4 (100 μg/ml). A: The protein expression level of pSTAT3 was detected by Western blot in treated AN3CA cells (*P < 0.05, ***P < 0.001). B: The protein expression level of pSTAT3 was detected by Western blot in treated HEC-1B cells (*P < 0.05, ***P < 0.001). C: The protein expression level of STAT3 was detected by Western blot in treated AN3CA cells. D: The protein expression level of STAT3 was detected by Western blot in treated HEC-1B cells. E: The underlying mechanism of CCL4 induced growth of EC cells.
Discussion
Endometrial carcinoma is a type of uterine cancer, and most cases of EC are diagnosed in the early clinical stages because of abnormal uterine bleeding. The tumor is confined to the uterus for most patients, and the treatments are curative. However, there are currently no effective treatments for patients whose tumors have progressed outside of the uterus [20]. Although the pathogenesis of EC is still unclear, increased evidence has indicated that chemokines may play a critical role in the progression and metastasis of this disease [21-23]. In the current study, we found that CCL4 upregulated the expression level of VEGF-A through the STAT3 pathway in EC, which is responsible for the increased tumor angiogenesis and invasive ability.
Chemokines are thought to be a connection between inflammation and cancer [24,25], and sufficient evidence has suggested that the chemokine system is involved in the pathogenesis of several cancers, including ovarian cancer, gastric cancer and prostate cancer [26-28]. Previous studies have demonstrated that CCL3 could promote tumor growth by increasing the VEGF-A production in human osteosarcoma cells [10], and CCL2 may promote the progression of human endometrial cancer by inhibiting the expression of LKB1, a kind of tumor suppressor [20]. Overexpression of CCL4 has been found in prostate cancer, and it plays a pivotal role in the progression of prostate tumors [29]. In our research, we found that the expressions of CCL4, CCR5 (the receptor of CCL4), and VEGF-A are upregulated in EC patients compared with normal specimens, and the production of CCL4, CCR5 and VEGF-A is associated with the clinical grade of the tumors in EC using an immunohistochemistry assay. We further confirmed the increase of CCL4 and VEGF-A using qRT-PCR. In addition, we demonstrated that the mRNA expression level of CCL4 is positively correlated with VEGF-A in EC patients.
CC-chemokine receptor 5 (CCR5) is a G protein coupled receptor that functions as a chemokine receptor by binding to several chemokines, including CCL3, CCL4 and others [30]. Increasing evidence has supported that CCR5 is expressed on the surface of some tumor cells and is involved in angiogenesis via VEGF-A [10,31]. In this research, we found that increased CCL4 expression was positively correlated with the invasive ability of AN3CA and HEC-1B, and we also found that the antibody of CCR5 or VEGF-A could block this effect induced by CCL4. Moreover, ELISA and qRT-PCR results indicated that CCL4 promoted the expression level of VEGF-A in AN3CA and HEC-1B cells, and the upregulation of VEGF-A was also reversed by the antibodies of CCR5 or VEGF-A. These data suggested that CCL4 promoted the invasive ability and VEGF-A production by interacting with CCR5. VEGF-A plays a critical role in the growth and progression of tumor cells and is considered the most notable mediator of angiogenesis in various cancers [32,33]. The tube formation assay demonstrated that CCL4 promoted angiogenesis by upregulating the expression level of VEGF-A.
To further confirm the effects of CCL4 in EC cells, we knocked down the expression of CCL4 in HEC-1B cells by shRNA, and then we transferred the cells into nude mice to establish a tumor xenograft model in vivo. The results showed that silenced CCL4 inhibited the growth of EC tumors in nude mice.
Signal transducers and activators of transcription (STAT3) is a member of the STAT family, which acts as transcription factors and are activated by the phosphorylation of tyrosine [34]. Activated STAT3 has been found in several cancers, including colorectal cancer, lung cancer and endometrial cancer [19,35,36]. In our results, we demonstrated that STAT3 was activated after exposure to CCL4 (30, 60, 100 μg/ml) in AN3CA and HEC-1B cells, and the CCL4 (100 μg/ml) induced upregulation of pSTAT3 was reversed by the application of VEGF-A neutralizing antibody. These results suggested that the phosphorylation of STAT3 may be involved in the CCL4-VEGF-A pathway, which is responsible for the progression of EC cells.
In conclusion, CCL4 was highly expressed in the EC cells, and it promoted the expression of VEGF-A by activating STAT3 via binding to CCR5. Additionally, VEGF-A promoted the invasive ability and angiogenesis of the EC cells. These results support the possibility that CCL4 may be a novel therapeutic target in EC.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No 81402165).
Disclosure of conflict of interest
None.
References
- 1.Jick H, Walker AM, Rothman KJ. The epidemic of endometrial cancer: a commentary. Am J Public Health. 1980;70:264–267. doi: 10.2105/ajph.70.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trimble EL, Harlan LC, Clegg LX, Stevens JL. Pre-operative imaging, surgery and adjuvant therapy for women diagnosed with cancer of the corpus uteri in community practice in the United States. Gynecol Oncol. 2005;96:741–748. doi: 10.1016/j.ygyno.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson MJ, Thiel KW, Leslie KK. Past, present, and future of hormonal therapy in recurrent endometrial cancer. Int J Womens Health. 2014;6:429–435. doi: 10.2147/IJWH.S40942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gadducci A, Cosio S, Genazzani AR. Old and new perspectives in the pharmacological treatment of advanced or recurrent endome-trial cancer: hormonal therapy, chemotherapy and molecularly targeted therapies. Crit Rev Oncol Hematol. 2006;58:242–256. doi: 10.1016/j.critrevonc.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Gullino PM. Angiogenesis and oncogenesis. J Natl Cancer Inst. 1978;61:639–643. [PubMed] [Google Scholar]
- 8.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto K, Ema M. Roles of VEGF-A signalling in development, regeneration, and tumours. J Biochem. 2014;156:1–10. doi: 10.1093/jb/mvu031. [DOI] [PubMed] [Google Scholar]
- 10.Liao YY, Tsai HC, Chou PY, Wang SW, Chen HT, Lin YM, Chiang IP, Chang TM, Hsu SK, Chou MC, Tang CH, Fong YC. CCL3 promotes angiogenesis by dysregulation of miR-374b/VEGF-A axis in human osteosarcoma cells. Oncotarget. 2016;7:4310–4325. doi: 10.18632/oncotarget.6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lira SA, Furtado GC. The biology of chemokines and their receptors. Immunol Res. 2012;54:111–120. doi: 10.1007/s12026-012-8313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 13.Locati M, Deuschle U, Massardi ML, Martinez FO, Sironi M, Sozzani S, Bartfai T, Mantovani A. Analysis of the gene expression profile activated by the CC chemokine ligand 5/RANTES and by lipopolysaccharide in human monocytes. J Immunol. 2002;168:3557–3562. doi: 10.4049/jimmunol.168.7.3557. [DOI] [PubMed] [Google Scholar]
- 14.Erreni M, Bianchi P, Laghi L, Mirolo M, Fabbri M, Locati M, Mantovani A, Allavena P. Expression of chemokines and chemokine receptors in human colon cancer. Methods Enzymol. 2009;460:105–121. doi: 10.1016/S0076-6879(09)05205-7. [DOI] [PubMed] [Google Scholar]
- 15.Milliken D, Scotton C, Raju S, Balkwill F, Wilson J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res. 2002;8:1108–1114. [PubMed] [Google Scholar]
- 16.Blum DL, Koyama T, M’Koma AE, Iturregui JM, Martinez-Ferrer M, Uwamariya C, Smith JA Jr, Clark PE, Bhowmick NA. Chemokine markers predict biochemical recurrence of prostate cancer following prostatectomy. Clin Cancer Res. 2008;14:7790–7797. doi: 10.1158/1078-0432.CCR-08-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma YR, Zhang S, Sun Y, Liu YY, Song Q, Hao YW. [MIP-1alpha promotes the migration ability of Jurkat cell through human brain microvascular endothelial cell monolayer] . Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014;22:35–39. doi: 10.7534/j.issn.1009-2137.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Ding L, Li B, Zhao Y, Fu YF, Hu EL, Hu QG, Ni YH, Hou YY. Serum CCL2 and CCL3 as potential biomarkers for the diagnosis of oral squamous cell carcinoma. Tumour Biol. 2014;35:10539–10546. doi: 10.1007/s13277-014-2306-1. [DOI] [PubMed] [Google Scholar]
- 19.Zhu M, Che Q, Liao Y, Wang H, Wang J, Chen Z, Wang F, Dai C, Wan X. Oncostatin M activates STAT3 to promote endometrial cancer invasion and angiogenesis. Oncol Rep. 2015;34:129–138. doi: 10.3892/or.2015.3951. [DOI] [PubMed] [Google Scholar]
- 20.Pena CG, Nakada Y, Saatcioglu HD, Aloisio GM, Cuevas I, Zhang S, Miller DS, Lea JS, Wong KK, DeBerardinis RJ, Amelio AL, Brekken RA, Castrillon DH. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Invest. 2015;125:4063–4076. doi: 10.1172/JCI82152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace AE, Sales KJ, Catalano RD, Anderson RA, Williams AR, Wilson MR, Schwarze J, Wang H, Rossi AG, Jabbour HN. Prostaglandin F2alpha-F-prostanoid receptor signaling promotes neutrophil chemotaxis via chemokine (C-X-C motif) ligand 1 in endometrial adenocarcinoma. Cancer Res. 2009;69:5726–5733. doi: 10.1158/0008-5472.CAN-09-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakane R, Tsubamoto H, Sakata K, Inoue K, Ogino M, Shibahara H, Hao H, Hirota S. Expression of chemokine ligand 18 in stage IA low-grade endometrial cancer. Anticancer Res. 2014;34:5331–5336. [PubMed] [Google Scholar]
- 23.Attar R, Agachan B, Kuran SB, Cacina C, Sozen S, Yurdum LM, Attar E, Isbir T. Association of CCL2 and CCR2 gene variants with endometrial cancer in Turkish women. In Vivo. 2010;24:243–248. [PubMed] [Google Scholar]
- 24.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 25.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son DS, Parl AK, Rice VM, Khabele D. Keratinocyte chemoattractant (KC)/human growthregulated oncogene (GRO) chemokines and pro-inflammatory chemokine networks in mouse and human ovarian epithelial cancer cells. Cancer Biol Ther. 2007;6:1302–1312. doi: 10.4161/cbt.6.8.4506. [DOI] [PubMed] [Google Scholar]
- 27.Leung SY, Yuen ST, Chu KM, Mathy JA, Li R, Chan AS, Law S, Wong J, Chen X, So S. Expression profiling identifies chemokine (C-C motif) ligand 18 as an independent prognostic indicator in gastric cancer. Gastroenterology. 2004;127:457–469. doi: 10.1053/j.gastro.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102:522–528. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang LY, Izumi K, Lai KP, Liang L, Li L, Miyamoto H, Lin WJ, Chang C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013;73:5633–5646. doi: 10.1158/0008-5472.CAN-12-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onuffer JJ, Horuk R. Chemokines, chemokine receptors and small-molecule antagonists: recent developments. Trends Pharmacol Sci. 2002;23:459–467. doi: 10.1016/s0165-6147(02)02064-3. [DOI] [PubMed] [Google Scholar]
- 31.Barcelos LS, Coelho AM, Russo RC, Guabiraba R, Souza AL, Bruno-Lima G Jr, Proudfoot AE, Andrade SP, Teixeira MM. Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc Res. 2009;78:148–154. doi: 10.1016/j.mvr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 33.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 34.Darnell JE Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 35.Morikawa T, Baba Y, Yamauchi M, Kuchiba A, Nosho K, Shima K, Tanaka N, Huttenhower C, Frank DA, Fuchs CS, Ogino S. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin Cancer Res. 2011;17:1452–1462. doi: 10.1158/1078-0432.CCR-10-2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang R, Jin Z, Liu Z, Sun L, Wang L, Li K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol Diagn Ther. 2011;15:347–352. doi: 10.1007/BF03256470. [DOI] [PubMed] [Google Scholar]