

Abstract
Non-coding RNAs are critical regulators of tumor biology. nc886, a recently identified non-coding RNA, is overexpressed in some tumors, but undetected in others. However, the precise role of nc886 remains unclear in cervical cancers. In this study, we found that nc886, major vault protein (MVP), and E2F1 exhibited coordinate expression as they were silenced in normal tissues but overexpressed in cervical cancer tissues. We subsequently demonstrate that nc886 upregulation was a critical response to chemotherapy treatment of cervical cancer cells. Mechanistically, inhibition of nc886 increased chemosensitivity, induced apoptosis, and suppressed the protein expression of MVP, a critical regulator of drug resistance. Furthermore, we identify E2F1 as a key transcription regulator of nc886 that directly interacts and modulates promoter activity. Taken together, we demonstrate that E2F1 sufficiently promotes nc886 transcription and in turn MVP expression to drive drug resistance in cervical cancer cells.
Keywords: Cervical cancer, drug resistance, Nc886, E2F1, MVP
Introduction
Cervical cancer is the second most common type of cancer in women with an estimated 500,000 new cases diagnosed and 270,000 deaths each year [1]. Surgical resection in combination with postoperative radiotherapy is the primary treatment modality for patients with cervical cancer, while chemotherapy is generally utilized to treat patients that present with metastatic or recurrence disease [2]. Persistent infection with high-risk human papillomavirus (HPV) is considered as one of the main causes of cervical cancer [3]. Investigations have shown that upon infection, HPV E6 and E7 oncoproteins promote cell growth and inhibit apoptosis by targeting p53 and pRb tumor suppressors, respectively [4,5]. In addition to viral antigens, numerous genetic and epigenetic alterations have been shown to transform cervical cells and may drive carcinogenesis [6]. Targeting known and elucidating unknown molecular mechanisms that contribute to carcinogenesis is critical in developing novel therapeutic agents for cervical cancer patients.
Protein coding open reading frames comprise 1%-2% of the human genome. Expectedly, most of the transcribed mammalian genome consists of non-coding transcripts and this includes non-coding RNAs (ncRNAs) [7]. Increasing evidence suggests that a great number of ncRNAs regulate cellular biology (e.g., cell proliferation, apoptosis and differentiation) by suppressing the expression of target genes [8,9]. These ncRNAs, either small (<50 nucleotides) or long (>200 nucleotides) in length, are frequently deregulated in human tumors and are associated with cancer initiation and progression [10]. nc886 (also known as pre-miR-886 or vtRNA2-1) is a recently identified regulatory ncRNA that is composed of 102 nucleotides and located on chromosome 5q31.1 [11]. Increasing evidence has shown that nc886 plays an important role in several human cancers, including small cell lung cancer, cholangiocarcinoma, and esophageal cancer [12-15]. Our previous work has also demonstrated that nc886 is upregulated in cervical squamous cell carcinomas (CSCC) and reduces CSCC cell apoptosis by down-regulating Bax protein expression [16]. However, it remains unclear if the anti-apoptotic functions of nc886 regulates chemosensitivity of cervical cancers and if targeting its activity would affect chemotherapy resistant patients. Therefore, this study examined the potential role of nc886 in chemotherapeutics resistance of cervical cancer cells as well as underlying molecular mechanism associated with this phenotype.
Materials and methods
Patients
The cervical cancer tissues (10 early-stage tumor and 10 advanced-stage tumor) and 10 matched normal tissues were obtained from the Clinical Laboratory of Beijing Tiantan Hospital, Beijing. This study was approved by the Ethics Committee of Beijing Tiantan Hospital, and informed consent was obtained from each patient.
Cell culture
SiHa and HeLa cell lines were purchased from the American Type Culture Collection (ATCC, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, USA) with 10% FBS at 37°C and 5% CO2.
Real-time PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, USA). 2 μg of total RNA was used to synthesize cDNA using Reverse Transcription kit (Ferments, USA). Real time PCR reactions were performed using Brilliant II SYBR Green Master Mix (Stratagene, USA). The expression of nc886 was normalized to the level of human U6 snRNA. The sequences of the forward and reverse primers for human nc886 were 5’-CGGGTCGGAGTTAGCTCA-3’ and 5’-GTGCAGGGTCCGAGGT-3’, respectively, and those for human U6 snRNA were 5’-ATTGGAACGATACAGAGAAGATT-3’ and 5’-GGAACGCTTCACGAATTTG-3’, respectively. Melting curves for each PCR reaction were generated to ensure the purity of the amplification products. PCR was performed in triplicate for each sample. The fold change for nc886 was calculated by the 2-ΔΔCt method [17].
Dimethyl thiazolyl diphenyl tetrazolium (MTT) assay
Cells (5000 cells per well) were seeded in 100 μL of media in 96-well plates and transfected with anti-nc886 (50 nM) or negative control (50 nM). Every 24 h post transfection, 20 μL of MTT reagent (Solarbio, China) was added to wells followed by a 4 h incubation. After removal of medium, 200 μL dimethyl sulfoxide (DMSO) was added to dissolve the formazan and the absorbance was measured at 490 nm.
Apoptosis assay
Flow cytometry was performed to detect apoptosis after fixing and staining cells with an annexin V-fluorescein-5-isothiocyanate Apoptosis Detection Kit (Biovision, USA). Briefly, SiHa cells were transfected with anti-nc886 (50 nM) or a negative control (50 nM). Forty-eight hours after transfection, cells were incubated with annexin V-FITC and propidium iodide (PI) for 30 min at 4°C in the dark. Cell apoptosis was analyzed by using flow cytometry and the percentages of apoptotic cells was subsequently calculated.
Western blot
Cells were lysed in RIPA lysis buffer and the protein concentration of the extracts was determined using a BCA assay (Pierce, Rockford, IL, USA). A total of 30 μg of proteins were subject to SDS-PAGE and resulting membranes were incubated overnight at 4°C with antibodies to MVP (Santa Cruz, CA, USA) followed by HRP-conjugated anti-rabbit IgG for 1 h. β-actin served as an internal control (Santa Cruz, USA). The protein bands were developed using an enhanced chemiluminescence reagent (Pierce, USA).
Luciferase reporter assay
The full-length nc886 promoter and mutant luciferase reporter plasmids were constructed based on the pGL3 promoter vector (Promega, San Luis Obispo, CA, USA). Cells were transfected with 100 ng of the luciferase reporter plasmids and 40 nM of full-length or mutant nc886 promoter constructs using Lipofectamine 2000 (Invitrogen, USA). Cells were harvested 24 h after transfection and luciferase activity was determined using the Promega Dual-Luciferase™ reporter assay system (San Luis Obispo, CA, USA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was carried out as described previously [18]. Immunoprecipitation was performed overnight at 4°C using anti-E2F1 antibody or mouse IgG control. DNA was analyzed by real time PCR directed to specific regions of nc886 promoter and results were normalized to DNA input controls.
Fluorescent in situ hybridization (FISH)
FISH was performed to detect the expression of MVP, E2F1, and nc886. The slides were dewaxed in xylene, washed in ethanol and water. After air-drying the slides, the probe was applied and slides were denatured at 73°C for 8 minutes. Next, the slides were hybridized at 37°C overnight and then washed in SCC buffer for 5 min. After blocking the slides, they were incubated with antibodies against MVP and E2F1 followed by incubation with goat anti-rabbit IgG-Cy3. The slides were then dried, counterstained and observed under a confocal microscopy (LSM 710, Carl Zeiss, Germany).
Statistical analysis
All the results are expressed as means ± standard deviation (S.D.). Data were analyzed by SPSS 17.0 (SPSS Inc., Chicago, IL, USA). When comparing two groups, Student’s t-test was used to calculate the differences. When comparing more than two groups, a one-way analysis of variance (ANOVA) was used followed by a LSD test. All p-values were two-sided, P<0.05 was considered statistically significant.
Results
Regulation of nc886 expression by chemotherapeutic agents in cervical carcinoma cells
In our previous study, we found that nc886 inhibited apoptosis of cervical carcinoma cells by targeted suppression of Bax expression [16]. To examine the potential drug-resistance role of nc866 in cervical cancer, we further explored the relationship between nc886 and chemotherapeutic agents. Cervical cancer SiHa cells were exposed to paclitaxel or VP16 (etoposide) at different concentrations and we utilized Real time PCR to determine expression of nc886 at each respective dose. Results demonstrate that nc886 expression was upregulated by paclitaxel in a dose-dependent manner (Figure 1A). Similarly, nc886 expression in SiHa cells was also increased upon VP16 treatment in a dose-dependent fashion (Figure 1B). Our current and previous results demonstrate nc886 expression is a mechanistic response to chemotherapy and suggests that nc886 potentially promotes chemotherapeutic resistance of cervical cancer cells due to its anti-apoptotic properties [16].
Figure 1.
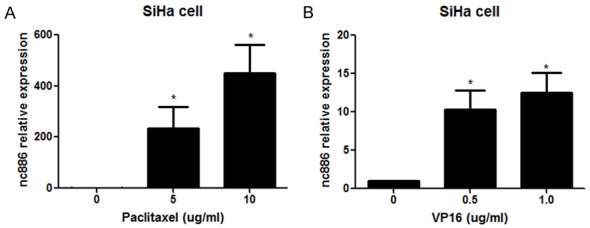
Expression of nc886 in SiHa cells exposed to chemotherapeutic agents. SiHa cells were exposed to chemotherapeutic agents including paclitaxel (5 ug/ml and 10 ug/ml) (A) and VP16 (0.5 ug/ml and 1.0 ug/ml) (B). After 48 h, real time PCR was performed to analyze the expression of nc886 in SiHa cells. *P<0.05.
nc886 suppression induces apoptosis of cervical cancer cells upon chemotherapy
To explore the potential of ablating nc866-mediated chemotherapy resistance in cervical cancer cells, we next suppressed nc886 and determined its effects on SiHa cells. Utilizing MTT to examine relative cell viability, we demonstrate that targeted suppression of nc886 expression increased the chemosensitivity of SiHa cells treated by paclitaxel in a time-dependent manner (Figure 2A). We next determined if defects in viability upon nc866 suppression was specifically due to apoptosis. Upon transfection of SiHa cells with anti-nc866, we observe through flow cytometry analysis that nc886 suppression increased the population of apoptotic cells upon exposure to paclitaxel (Figure 2B). Together, our results indicate nc886 expression regulates chemosensitivity of cervical cancer cells.
Figure 2.
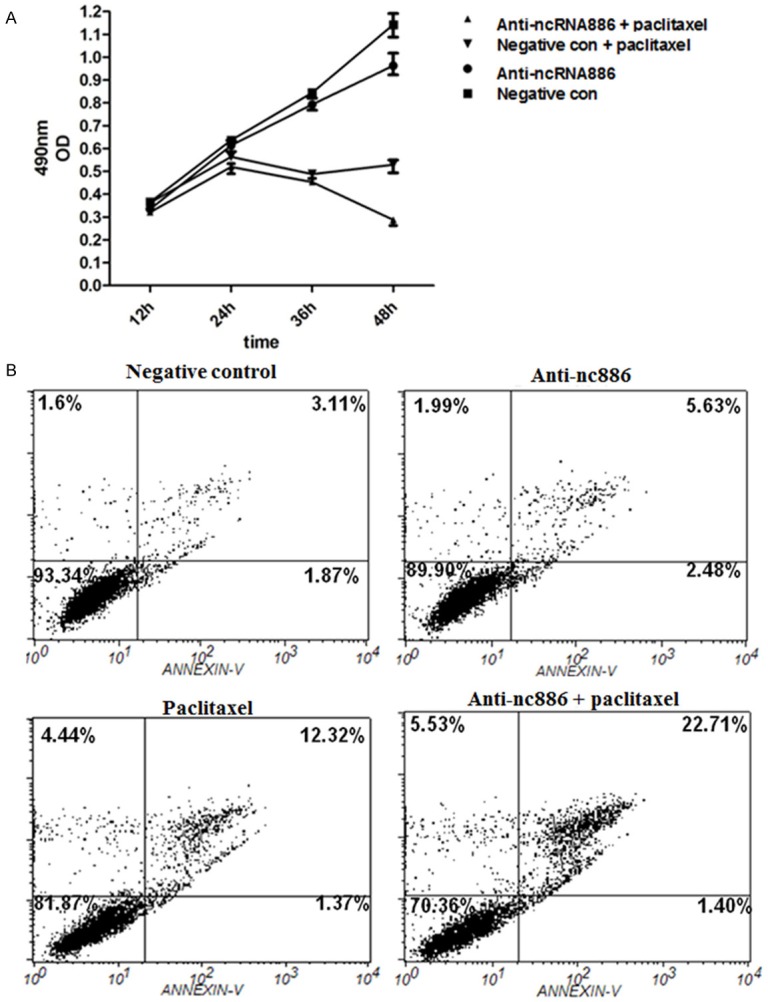
Effects of nc886 on cervical cancer cell viability and apoptosis. SiHa cells treated with or without paclitaxel were transfected with anti-nc886 or negative control. A. MTT assay was performed to measure cell viability at 12 h, 24 h, 36 h, and 48 h post-transfection. B. Flow cytometry was applied to determine the apoptosis rate in SiHa cells in different groups.
nc886 regulates MVP expression in cervical cancer cells
We next examined a potential regulatory role of nc886 on MVP expression in cervical cancer cells. In anti-nc886 transfected SiHa cells, we first demonstrate through real time PCR that suppression of nc886 led to decreased expression of MVP transcripts (Figure 3A). To further confirm the negative regulation of regulation nc886 on MVP, we also observe that cells with anti-nc886 also had significantly reduced levels of MVP protein expression (Figure 3B). These results confirm nc866 levels are critical in regulating MVP expression in cervical cancer cells.
Figure 3.
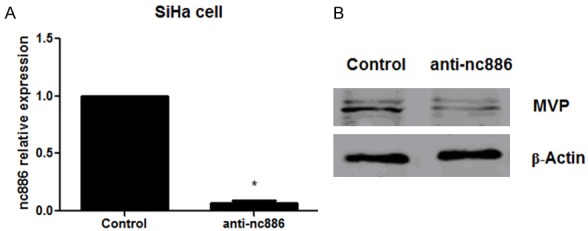
Effect of nc886 on the expression of MVP in cervical cancer cells. SiHa cells were transfected with anti-nc886 or negative control. Real time PCR and western blot were used to measure the expression of nc886 (A) and MVP (B), respectively. *P<0.05.
E2F1 regulates nc886 expression in cervical cancer cells
Given its role of nc886 in chemotherapy resistance, we proceeded to investigate upstream mechanisms that could critically regulate its expression in cervical cancer cells. Upon sequence and motif analysis (http://genome.ucsc.edu/cgi-bin/hgGateway), we identify an upstream promoter region of nc886 that contained a potential E2F1 binding site at -202 and -299 (Figure 4A). To begin determining the potential transcription regulatory role of E2F1, we next examined the effects of E2F1 overexpression on nc886. We transfected recombinant plasmids encoding E2F1 to overexpress it in cervical cancer cell lines SiHa and HeLa. In agreement with our hypothesis, relative to negative control transfected cells, we observe that E2F1 overexpression sufficiently promoted nc886 expression in SiHa (Figure 4B) and HeLa cells (Figure 4C). Studies have shown that caffeine inhibits expression of E2F1 [18]. We found that E2F1 activity was necessary for nc886 expression, as caffeine-treated SiHa cells exhibited a dose-dependent decrease of nc886 expression (Figure 4D). Overall, our results demonstrate that E2F1 is both necessary and sufficient in regulating nc886 expression in cervical cancer cells.
Figure 4.
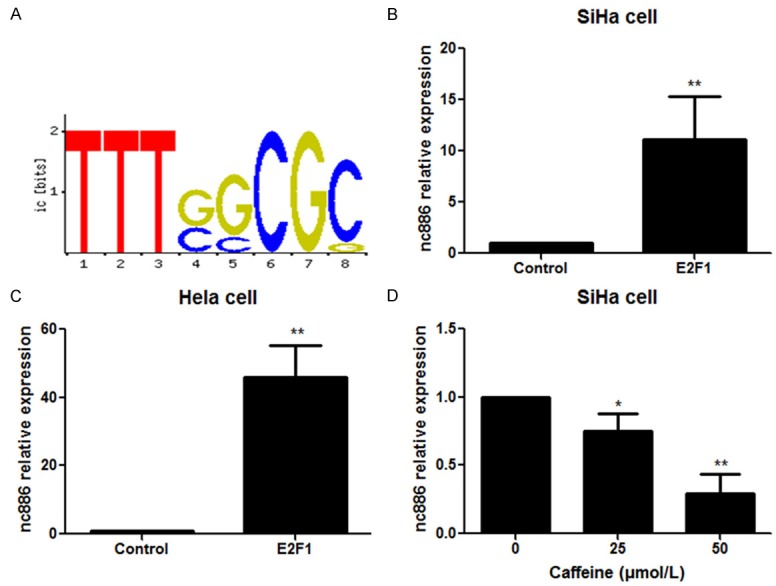
Effect of E2F1 on the expression of nc886 in cervical cancer cells. (A) Sequence analysis of nc886 promoter region. SiHa (B) and HeLa (C) cells were transfected with the recombinant plasmids encoding E2F1 followed by determination of nc886 expression. (D) SiHa cells were exposed to caffeine (25 nM and 50 nM) for 48 h and nc886 levels were then detected. *P<0.05, **P<0.01.
E2F1 directly interacts with the nc886 promoter
We next sought to experimentally determine if E2F1 specifically regulates nc886 transcription through direct interactions with its promoter. We first constructed different recombinant vectors that contained a luciferase gene preceded by either a full-length nc886 promoter or one in which we deleted E2F1 motifs (Figure 5A). Upon co-transfection of these luciferase constructs with plasmids encoding E2F1, we subsequently determined relative luciferase activity in the transfected cells. We observed higher luciferase activity in cells transfected with either a full-length nc886 promoter or the deletion 1 mutant (containing the binding sites) relative to those transfected with the deletion 2 mutant (lacking the binding sites) or negative control (pGL3-empty vector) (Figure 5B). In parallel experiments, we examined if E2F1 physically interacted at the nc886 promoter. We precipitated E2F1 and subsequently utilized real time PCR to determine enrichment of the nc886 promoter sequence relative to input. Relative to IgG controls, the E2F1 immuno-precipitant was significantly enriched of this sequence (Figure 5C). Together, our results demonstrates that E2F1 directly interacts with the nc886 promoter to regulate its transcription.
Figure 5.
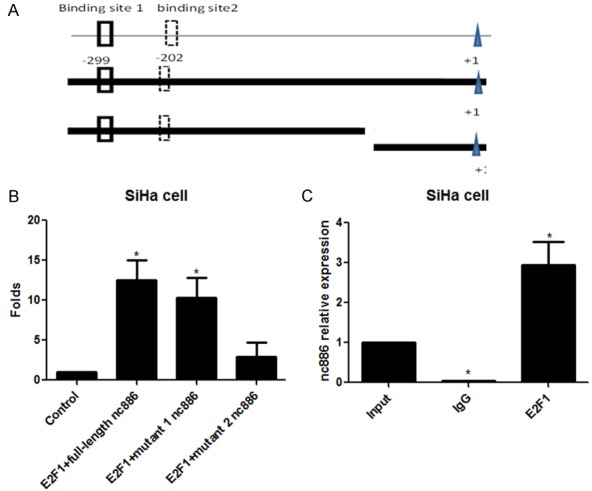
E2F1 directly interacted with nc886. A. Construction of recombinant plasmids containing full-length promoter or deleted promoter fragments of nc886. B. Luciferase assay was performed to measure the fluorescent activity after co-transfection with E2F1 and full-length or mutant nc886 promoter luciferase constructs. C. The relative enrichment of nc886 promoter sequence in E2F1-immunoprecipitants was determined by real time PCR and compared to IgG controls. *P<0.05.
Clinical examination of nc886, E2F1 and MVP expression in cervical cancer tissues
Finally, we aimed to clinically confirm the regulatory relationship between nc886, E2F1 and MVP in cervical cancer tissues. To do so, we utilized FISH to detect relative expression in normal, early and late stage cervical cancers. While nc886 was undetectable in normal tissues, we observed modest expression in the early-stage cervical cancer tissues but significant overexpression in the tissue derived from advanced tumors. We also note that E2F1 was expression was generally overexpressed in cancerous tissues relative to normal tissues. Lastly, MVP was hardly detectable in normal tissues. Similar to nc886, MVP expression was modest in early-stage but significantly expressed in the advanced-stage tumors (Figure 6). Taken together, our clinical analysis of cervical cancer tissues demonstrated a positive regulatory relationship between E2F1, nc886, and MVP expression levels.
Figure 6.
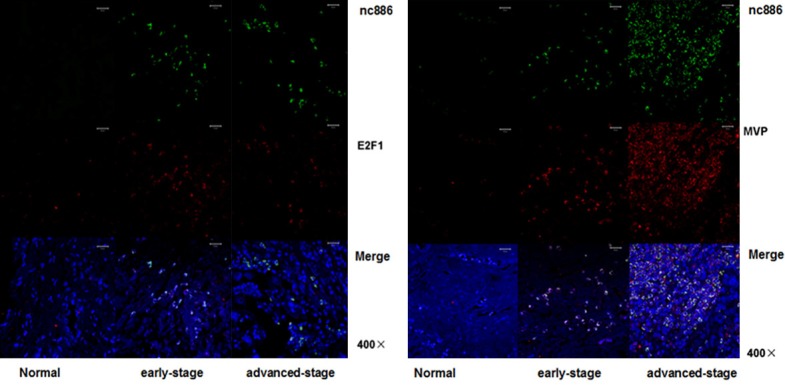
Expression of nc886, E2F1 and MVP in cervical cancer tissues. FISH was performed to detect the expression of nc886 (green), E2F1 (red), and MVP (red) in the normal tissue, early-stage and advanced-stage tumors.
Discussion
Our work identifies nc886 as a novel regulator of cervical cancer chemosensitivity and demonstrates it specifically regulates MVP expression upon treatment with chemotherapeutic agents. Additionally, we demonstrate E2F1 is necessary and sufficiently regulates nc886 expression through direct promoter interaction. Finally, the coordinate expression of nc886, MVP and E2F1 in cervical cancer tissues support our results in pre-clinical models.
nc886 was originally registered in the miRNA database because its mature products (miR-886-5p and -886-3p) was captured in high-throughput sequencing and formed a stem in a predicted stem-loop hairpin structure [17,19]. nc886 also has a proposed role as a vault RNA (vtRNA), which are component of the vault complex involved in chemotherapeutics resistance [20]. Studies have shown that nc886 is associated with carcinogenesis and progression of human cancers, including cervical cancer, cholangiocarcinoma, esophageal cancer, thyroid cancer, and small cell lung cancer [12-16,21]. As an example, Lee et al. reported that nc886 regulates gene expression through interaction with RNA-activated protein kinase (PKR), and nc886 inhibition suppressed cell proliferation in cervical cancer and colorectal cancer cells [22]. They also demonstrate that nc886 positively regulated proliferation, invasion and migration of thyroid cancer cells [21]. Conversely, another study demonstrated that CpG hypermethylation suppressed nc886 expression in human gastric cancer tissues and that ectopic expression of nc886 inhibited cell proliferation by downregulation of oncogenes including NF-κB, FOS, and MYC [23]. Together, these studies suggest that nc886 functions are either oncogenic or a tumor suppressive depending on the cellular context which likely dictates its signaling properties and target genes. Our previous study demonstrated that nc886 plays an oncogenic role in cervical cancers by downregulating the expression of the pro-apoptotic protein Bax [16]. This work demonstrates that nc886 was absent in normal cervical tissues, detectable in early-stage tumors while abundant in advanced-stage tumors. Moreover, we demonstrate that nc886 was elevated in cervical cancer cells after exposure to chemotherapeutic agents in a dose-dependent manner and that nc886 suppression increased chemosensitivity through apoptotic cell death. Our overall results demonstrate a critical role of nc886 in regulating therapeutic response and resistance of cervical cancer cells.
Major vault protein (MVP), also known as lung resistance protein (LRP), forms a hollow, barrel-like structure in the cell called the vault. It is widely expressed in normal tissues and plays a diverse role in intracellular signaling, cell growth/survival, and innate immunity [24,25]. Additionally, MVP is frequently elevated in human cancer cells resistant to chemotherapeutic agents [26]. It has been reported that siRNA-mediated knockdown of MVP increases doxorubicin sensitivity and its accumulation in the nuclear of human bladder cancer cells [27]. In our study, inhibition of nc886 significantly suppressed the protein expression of MVP in cervical cancer cells. In addition, MVP was rarely detected in normal cervical tissues, but was abundant in the advanced-stage cervical tumors. Our results provide support that MVP modulates nc886-mediated chemotherapy resistance in cervical cancer cells.
As a critical member of E2F family of transcription factors, E2F1 regulates cell proliferation, differentiation, cell cycle progression, DNA repair, and apoptosis [28]. E2F1 function is either oncogenic or a tumor suppressive depending on the cellular context and micro-environment [29]. Mechanistically, E2F1 interacts with critical epigenetic regulators to modulate histone modifications and gene expression [30,31]. For instance, E2F1 has been reported to function as a transcriptional activator by interacting with the histone acetyltransferase (HAT) [32]. In other studies, E2F1 acts as a transcriptional repressor either alone or in association with specific co-factors, such as HDAC1 and DNMT1 [33]. In this study, we found that E2F1 was elevated in cervical cancer tissues. Moreover, we identify that nc886 promoters contained functional E2F1 binding sites. Exogenous overexpression of E2F1 promoted the expression of nc886 in cervical cancer cells. Caffeine is an inhibitor of ataxia telangiectasia mutated (ATM) enzyme and can block E2F1 signaling and inhibits E2F1-mediated apoptosis [18]. Upon E2F1 inhibition with caffeine, we also suppressed nc886 expression in a dose-dependent manner. Lastly, from luciferase assays and ChIP real time PCR analysis, we also demonstrate that E2F1 directly interacts and regulates transcription at the nc886 promoter. Our overall results demonstrate that E2F1 is a key upstream regulator of nc886 in cervical cancer cells.
In conclusion, this study demonstrates that E2F1 regulates nc886 and in turn MVP to promote chemotherapy resistance. This novel mechanism informs of therapeutic strategies to treat cervical cancers that develop resistance.
Acknowledgements
The study was supported by National Natural Science Foundation (No. 81101969) and Beijing Natural Science Foundation (No. 7152051).
Disclosure of conflict of interest
None.
References
- 1.Wright TC, Stoler MH, Behrens CM, Sharma A, Zhang G, Wright TL. Primary cervical cancer screening with human papillomavirus: end of study results from the ATHENA study using HPV as the first-line screening test. Gynecol Oncol. 2015;136:189–97. doi: 10.1016/j.ygyno.2014.11.076. [DOI] [PubMed] [Google Scholar]
- 2.Elit LM, Hirte H. Management of advanced or recurrent cervical cancer: chemotherapy and beyond. Expert Rev Anticancer Ther. 2014;14:319–32. doi: 10.1586/14737140.2014.866041. [DOI] [PubMed] [Google Scholar]
- 3.Kessler TA. Cervical cancer: prevention and early detection. Semin Oncol Nurs. 2017;33:172–83. doi: 10.1016/j.soncn.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Park DJ, Wilczynski SP, Paquette RL, Miller CW, Koeffler HP. p53 mutations in HPV-negative cervical carcinoma. Oncogene. 1994;9:205–10. [PubMed] [Google Scholar]
- 5.Ip SM, Huang TG, Yeung WS, Ngan HY. pRb-expressing adenovirus Ad5-Rb attenuates the p53-induced apoptosis in cervical cancer cell lines. Eur J Cancer. 2001;37:2475–83. doi: 10.1016/s0959-8049(01)00308-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhao S. Specific type epigenetic changes in cervical cancers. Methods Mol Biol. 2015;1238:733–49. doi: 10.1007/978-1-4939-1804-1_38. [DOI] [PubMed] [Google Scholar]
- 7.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor MG, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 8.Mercer TR, Dinger ME, Mattick JS. Long noncoding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 9.Mehler MF. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog Neurobiol. 2008;86:305–41. doi: 10.1016/j.pneurobio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 11.Nandy C, Mrazek J, Stoiber H, Grasser FA, Huttenhofer A, Polacek N. Epstein-barr virusinduced expression of a novel human vault RNA. J Mol Biol. 2009;388:776–84. doi: 10.1016/j.jmb.2009.03.031. [DOI] [PubMed] [Google Scholar]
- 12.Zhang QD, Xu MY, Cai XB, Qu Y, Li ZH, Lu LG. Myofibroblastic transformation of rat hepatic stellate cells: the role of notch signaling and epithelial-mesenchymal transition regulation. Eur Rev Med Pharmacol Sci. 2015;19:4130–8. [PubMed] [Google Scholar]
- 13.Kunkeaw N, Jeon SH, Lee K, Johnson BH, Tanasanvimon S, Javle M, Pairojkul C, Chamgramol Y, Wongfieng W, Gong B, Leelayuwat C, Lee YS. Cell death/proliferation roles for nc886, a noncoding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene. 2013;32:3722–31. doi: 10.1038/onc.2012.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J, Song Y, Bi N, Shen J, Liu W, Fan J, Sun G, Tong T, He J, Shi Y, Zhang X, Lu N, He Y, Zhang H, Ma K, Luo X, Lv L, Deng H, Cheng J, Zhu J, Wang L, Zhan Q. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73:3326–35. doi: 10.1158/0008-5472.CAN-12-3055. [DOI] [PubMed] [Google Scholar]
- 15.Lee HS, Lee K, Jang HJ, Lee GK, Park JL, Kim SY, Kim SB, Johnson BH, Zo JI, Lee JS, Lee YS. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget. 2014;5:3472–81. doi: 10.18632/oncotarget.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li JH, Xiao X, Zhang YN, Wang YM, Feng LM, Wu YM, Zhang YX. MicroRNA miR-886-5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol Oncol. 2011;120:145–51. doi: 10.1016/j.ygyno.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 17.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogoff HA, Pickering MT, Debatis ME, Jones S, Kowalik TF. E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol Cell Biol. 2002;22:5308–18. doi: 10.1128/MCB.22.15.5308-5318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JH, Shao P, Zhou H, Chen YQ, Qu LH. deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 2010;38:D123–30. doi: 10.1093/nar/gkp943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger W, Steiner E, Grusch M, Elbling L, Micksche M. Vaults and the major vault protein: novel roles in signal pathway regulation and immunity. Cell Mol Life Sci. 2009;66:43–61. doi: 10.1007/s00018-008-8364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee EK, Hong SH, Shin S, Lee HS, Lee JS, Park EJ, Choi SS, Min JW, Park D, Hwang JA, Johnson BH, Jeon SH, Kim IH, Lee YS, Lee YS. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget. 2016;7:75000–12. doi: 10.18632/oncotarget.11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee K, Kunkeaw N, Jeon SH, Lee I, Johnson BH, Kang GY, Bang JY, Park HS, Leelayuwat C, Lee YS. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA. 2011;17:1076–89. doi: 10.1261/rna.2701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KS, Park JL, Lee K, Richardson LE, Johnson BH, Lee HS, Lee JS, Kim SB, Kwon OH, Song KS, Kim YS, Ashktorab H, Smoot DT, Jeon SH, Kim SY, Lee YS. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget. 2014;5:3944–55. doi: 10.18632/oncotarget.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rimsza LM, Campbell K, Dalton WS, Salmon S, Willcox G, Grogan TM. The major vault protein (MVP), a new multidrug resistance associated protein, is frequently expressed in multiple myeloma. Leuk Lymphoma. 1999;34:315–24. doi: 10.3109/10428199909050956. [DOI] [PubMed] [Google Scholar]
- 25.Das D, Wang YH, Hsieh CY, Suzuki YJ. Major vault protein regulates cell growth/survival signaling through oxidative modifications. Cell Signal. 2016;28:12–8. doi: 10.1016/j.cellsig.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park K. The role of major vault protein (MVP) in drug resistance. J Control Release. 2012;163:266. doi: 10.1016/j.jconrel.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Herlevsen M, Oxford G, Owens CR, Conaway M, Theodorescu D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol Cancer Ther. 2007;6:1804–13. doi: 10.1158/1535-7163.MCT-06-0372. [DOI] [PubMed] [Google Scholar]
- 28.Giovanni A, Keramaris E, Morris EJ, Hou ST, O’Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of B-amyloidtreated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–60. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- 29.Johnson DG. The paradox of E2F1: oncogene and tumor suppressor gene. Mol Carcinog. 2000;27:151–7. doi: 10.1002/(sici)1098-2744(200003)27:3<151::aid-mc1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 30.Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, Chen J. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025–31. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 31.Singh RK, Dagnino L. E2F1 interactions with hHR23A inhibit its degradation and promote DNA repair. Oncotarget. 2016;7:26275–92. doi: 10.18632/oncotarget.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, Livingston DM, Amati B. E2Fdependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol. 2004;24:4546–56. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]