

Abstract
Very few cases of gliosarcoma (GS) in the spinal cord with or without rhabdomyoblastic differentiation have been reported at young ages, leading to limited information on the clinical, pathological and prognosis of this type of tumors. We report a case of GS with rhabdomyoblastic differentiation in a 6-year-old girl in C1-C6 level spinal cord. This is, to the best of our knowledge, the first report of GS with rhabdomyoblastic differentiation primarily developed in spinal cord at such a young age. Histologically, GS is composed of both glioblastoma components and malignant mesenchymal components. In the present case, the mesenchymal portion displayed a typical pattern of rhabdoid morphology. The rhabdomyoblastic-differentiated cells were confirmed by desmin, MyoD1, myogenin and Vimentin immunopositivity. Loss of PTEN (phosphatase and tensin homolog) and amplification of EGFR (epidermal growth factor receptor) were not detected in both parts of GS (glioblastoma component and rhabdomyosarcoma component). Interestingly, in this case rhabdomyoblastic-differentiated cells (rhabdomyosarcoma component) were focally negative for integrase interaction 1 (INI-1) protein and glial cells (glioblastoma component) were positive, and monosomy 22 in the former and absence in the latter. The patient only received low-dose radiotherapy and survived only 6 months after diagnosis. GSs with rhabdomyoblastic differentiation have a worse prognosis than common GSs and high-dose radiotherapy is suggested to considerer.
Keywords: Gliosarcoma, spinal cord, malignant rhabdoid tumor, differential diagnosis
Introduction
GS is a malignant brain tumor formed by glioblastoma (anaplastic astrocytes, mostly showing the typical features of a glioblastoma) and malignant mesenchymal components, firstly described in 1895 by Stroebe [1]. Cases of GS are intracranial tumors and are prevalent in elderly individuals [2-4]. Rare cases of GS with rhabdomyoblastic differentiation have been reported [5,6], which are all located in cerebral hemisphere of in adults. We present a case of GS with rhabdomyoblastic differentiation which occurred in an unusual site (in the spinal cord) and in rare age (in a child) with aggressive clinical course.
Case report
A 6-year-old girl attended our hospital because of a 10-day history of weakness in the limbs and the symptoms tended to get worse progressively with feces and urine incontinence for 7 days. Neurological examination found that the muscle strength scored grade 1 in both upper extremities. And the muscle score was grade 2-3 in left lower extremity and grade 2 in right lower extremity. Muscular tension was low. Routine laboratory examinations were normal.
MRI revealed an intramedullary mass at levels of C1-C6. The mass showed homogeneous isointense on T1-weighted images and mild hyper-intensity on T2-weighted images (Figure 1A, 1B), with enhancement on enhanced T1-weighted MRI at levels of C1-C6 (Figure 1C). A surgery through posterior cervical midline approach was performed. During surgery, macroscopic appearance of the spinal lesion was a gray-brown mass with obscure boundaries. It felt soft in texture. Pathological examination of frozen section confirmed the presence of glioblastoma components with vascular proliferation (Figure 2A) and necrosis (Figure 2B). Glioblastoma was diagnosed according to the frozen section examination. Then, the tumor was almost totally resected. Neurologic symptoms of the patient were slightly relieved after surgery. Then the patient received a course of radiotherapy in the spinal cord with a total dose of 36 Gy. The girl only survived 6 months after diagnosis.
Figure 1.
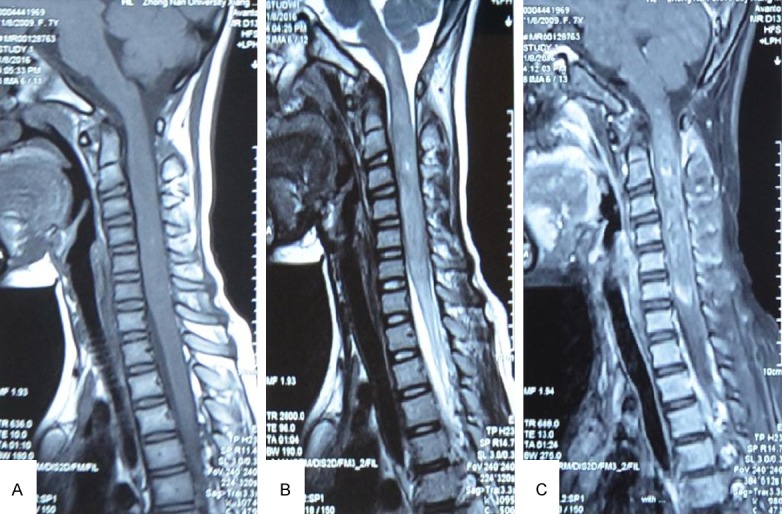
MR images disclosed a lesion in levels of C1-C6. A. MR images showed homogeneous isointense signal on T1-weighted MR images (sagittal). B. T2 revealed a subtle hyper-intensity signal (sagittal). C. The lesion was observed on markedly heterogeneous enhancement on enhanced T1-weighted MRI at levels of C1-C6 (sagittal).
Figure 2.
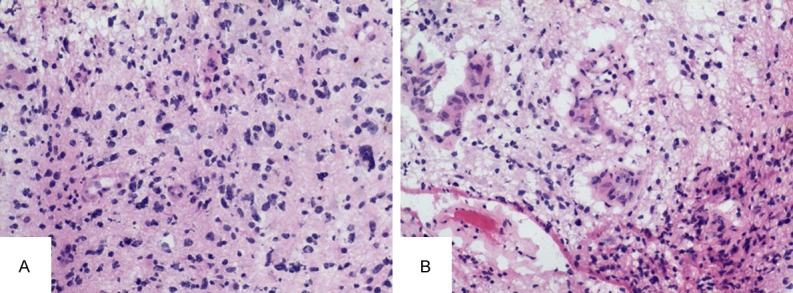
Histological features of frozen section. The presence of anaplastic astrocytes component with vascular proliferation (A) and necrosis (B).
Macroscopically, the tumor was soft, dark- red mass, with 4.3 cm × 2.5 cm × 1.3 cm in size. After formalin fixation, 4-μ-thick paraffin sections were stained routinely with HE staining.
Microscopically, the tumor included two distinct components (Figure 3A): glioblastoma component and malignant mesenchymal component. Pathological studies from the glioblastoma component revealed atypical astrocytes demonstrating pleomorphism and mitoses. Vascular proliferation (Figure 3B) and necrosis (Figure 3C) were found in glioblastoma part. Immunostaining of reticular fibers was negative for this component. The latter part, mesenchymal component, was consisted of large pleiomorphic cells arranged in sheet with round or oval nuclei and abundant cytoplasm (Figure 3D). A large number of multinucleated giant cells were found (Figure 3D). Some tumor cells exhibited typical rhabdomyoblastic features with eccentric nuclei, prominent nucleoli and abundant eosinophilic cytoplasm (Figure 3E). Mitosis was common. A rich network of fibrils could be seen in this area (Figure 3F). This part was considered to be of mesenchymal origin with rhabdomyoblastic differentiation.
Figure 3.

Histological features. The tumor consisted of two distinct components: glioblastoma and malignant mesenchymal component (A: Haematoxylin-eosin, magnification × 40). The vascular proliferation (B: × 200) and necrosis (C: × 200) were found in glioblastoma portion. The mesenchymal component consisted of large pleiomorphic cells arranged in sheet with round or oval nuclei and abundant cytoplasm (D: × 200). Part of tumor cells exhibited typical rhabdoid morphology with eccentric nuclei, prominent nucleoli and abundant eosinophilic cytoplasm (E: × 400). The results demonstrated a rich network of fibrils in mesenchymal area (F: × 200).
IHC staining was performed with monoclonal antibodies against glial fibriliary acidic protein, (GFAP, 1:500), Actin (1:400), pan-cytokeratin (CK, 1:250), Desmin (1:300), EMA (1:300), INI-1 (1:300), IDH1-R132H (1:350), MyoD1 (1:250), Myoglobin (1:200), Myogenin (1:150), Olig-2 (1:300), TP53 (1:300), S-100 (1:200), Vimentin (1:200) and MIB-1 respectively. All these antibodies were purchased from DAKO GmbH, Germany except IDH1-R132H and INI-1, which was obtained from DIANOVA and Cell Marque, respectively. In addition, positive and negative controls were also included and evaluated appropriately for each procedure. Besides, reticular fiber staining was also performed.
IHC staining demonstrated cytoplasmic expre-ssion of GFAP in the typical glioblastoma cells (Figure 4A), but not in the rhabdomyoblastic differentiation tumor cell areas. The atypical glioblastoma area was also strong immune-positive for Olig-2 (Figure 4B), S-100 (Figure 4C), TP53 (Figure 4D) and INI-1 protein. In contrast, cells in mesenchymal component were not appeared to be positive for GFAP and S-100, and only very few cells were positive for GFAP (Figure 4E) and S-100 (Figure 4F). The mesenchymal component was diffusely desmin (Figure 4G), Vimentin (Figure 4H), MyoD1 (Figure 4I), Actin (Figure 4J), Myogenin (Figure 4K), TP53 (Figure 4L) and focally EMA (Figure 4M), Myoglobin (Figure 4N), CK (Figure 4O) positive. Importantly, the rhabdomyoblastic-differentiated tumor cell areas showed focal loss of INI-1 protein immunostaining (Figure 4P).
Figure 4.

Results of immunohistochemistry. GFAP was positive in the glial part (A: × 200). Olig-2 (B: × 200), S-100 (C: × 200) and P53 (D: × 200) were diffusely positive in glial part. In contrast, all the cells were not appeared to be positive for GFAP and S-100, only very few cells were positive for GFAP (E: × 200) and S-100 (F: × 200) in mesenchymal area. The mesenchymal component was diffusely desmin (G: × 200), Vimentin (H: × 200), MyoD1 (I: × 200), Actin (J: × 200), Myogenin (K: × 200), TP53 (L: × 200) and focally EMA (M: × 200), Myoglobin (N: × 200), CK (O: × 200) positive. The rhabdoid tumor area showed focal loss of INI-1 protein immunostaining (P: × 100).
The proliferative labelling index assessed by MIB-1 immunostaining was 20-25% in the glioblastoma part and 40% in the mesenchymal origin with rhabdomyoblastic differentiation rhabdomyosarcoma part.
Dual-color FISH were performed using LSI PTEN/CEP 10 dual color probe (Vysis/Abbott Molecular) and dual TUPLE1/ARSA (Vysis/Abbott Molecular) for losses of PTEN and INI-1 respectively. The EGFR gene copy number alterations were performed using LSI EGFR/CEP 7 (Vysis/Abbott Molecular). Fluorescent signals analysis was performed as previously described [10].
FISH results revealed that rhabdoid part of tumor cells showed deletion of INI-1 (Figure 5A), but without loss of PTEN nor amplification of EGFR. The glioblastoma part of cells did not show deletion of ini-1 (Figure 5B). No loss/amplification of PTEN/EGFR was found in of glioblastoma part cells.
Figure 5.

FISH results of ini-1. FISH results revealed that the part of rhabdoid tumor cells showed deletion of ini-1 (A), but the part of glioblastoma cells did not (B).
Discussion
Typically, malignant mesenchymal components of GS resemble fibrosarcoma or malignant fibrous histiocytoma, with occasional presence of features of osteosarcoma, chondrosarcoma, liposarcoma or leiomyosarcoma [7-10]. Very few cases of GS with rhabdomyoblastic differentiation have been reported [5,6]. Previously published literatures have not provided much pathological and prognostic information for this kind of tumor. Considering the rare incidence of such cases, we herein report the clinical, pathological and prognosis of a case of GS with rhabdomyoblastic differentiation in a 6-year-old girl, occurring in C1-C6 level of spinal cord.
Histologically, the GS contains glial and sarcoma components. In this case, the glial component typically resembles glioblastoma. The mesenchymal portion is consisted of large round tumor cells, exhibiting typical rhabdomyoblastic differentiation with an eccentric nucleus and abundant eosinophilic cytoplasm. The histogenesis of the rhabdomyoblastic differentiation cell origin is unknown but it might be of mesenchymal, neuroectomesenchymal or meningeal lineage [11-14]. The rhabdomyoblastic-differentiated cells in different tumors always have their special immunohistochemical features. When rhabdomyoblastic differentiation is presented in a GS, rhabdomyosarcoma differentiation cells showed positive for of desmin, MyoD1, myogenin and Vimentin [5,6]. Beyond that, cells in this area were very rich in reticulin fibers. The rhabdomyoblastic differentiation cells in this case showed diffuse immunopositivity for desmin, Vimentin, MyoD1, Myogenin and focally Myoglobin, CK, EMA and were immune-negative for GFAP and S-100, which is in agreement with previous reports [5,6]. The individual rhabdomyoblastic-differentiated cells are surrounded by reticulin fibers. In addition, the rhabdomyoblastic-differentiated cells were focally negative for INI-1. The present case was diagnosed as GS with rhabdomyoblastic differentiation based on microscopic features and immunohistochemical features.
Limited molecular genetics characteristics have been reported about GS with rhabdomyoblastic differentiation. We examined IDH1 R132H mutations, p53 mutations, loss of chromosome 10q (PTEN), loss of INI-1 and gene amplification of EGFR in glioma area and rhabdomyosarcoma area. The LOH of PTEN, EGFR gene amplification, IDH1 R132H mutation were not found in both parts of this tumor. However, p53 mutations were detected in both parts. Interestingly, in this case, loss of INI-1 as well as monosomy of chromosome 22 was presented in rhabdomyosarcoma part, but not in the glioblastoma part. A recent study [15] found that focal rhabdomyoblastic-differentiated areas in the rhabdoid GBMs (R-GBMs) showed the focal loss of INI-1 expression. The authors suggested that the plausible explanation for focal loss of INI-1 expression was that this represented a second genetic hit in the INI-1 gene in a subset of the rhabdomyoblastic-differentiated tumor cells [15]. In our case, rhabdomyoblastic differentiation cells in GS also manifested focal loss of INI-1 expression. This perhaps is the first discovery of focal loss of INI-1 expression of rhabdomyoblastic differentiation cells in GS. We infer a second hit have also occurred in the rhabdomyoblastic-differentiated tumor cells of this case.
The differential diagnosis for this case mainly includes R-GBM and AT/RTs. Although R-GBM, AT/RT and GS with rhabdomyoblastic differentiation have overlapping pathologic features, they also have unique clinic-pathologic and immunohistochemical features, respectively that are extremely useful in differential diagnosis. In R-GBM, nuclear pleomorphism is more pronounced than GS with rhabdomyoblastic differentiation. And GFAP immunostaining is positive [11,14,15]. In contrast, GFAP immunostaining is negative in rhabdomyoblastic differentiation tumor cells of GS. Moreover, individual rhabdomyoblastic differentiation tumor cells of GS are usually surrounded by reticulin fibers, while we don’t see this pattern in R-GBM.
AT/RTs are polyphenotypic tumors, which are frequently immune-reactive for cytokeratin, EMA, GFAP, smooth muscle actin, vimentin and neurofilament. Histologically, AT/RTs contain primitive neuroectodermal, malignant mesenchymal components and some rhabdomyoblastic differentiation tumor cells. In addition, AT/RTs mainly occur in the posterior fossa of infants or children. Importantly, INI-1 immunohistochemistry is always completely negative in most of AT/RTs but mostly retained in GS.
It remains unclear that whether GS with rhabdomyoblastic differentiation should be treated as a rhabdoid tumor or as a GS. The optimal treatment method has not yet been determined. Focal radiotherapy is recommended for infants and young children to treat malignant tumors with rhabdomyoblastic differentiation [16]. Salvati M et al. [17] believed that chemotherapy given after postoperative radiotherapy in patients was adjuvant treatment for childhood’s GS. They also believed that doses lower than 60 Gy were not sufficient to guarantee an efficacious local treatment of the residual childhood’s GS [17]. Some reports showed that histological types of mesenchymal component were tightly associated with prognosis [18,19]. Tumor with rhabdomyoblastic differentiation showed significant associations with malignant progression [20]. We recommended considering high-dose radiotherapy and chemotherapy with temozolomide as adjuvant treatment for the patient but got rejected by the parents. Subsequent follow-up revealed that the patient underwent low-dose radiotherapy (total dose of 36 Gy) without concomitant temozolomide and died of respiratory failure with tumor recurrence 6 months after the operation. Although quite small numbers of GS in children have been described, relatively long survival times have been observed (24 ms, Salvati et al. [16]; 34 ms, Ono et al. [21]). This case survived significantly shorter than the median survival of common GSs. GS with rhabdomyoblastic differentiation appears to be more aggressive than most other GSs, especially at a young age. We think that the low-dose radiotherapy (36 Gy) is not sufficient to prevent or impede tumor recurrence. On the other hand, possible radiotherapy side effects in the children must be weighed against the benefit of giving high-dose radiotherapy in children who are expected to experience long-term survivals. More cases are needed to corroborate our inference.
Acknowledgements
This work was supported by the Joint Fund for Basic and Clinical Research Cooperation Project of Capital Medical University (Grant No. 16JL47).
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Disclosure of conflict of interest
None.
References
- 1.Stroebe H. Uber entstehung und bau der gehirngliome. Beitr Pathol Anat Allg Pathol. 1895;18:405–486. [Google Scholar]
- 2.Moscote-Salazara LR, Alcalá-Cerra G, Gutiérez-Paternina JJ, Penagos Gonzélezc PJ, Zubieta Vega C, Chater-Cure G, Alberto Meneses C, Saenz M. Pediatric gliosarcoma: case report and literature review. Bol Asoc Med P R. 2014;106:43–47. [PubMed] [Google Scholar]
- 3.Savant HV, Balasubramaniam S, Mahajan V. Giant parietal lobe infantile gliosarcoma in a 5-year-old child. Pediatr Neurosci. 2015;10:159–161. doi: 10.4103/1817-1745.159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang PF, Liu N, Song HW, Yao K, Jiang T, Li SW, Yan CX. IDH-1 R132H mutation status in diffuse glioma patients: implications for classification. Oncotarget. 2016;7:31393–400. doi: 10.18632/oncotarget.8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sethi S, Siraj F, Roy S. Gliosarcoma with rhabdomyomatous differentiation: a case report. Indian J Cancer. 2011;48:129–131. doi: 10.4103/0019-509X.76643. [DOI] [PubMed] [Google Scholar]
- 6.Svajdler M Jr, Rychlý B, Gajdoš M, Pataky F, Fröhlichová L, Perry A. Gliosarcoma with alveolar rhabdomyosarcoma-like component: report of a case with a hitherto undescribed sarcomatous component. Cesk Patol. 2012;48:210–214. [PubMed] [Google Scholar]
- 7.Charfi S, Ayadi L, Khabir A, Daoud E, Kallel R, Kharrat O, Mnif Z, Boudawara Z, Makni S, Boudawara T. Gliosarcoma with osteosarcomatous features: a short illustrated review. Acta Neurochir. 2009;151:809–813. doi: 10.1007/s00701-009-0341-2. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee AK, Sharma BS, Kak VK, Ghatak NR. Gliosarcoma with cartilage formation. Cancer. 1989;63:518–523. doi: 10.1002/1097-0142(19890201)63:3<518::aid-cncr2820630320>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Vlodavsky E, Konstantinesku M, Soustiel JF. Gliosarcoma with liposarcomatous differentiation: the new member of the lipid-containing brain tumors family. Arch Pathol Lab Med. 2006;130:381–384. doi: 10.5858/2006-130-381-GWLDTN. [DOI] [PubMed] [Google Scholar]
- 10.Khanna M, Siraj F, Chopra P, Bhalla S, Roy S. Gliosarcoma with prominent smooth muscle component (gliomyosarcoma): a report of 10 cases. Indian J Pathol Microbiol. 2011;54:51–54. doi: 10.4103/0377-4929.77324. [DOI] [PubMed] [Google Scholar]
- 11.Kleinschmidt-DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO. Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 but not claudin 6. Am J Surg Pathol. 2010;34:341–354. doi: 10.1097/PAS.0b013e3181ce107b. [DOI] [PubMed] [Google Scholar]
- 12.Chen SC, Lin DS, Lee CC, Hung SC, Chen YW, Hsu SP, Lin CF, Wong TT, Chen MH, Chen HH. Rhabdoid glioblastoma: a recently recognized subtype of glioblastoma. Acta Neurochir. 2013;155:1443–1448. doi: 10.1007/s00701-013-1793-y. [DOI] [PubMed] [Google Scholar]
- 13.He MX, Wang JJ. Rhabdoid glioblastoma: case report and literature review. Neuropathology. 2011;31:421–426. doi: 10.1111/j.1440-1789.2010.01166.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee JY, Kim IK, Phi JH, Wang KC, Cho BK, Park SH, Ahn HS, Kim IH, Kim SK. Atypical teratoid/rhabdoid tumors: the need for more active therapeutic measures in younger patients. J Neurooncol. 2012;107:413–419. doi: 10.1007/s11060-011-0769-0. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y, Xie Q, Gong Y, Mao Y, Zhong P, Che X, Jiang C, Huang F, Zheng K, Li S, Gu Y, Bao W, Yang B, Wu J, Wang Y, Chen H, Xie L, Zheng M, Tang H, Wang D, Zhu H, Chen X. Clinicopathological analysis of rhabdoid meningiomas: report of 12 cases and a systematic review of the literature. World Neurosurg. 2013;79:724–732. doi: 10.1016/j.wneu.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, Wallace D, Gilbertson RJ, Gajjar A. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J. Clin. Oncol. 2005;23:1491–9. doi: 10.1200/JCO.2005.05.187. [DOI] [PubMed] [Google Scholar]
- 17.Salvati M, Lenzi J, Brogna C, Frati A, Piccirilli M, Giangaspero F, Raco A. Childhood’s gliosarcomas: pathological and therapeutical considerations on three cases and critical review of the literature. Childs Nerv Syst. 2006;22:1301–1306. doi: 10.1007/s000381-006-0057-z. [DOI] [PubMed] [Google Scholar]
- 18.Yao K, Qi XL, Mei X, Jiang T. Gliosarcoma with primitive neuroectodermal, osseous, cartilage and adipocyte differentiation: a case report. Int J Clin Exp Pathol. 2015;8:2079–2084. [PMC free article] [PubMed] [Google Scholar]
- 19.Charfi S, Ayadi L, Khabir A, Daoud E, Kallel R, Kharrat O, Mnif Z, Boudawara Z, Makni S, Boudawara T. Gliosarcoma with osteosarcomatous features: a short illustrated review. Acta Neurochir. 2009;151:809–813. doi: 10.1007/s00701-009-0341-2. [DOI] [PubMed] [Google Scholar]
- 20.Can Z, Saray A, Yilmaz S, Erçöçen AR, Emiroğlu M. Malignant triton tumor of the maxilla: a patient report. Ann Plast Surg. 1999;42:96–9. doi: 10.1097/00000637-199901000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Ono N, Nakamura M, Inoue HK, Tamura M, Murata M. Congenital gliosarcoma: so-called sarcoglioma. Child’s Nerv Syst. 1990;6:416–420. doi: 10.1007/BF00302231. [DOI] [PubMed] [Google Scholar]