

Abstract
Prostate cancer is one of the most common male malignancies and remains the second leading cause for cancer-specific mortalities in men. Cisplatin is commonly used as a chemotherapeutic agent against advanced cancers, and is now used in metastatic prostate cancers. Cisplatin exerts its cytotoxic effects by cross-linking genomic DNA (gDNA) which induces DNA damage on rapidly dividing cancer cells. However, cisplatin leads to systemic side effects and some patients never respond. Our previous report demonstrated an oncogenic role of miR-181a in human prostate cancer. In this study, we investigate the mechanistic potential of miR-181a in regulating cisplatin sensitivity in this context. We report that cisplatin treatment significantly enhanced miR-181a expression and that exogenous overexpression of miR-181a decreased sensitivity of prostate cancer cells to cisplatin. Additionally, we observed that cisplatin-resistant prostate cancer cells harbored high levels of miR-181a expression. Mechanistically, we demonstrate the pro-apoptotic protein, BAX, is typically enhanced by cisplatin treatment but its suppression promoted resistance. Here we demonstrate miR-181a regulation of BAX was mediated through a complimentary interaction with the 3’UTR of the BAX transcript. We subsequently show that BAX expression restored cisplatin sensitivity in miR-181a overexpressing prostate cancer cells. In parallel, we demonstrate inhibition of miR-181a restored BAX expression as well as cisplatin sensitivity in resistant cells. This study suggests that miR-181a is a potential therapeutic target for prostate cancers that are resistant to cisplatin.
Keywords: Prostate cancer cells miR-181, BAX downregulation
Introduction
Prostate cancer remains one of the most common types of cancer in males and treatments have limited success for locally advanced or metastatic disease [1]. Prostate cancer is a major public health concern, as the molecular pathogenesis of prostate cancer is complicated, this in turn has presented challenges for predicting clinical responses to modern therapeutics [2]. While modern therapeutics, such as androgen therapies have improved the outcome of patients even with advanced disease, some never respond or quickly become resistant. For such patients, there are no durable treatment options, and much research is focused in developing the next lines of therapy. Identification of novel molecular mechanisms remains a viable approach that will contribute to development of strategies for the diagnosis, treatment, and evaluating overall prognosis for prostate cancer patients.
MicroRNAs (miRNAs) are a conserved class of small (18-22 nt), non-coding RNAs that directly bind to the complementary sequence in 3’UTR region of their mRNA targets, resulting in mRNA degradation or translation repression [5]. Accumulating evidence implicate miRNA as critical regulators of carcinogenesis and tumor progression in numerous types of human cancers [6,7]. Our previous study has reported miR-181a is upregulated in prostate tumor cells [8], suggesting an oncogenic role of miR-181a in this context. Moreover, previous studies have demonstrated that the upregulation of hepatic miR-181 promotes the growth, clonogenic survival, migration, and invasion of hepatocellular carcinoma cells [9]. Furthermore, it has been reported that miR-181 is significantly associated with overall survival in hematological malignancies and may be an important clinical prognostic factor for patients with hepatocellular carcinoma [10]. While the oncogenic roles of miR-181 have been demonstrated in several cancer types, and we observe its expression in prostate cancers, its specific mechanistic actions in prostate cancer have not been elucidated.
Cisplatin is a frequently used chemotherapeutic agent that has shown efficacy across cancer types; it mechanistically cross-links genomic DNA (gDNA) which results in DNA damage to rapidly dividing cancer cells [11]. While effective in many settings, cisplatin has systemic side effects, and several cancers have demonstrated clinical resistance to such therapy [12]. As cisplatin is implemented in the treatment of advanced prostate cancer patients that do not respond to other therapies, we sought to examine mechanisms that regulate cisplatin sensitivity of prostate cancer cells. In this study, we have investigated the role of the prostate cancer associated miR-181a in regulating cisplatin sensitivity. As we find that miR-181a regulates cisplatin sensitivity of prostate cancer cells, we proceeded to examine the targets of miR-181 to characterize the potential underlying mechanisms of cisplatin sensitivity in prostate cancers.
Materials and methods
Cell cultures
Human prostate cancer cell lines, PC3 and DU145 cells were provided by the Institute of Biochemistry and Cell Biology of Chinese Academy of Science (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies), 100 IU/ml penicillin and 100 µg/ml streptomycin sulfate. Cells were incubated at 37°C with 5% CO2.
microRNA mimics and transfection
Human miR-181a mimics, miR-181a inhibitor and negative controls were purchased from Qiagen (Shanghai, China). Transfections of PC3 and DU145 cells were performed using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA), following the manufacturer’s instructions. Transfections were performed at 100 nM for 72 hours.
RNA extraction and qRT-PCR
The measurement of miR-181a expression was performed as previously reported [10]. Briefly, total RNA was isolated using TRIzol reagent (Invitrogen, Shanghai, China). Reverse transcription was performed using the PrimeScript RT reagent kit (TaKaRa, Dalian, China) following the manufacturer’s instructions. Real-time quantitative PCR analyses were performed using SYBR Premix Ex Taq (TaKaRa). For miRNA expression, RNA was first reverse transcribed using a specific primer. The expression of miRNAs was then quantified using TaqMan probes, and the expression levels of miRNAs were normalized using U6 small nuclear RNA utilizing TaqMan miRNA assays (Applied Biosystems, Foster City, CA, USA). MiR-181a inhibitor was chemically synthesized by GenePharma (Shanghai, China). The miR-181a inhibitor consisted of sequence-specific single-stranded RNA molecules with optimized nucleic acids designed to specifically target and knockdown the miR-181a molecules in cells. Sequence was 5’-ACUCACCGACAGCGUUGAAUGUU-3’ and inhibitor NC Sequence was 5’-CAGUACUUUUGUGUAGUACAA-3’.
Luciferase reporter assay
cDNA fragments corresponding to the entire 3’-untranslated region (UTR) of BAX were amplified by PCR from the total RNA extracted from PC3 cells with KpnI and EcoRI linkers. The PCR products were cloned downstream of the Renilla luciferase open reading frame of the pMir-Report (Qiagen), which also contained a constitutively expressed firefly luciferase gene that was used to normalize the transfections. For the luciferase reporter assays, the cells were seeded in 24-well plates and harvested 48 h after transfection. The wild-type or mutant 3’-untranslated region fragments from the human BAX gene were cloned into pMir-Report (Qiagen). Mutations were introduced in potential miR-181a binding sites using a site-directed mutagenesis kit (Qiagen) [8]. Luciferase values were determined using the Dual-Luciferase Reporter assay system (Promega Corporation, Madison, WI, USA). All experiments were repeated three times.
Cell survival assay
The viability of prostate cells was determined by assaying the reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-di-phenylte-trazolium bromide (MTT; Beyotime Company, Shanghai, China) to formazan [8]. For the analysis of cell survival rate in response to cisplatin, equal number cells were seeded onto 24-well plates for overnight, then cells were treated with cisplatin for 72 hours. Absorbance was measured at 570 nm using the Spectra Max 190 ELISA reader (Molecular Devices, Sunnyvale, CA, USA). All experiments were repeated three times.
Western blot analysis
Total cell protein was extracted using RIPA buffer. Protein concentrations were determined by Bradford assay. Protein extracts were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto a polyvinylidene difluoride membrane. After blocking with 5% non-fat milk in phosphate-buffered saline, the membranes were immunoblotted with primary antibodies as indicated at 1:1000, followed by horseradish peroxidase-linked secondary antibodies at 1:3000 (Cell Signaling Technology, Inc., Danvers, MA, USA). The signals were detected using a chemiluminescence detection kit (Millipore, Billerica, MA, USA). Protein levels were normalized against those of β-actin (Santa Cruz Biotechnology, Inc.).
Statistical analysis
Differences between groups were analyzed using a Student’s t-test and expressed as the mean ± standard deviation from three independent experiments. P < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism version 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
MiR-181a contributes to the cisplatin resistance in prostate cancer cells
Our previous study demonstrated miR-181a was significantly upregulated in human prostate cancer cells as well as prostate cancer tissues relative to adjacent normal tissues [8]; these findings implicated an oncogenic role of miR-181a in human prostate cancer. In this study, we investigate the miR-181a regulation of cisplatin sensitivity. Cisplatin has been given to patients that fail to respond to other commonly used therapeutic agents in prostate cancer. We measured the expressions of miR-181a in response to cisplatin treatments at 0, 2.5, 5, and 10 µM. Interestingly, our results showed cisplatin treatments significantly enhanced miR-181a expression in DU145 and PC3 prostate cancer cell lines (Figure 1A), associating miR-181a as a cisplatin response mechanism in prostate cancer cells. To examine if miR-181a regulates cisplatin sensitivity, we exogenously overexpressed miR-181a in DU145 and PC3 cells (Figure 1B). As expected, DU145 and PC3 cells overexpressing miR-181a demonstrated enhanced resistance to cisplatin relative to cells transfected with controls (Figure 1C, 1D). The IC50 of DU145 control cells was 38.5 μM at 72 hours, lower than that of the DU145 cells expressing miR-181a (56.3 μM). Similarly, we observed an increase in IC50 of PC3 cells transfected with miR-181a cells (32.4 μM) relative to controls (18.7 μM). These results indicate miR-181a expression contributes to cisplatin resistance in prostate cancer cells.
Figure 1.
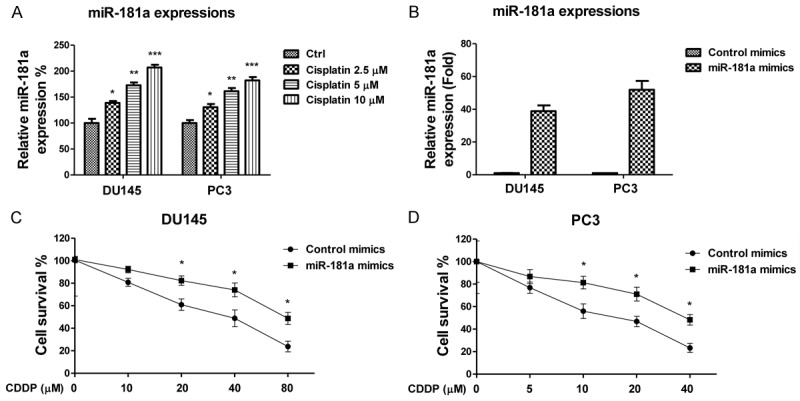
MiR-181a contribute to cisplatin resistance. A. DU145 and PC3 cells were treated with cisplatin at concentrations of 0, 2.5, 5 or 10 μM for 72 hours. Expression levels of miR-181 were measured by qRT-PCR. B. DU145 and PC3 cells were transfected with control or miR-181a. After 48 hours the expression of miR-181 was measured by qRT-PCR. C. DU145; D. PC3 cells were transfected with control or miR-181a. After 48 hours, cells were treated with the indicated concentrations of cisplatin for 72 hours. The cell viabilities were measured. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Cisplatin resistant prostate cancer cells exhibit upregulated miR-181a and downregulated BAX expression
To investigate the underlying mechanisms for the miR-181a modulated cisplatin resistance, we established a cisplatin resistant prostate cancer cell line from PC3 cells. Cells treated with elevated concentrations of cisplatin were cultured for the purpose of selecting resistant clones. Figure 2A demonstrates the resistant cell line, PC3 CisR cells, tolerate higher concentrations of cisplatin treatment relative to parental cells. The IC50 of PC3 CisR cells was (57.5 μM), which is significantly higher than that of the PC3 parental cells (19.6 μM). As we expected, miR-181a was also significantly upregulated in PC3 CisR cells (Figure 2B), supporting our findings that miR-181a expression promotes cisplatin resistance in PC3 and DU145 cells. BAX functions as an apoptotic activator through the formation of heterodimer with BCL2, resulting in triggering mitochondrial apoptosis pathway [13]. We compared the expressions of BCL2 in PC3 parental and CisR cells. Results demonstrated BAX expression was downregulated in PC3 CisR cells (Figure 3A). Additionally, we found that cisplatin treatments induced BAX expression (Figure 3B), reinforcing our hypothesis that miR-181a suppresses BAX to promote cisplatin resistance. Finally, to confirm the role of BAX in cisplatin sensitivity, we transfected siRNA targeting BAX into PC3 cells to specifically suppress its expression (Figure 3C). As we expected, BAX suppression promoted cisplatin-resistance of PC3 cells (Figure 3D).
Figure 2.
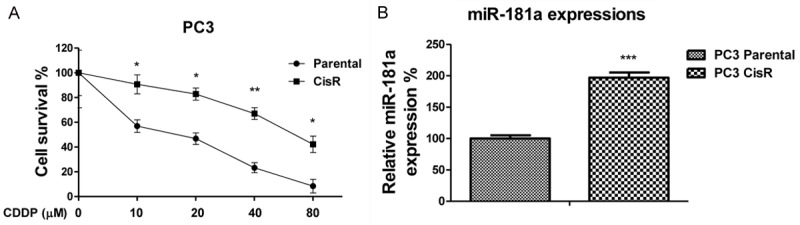
MiR-181a is upregulated in cisplatin resistant prostate cancer cells. A. The cell viabilities were assayed and shown for parental PC3 and cisplatin resistant cells. B. The expressions of miR-181a was assayed and shown in parental PC3 and cisplatin resistant cells. *, P < 0.05; **, P < 0.01.
Figure 3.
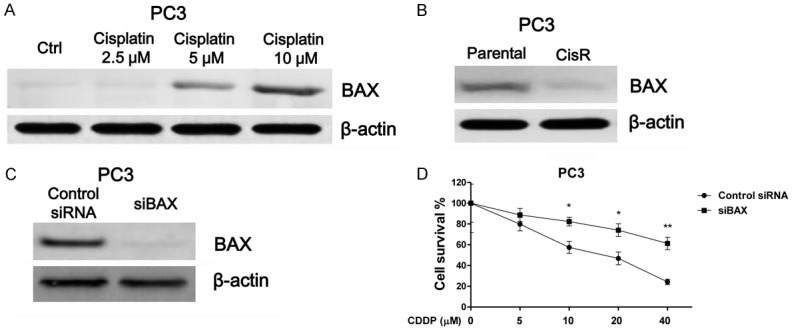
BAX is downregulated in cisplatin resistant prostate cancer cells. A. PC3 cells were treated with cisplatin at 0, 2.5, 5 or 10 μM for 72 hours. The expression of BAX was analyzed by Western blots. β-actin was used as a loading control. B. The expression of BAX was analyzed by Western blot in parental PC3 and cisplatin resistant cells. C. PC3 cells were transfected with control or siRNA targeting BAX, and after 48 hours BAX expression was measured by Western blots. D. PC3 cells were transfected with control siRNA targeting BAX, after 48 hours, cells were treated with cisplatin at 0, 5, 10, 20 or 40 μM for 72 hours. Cell viabilities were subsequently measured and shown. *, P < 0.05; **, P < 0.01.
MiR-181a directly targets BAX
To delineate the underlying correlation between miR-181a and BAX in cisplatin resistant prostate cancer cells, we searched for potential targets of miR-181a through the bioinformatics program, TargetScan. Intriguingly, we found a miR-181a targeting sequence had hypothetical complementarily interactions with the 3’UTR of BAX (Figure 4A). To test whether miR-181a could directly target BAX in prostate cancer cells, we transfected miR-181a mimics to overexpress it in PC3 cells and found overexpression of miR-181a inhibited BAX protein expression (Figure 4B). Additionally, luciferase assays illustrated overexpression of miR-181a significantly inhibited activity of a luciferase reporter fused to wild-type BAX miR-181a-targeting sequence, but not mutated sequence (Figure 4C). Taken together, these data demonstrated that BAX is a directly regulated by miR-181a in prostate cancer cells.
Figure 4.
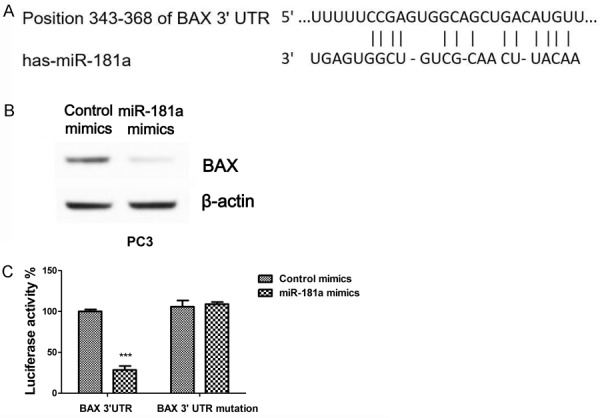
MiR-181 directly targets BAX. A. Prediction of miR-181a binding sites on the 3’-UTRs of human BAX gene using TargetScan software. B. Protein levels of BAX were analyzed using Western blot analysis in PC3 cells transfected with miR-181a or control after 48 h. C. Luciferase reporter assays in PC3 cells. Cells were co-transfected with either wild-type or mutant BAX 3’-UTR-reporter constructs along with either miR-181a or control. ***, P < 0.001.
MiR-181a promotes cisplatin resistance of prostate cancer cells through inhibition of BAX
To determine whether miR-181a-mediated cisplatin resistance through BAX suppression, we transfected PC3 cells with miR-181a alone or in combination with overexpressed BAX from an exogenous construct. Results showed co-transfection of miR-181a with BAX rescued the miR-181a-dependent BAX downregulation (Figure 5A). Additionally, co-transfection of BAX significantly attenuated the cisplatin resistance mediated by miR-181a overexpression in PC3 cells (Figure 5B). These observations support our hypothesis that miR-181a modulated cisplatin resistance through suppression of BAX.
Figure 5.
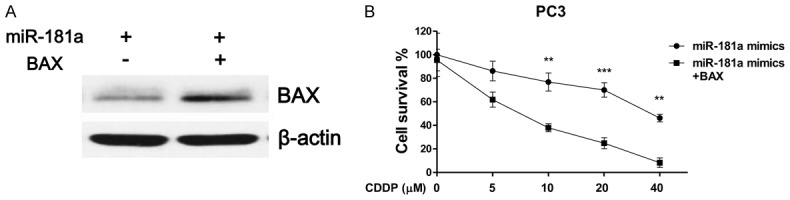
Restoration of BAX recovers cisplatin sensitivity. A. PC3 cells were transfected with miR-181a alone or co-transfected with miR-181a and BAX. BAX expression was analyzed by Western blots after 48 hours, B. PC3 cells were transfected with miR-181a alone or co-transfected with miR-181a and BAX, after 48 hours cells were treated with cisplatin at 0, 5, 10, 20 or 40 μM for 72 hours, cell viability was measured. **, P < 0.01; ***, P < 0.001.
Inhibition of miR-181a sensitizes prostate cancer cells to cisplatin
To further demonstrate the regulation of miR-181a on cisplatin sensitivity, we next determined the cisplatin sensitivity of prostate cancer cells upon miR-181a suppression. To do so, we transfected a miR-181a inhibitor into PC3 CisR cells (Figure 6A). Inhibition of miR-181a rescued BAX expression (Figure 6B) and more importantly, we observed that inhibition re-sensitized PC3 CisR cells to cisplatin (Figure 6C). These results confirmed that miR-181a negatively regulates BAX to promote resistance; suggesting miR-181a inhibition could serve as a therapeutic target against cisplatin resistant prostate cancers.
Figure 6.
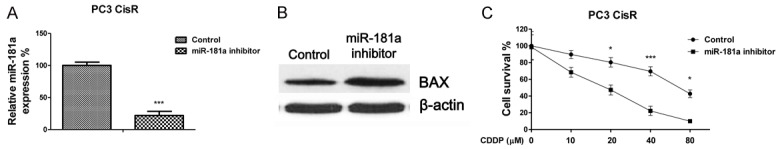
Inhibition of miR-181a re-sensitizes resistant cells to cisplatin through BAX upregulation. (A) PC3 cisplatin resistant cells were transfected with control or a miR-181a inhibitor. After 48 hours, the expression of miR-181a or (B) BAX was determined and shown. (C) PC3 cisplatin resistant cells were transfected with control or a miR-181a inhibitor, after 48 hours, cells were treated with cisplatin at 0, 10, 20, 40 or 80 μM for 72 hours, cell viability was measured. *, P < 0.05; ***, P < 0.001.
Discussion
It has been previously demonstrated that miRNAs are associated with prostate cancer progression and prognosis [14]. Moreover, mRNAs are dysregulated in prostate cancer tissues or cell lines. In a previous study from our lab, we reported miR-181a was upregulated in human prostate cancer cells and tissue [8]. In addition, miR-181 overexpression was observed to promoted the growth of LNCaP tumors in nude mice [8], revealing an oncogenic role of miR-181a in the development of prostate cancer. This study demonstrated that miR-181a contributes to cisplatin resistance in human prostate cancer cells by directly suppressing the pro-apoptotic protein, BAX. Our results present an opportunity for miRNA-based therapeutics against chemo resistant prostate cancers.
Cancer cells at various stages can acquire resistance to chemotherapy [15]. Additionally, chemoresistance remains a challenging issue as it prevents effective treatment of patients in the clinic. Cisplatin is now given to prostate cancer patients that fail several lines of therapy, thus understanding how resistance occurs in prostate cancers is essential. Evidence support that dysregulation of miRNAs is associated with the development of chemoresistance in multiple cancers [16]. Studies have shown that mechanisms of cisplatin resistance include increase of drug efflux, alterations in drug target, cell cycle regulation, DNA repair, and evasion of apoptosis [11]. In this study, we demonstrated miR-181a directly regulates BAX transcripts and expression to mediate cisplatin resistance. Recent studies in cervical squamous cell carcinoma [17] and breast cancer [18] demonstrated similar observations as our study in that miR-181a promoted cancer cell resistance to cisplatin or Doxorubicin. BAX is a Bcl-2 family effector that promotes apoptosis upon stress stimulation upon mitochondrial outer membrane permeabilization and subsequent cytochrome C release [13]. Our results illustrate BAX regulation by miR-181a directly regulated chemosensitivity of prostate cancer cells. Moreover, our findings demonstrate that miR-181a inhibition enhanced apoptosis in cisplatin resistant prostate cancer cells and restored BAX expression; these observations are consistent with previous reports in which BAX plays a critical role in mediating chemoresistance [19].
In conclusion, this study provides a novel role for miR-181a in prostate cancer cells, as it mediates chemoresistance through direct regulation of BAX transcripts. Our results also suggest that miR-181a is potentially a therapeutic target for prostate cancer patients that are resistant to cisplatin.
Acknowledgements
The project was supported by Shanghai Science and Technology Committee (No.14ZR1404600).
Disclosure of conflict of interest
None.
References
- 1.Emery JD, Shaw K, Williams B, Mazza D, Fallon-Ferguson J, Varlow M, Trevena LJ. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol. 2014;11:38–48. doi: 10.1038/nrclinonc.2013.212. [DOI] [PubMed] [Google Scholar]
- 2.Hutchinson L. Genetics: tracking clonal origin of prostate cancer. Nat Rev Clin Oncol. 2014;11:4. doi: 10.1038/nrclinonc.2013.220. [DOI] [PubMed] [Google Scholar]
- 3.Trewartha D, Carter K. Advances in prostate cancer treatment. Nat Rev Drug Discov. 2013;12:823–4. doi: 10.1038/nrd4068. [DOI] [PubMed] [Google Scholar]
- 4.Bhosale RR, Gangadharappa HV, Hani U, Ali M Osmani R, Vaghela R, Kulkarni PK, Koganti VS. Current perspectives on novel drug delivery systems and therapies for management of prostate cancer: an inclusive review. Curr Drug Targets. 2017;18:1233–1249. doi: 10.2174/1389450117666160613103705. [DOI] [PubMed] [Google Scholar]
- 5.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–65. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nat Rev Genet. 2013;14:535–48. doi: 10.1038/nrg3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–33. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong SJ, Liu J, Wang X, Qu LX. microRNA-181 promotes prostate cancer cell proliferation by regulating DAX-1 expression. Exp Ther Med. 2014;8:1296–300. doi: 10.3892/etm.2014.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azumi J, Tsubota T, Sakabe T, Shiota G. miR-181a induces sorafenib resistance of hepatocellular carcinoma cells through downregulation of RASSF1 expression. Cancer Sci. 2016;107:1256–62. doi: 10.1111/cas.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou C, Li Y, Cao Y, Zhang J, Jiang J, Sheng Y, Wang S, Huang A, Tang H. Up-regulated MicroRNA-181a induces carcinogenesis in hepatitis B virus-related hepatocellular carcinoma by targeting E2F5. BMC Cancer. 2014;14:97. doi: 10.1186/1471-2407-14-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. doi: 10.1038/cddis.2013.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of bax and bak activation and action. Biochim Biophys Acta. 2011;1813:521–31. doi: 10.1016/j.bbamcr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 14.Jackson BL, Grabowska A, Ratan HL. MicroRNA in prostate cancer: functional importance and potential as circulating biomarkers. BMC Cancer. 2014;14:930. doi: 10.1186/1471-2407-14-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Meng Y, Liu N, Wen XF, Yang T. Insights into chemoresistance of prostate cancer. Int J Biol Sci. 2015;11:1160–70. doi: 10.7150/ijbs.11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magee P, Shi L, Garofalo M. Role of microRNAs in chemoresistance. Ann Transl Med. 2015;3:332. doi: 10.3978/j.issn.2305-5839.2015.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Ke G, Han D, Liang S, Yang G, Wu X. MicroRNA-181a enhances the chemoresistance of human cervical squamous cell carcinoma to cisplatin by targeting PRKCD. Exp Cell Res. 2014;320:12–20. doi: 10.1016/j.yexcr.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 18.Niu J, Xue A, Chi Y, Xue J, Wang W, Zhao Z, Fan M, Yang CH, Shao ZM, Pfeffer LM, Wu J, Wu ZH. Induction of miRNA-181a by genotoxic treatments promotes chemotherapeutic resistance and metastasis in breast cancer. Oncogene. 2016;35:1302–13. doi: 10.1038/onc.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepper C, Bentley P, Hoy T. Regulation of clinical chemoresistance by bcl-2 and bax oncoproteins in B-cell chronic lymphocytic leukemia. Br J Haematol. 1996;95:513–7. doi: 10.1046/j.1365-2141.1996.d01-1927.x. [DOI] [PubMed] [Google Scholar]