

Abstract
MicroRNAs and long non coding RNAs (lncRNA) shows an encouraging trend in the therapeutics of Osteoarthritis (OA), but there are limited studies showing the functional role of lncRNAs and miRs in OA pathogenesis. The study investigated the role of HOTAIR and its predicted target miR-17-3p in pathogenesis and identification of novel drug target of OA. LncRNAs were identified qualitatively and quantitatively by qRT-PCR, ELISA and Western blot analysis in C28/I2 cells. Results showed that LPS induced cell injury, cell apoptosis and influx of inflammatory cytokines (P<0.001) in C28/I2 cells. Over-expression of HOTAIR aggravated LPS-induced cell viability inhibition, cell apoptosis and inflammatory cytokines influx, while suppression of HOTAIR alleviated the injury (P<0.05). MiR-17-3p is a target of HOTAIR. Suppression of HOTAIR reduced LPS-induced- cell proliferation inhibition, cell apoptosis and inflammatory cytokines influx by overexpression of miR-17-3p, while suppression of miR-17-3p alleviated the injury in C28/I2 cells (P<0.05). Erythroblast transformation-specific translocation variant 1 (ETV1) is a target of miR-17-3p. Overexpression of ETV1 promoted LPS-induced-cell proliferation inhibition, cell apoptosis and inflammatory cytokines influx, while suppression of ETV1 alleviated the injury in C28/I2 cells (P<0.01). Signaling of LPS-induced cell injury was found to be via ETV1 by activation of MAPK/c-Jun and NF-κB pathways. In conclusion, LPS-induced cell injury was found to be mediated via suppression of miR-17-3p and promotion of ETV1 expression by activation of MAPK/c-Jun and NF-κB pathways. HOTAIR and miR-17-3p can be a potential therapeutic target in OA patients.
Keywords: Osteoarthritis, miR-17-3p, HOTAIR, lncRNA, ETV1
Introduction
Osteoarthritis (OA) is a degenerative disease that involves the entire synovial joint, encompassing the cartilage, synovium, and the underlying bony structure. Gradual loss of articular cartilage is attributed to the independent capacities of the cells of these tissues to initiate and respond to the injury in the joint [1]. In OA, the degeneration of cartilage happens in 2 phases-a biosynthetic phase and a degradative phase [2]. The biosynthetic phase is marked by the attempt of chondrocytes to repair the extracellular matrix. In the degradative phase the enzymes produced by chondrocytes, such as matrix metalloproteinases (MMPs), disintegrin, and metalloprotease with thrombospondin motifs (ADAMTSs) [2], digest the matrix resulting in the inhibition of matrix synthesis and consequent erosion of matrix [1]. Since OA lesions are localized on weight bearing cartilages and trauma sites, chondrocytes are generally accepted as the target of the resultant biomechanical factors contributing to alterations in the normal functioning of these cells [3]. Various biochemical and genetic factors too cause these cellular alterations [3]. Therefore, it is very important to study the mechanism of inflammatory injury of cartilage cells in order to understand the underlying pathogenesis and to identify novel drug targets for osteoarthritis.
Epigenetic effects such as histone modification, DNA methylations and noncoding RNAs are likely to be implicated in OA [4,5]. Non coding RNAs are particularly shown to play an important role in development of OA [6]. Generally, non coding RNAs are of various types, short non coding RNAs (micro RNA), long non coding RNAs (lncRNAs) and the classic rRNAs, tRNAs and snRNAs. MicroRNAs are the most well studied non coding RNAs shown to be differentially expressed in osteoarthritic and normal cartilage [7].
In the human genome, a relatively large amount of sequence generates non-protein-coding RNA that are crucial in translation, RNA splicing, and gene regulation [2]. The long non-coding RNAs (lncRNAs) are this class of non-coding RNAs that have more than 200 nucleotides without an open reading frame [2]. They regulate key cellular processes such as cell proliferation, apoptosis, and cell differentiation [8]. Some examples are of lncRNA H19 (an attractive marker for cell anabolism in cartilage and cultured chondrocytes) and lnc RNA maternally expresses gene 3 (MEG3) which was shown to be downregulated in OA cartilage samples than when compared to the normal control and MEG 3 could regulates angiogenesis contributing to OA development [8]. Elevated levels of lncRNA prostate cancer gene expression marker 1 (PCGEM1) was seen in osteoarthritic synoviocytes [8]. lncRNA growth arrest-specific 5 (GAS5) was found to be overexpressed in chondrocytes of OA patients when compared to normal chondrocytes [9]. Studies done on temporomandibular joint in OA patients show a significant upregulation in HOTAIR in the synovial fluids when compared to normal controls [2]. Similarly, HOTAIR levels were high in the synovial fluids of temporomandibular joint in animal (rabbit) model of OA when compared to the normal rabbits [2]. Additionally, interleukin (IL)-1β treatment in chondrocytes isolated from the temporomandibular joint condylar cartilage of New Zealand white rabbits dramatically enhanced the expression of MMP-1, MMP-3, and MMP-9. These effects were reversed by HOTAIR knockdown [2]. Thus it can be said that differently expressed lncRNAs are associated in the pathogenesis of OA, especially HOTAIR which is involved in the upregulation of MMPs contributing to OA. Further exploration of these non protein coding RNAs can help in understanding this potential target for OA therapy [8].
Though there are no studies that report the direct association of miRNAs and OA. Suarez et al demonstrated that miRNA regulated TNF-mediated inflammation in the endothelial cells Transfections with the mimics of these miRNAs reduced neutrophil adhesion to endothelial cells, suggesting a negative feedback inhibition of inflammation by miRNAs [10].
Therefore, the present study has been undertaken to study the differentially expressed lncRNA (HOTAIR) and its predicted target genes in human articular chondrocyte C28/I2 cells using quantitative reverse transcription polymerase chain reaction (qRT-PCR), western blot and ELISA.
Materials and methods
Cell culture and treatment
Human cartilage C28/I2 cells were purchased from the American Type Culture Collection (Manassas, VA). RPMI-1640 medium supplemented with 10% FBS was used to culture the cells in a humidified incubator containing 5% CO2 at 37°C. The purified cells were frozen between fifth and tenth passages and were maintained in growth medium in a 75 cm2 flask. It was made sure that fresh medium was added to cells every 3 days until confluence was achieved. All experiments were done using cells at passage 10 or below. The cells were treated by LPS for 12 h.
CCK-8 assay
Cells were seeded in 96-well plate with 5000 cells/well. The cell proliferation was determined by a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD). Briefly, the CCK-8 solution was added to the culture medium after stimulation, and the cultures were incubated for 1 hour at 37°C in humidified 95% air and 5% CO2. A microplate reader (Bio-Rad, Hercules, CA) was used to measure the absorbance at 450 nm using a Microplate Reader.
Apoptosis assay
The cell apoptosis assay was conducted using propidium iodide (PI) and fluorescein isothiocynate (FITC)-conjugated Annexin V staining. The cells were washed in phosphate buffered saline (PBS) and fixed in 70% ethanol. Further, the fixed cells were washed in PBS twice and stained in PI/FITC-Annexin V in the presence of 50 μg/ml RNase A (Sigma-Aldrich), and then incubated for 1 h at room temperature in the dark. Flow cytometry analysis was done by using a FACS can (Beckman Coulter, Fullerton, CA, USA). The data were analyzed by using FlowJo software.
qRT-PCR
The total RNA was extracted from cells using Trizol reagent (Life Technologies Corporation, Carlsbad, CA, USA) according to the manufacturer’s instructions.
The real time PCR analysis was done using One Step SYBR® PrimeScript® PLUS RT-RNA PCR Kit (TaKaRa Biotechnology, Dalian, China) to test the expression levels of HOTAIR. Taqman MicroRNA Reverse Transcription Kit and Taqman Universal Master Mix II with the TaqMan MicroRNA Assay of miR-17-3p and U6 (Applied Biosystems, Foster City, CA, USA) were used for testing the expression levels of miR-17-3p in cells. Runx2 was tested using RNA PCR Kit (AMV) Ver.3.0 (TaKaRa Biotechnology, Dalian, China). The GAPDH was used as the internal control in this study for normalizing fold changes by relative quantification (2-ΔΔCt) method.
Transfection and generation of stably transfected cell lines
Short-hairpin RNA directed against human lncRNA HOTAIR was ligated into the U6/GFP/Neo plasmid (GenePharma, Shanghai, China) and was referred as to sh-HOTAIR. The complete length of HOTAIR and ETV1 sequences and short-hairpin RNA directed against ETV1 was constructed in pEX-2 and U6/GFP/Neo plasmids (GenePharma), respectively. They were referred to as pEX-HOTAIR, pEX-ETV1 and sh-ETV1. The lipofectamine3000 reagent (Life Technologies Corporation, Carlsbad, CA, USA) was used for cells transfection according to the manufacturer’s instructions. The plasmid carrying a non-targeting sequence was used as a negative control (NC) of sh-HOTAIR and sh-ETV1 that was referred to as sh-NC. The stably transfected cells were selected by the culture medium containing 0.5 mg/ml G418 (Sigma-Aldrich, St Louis, MO, USA) and G418-resistant cell clones were established after approximately 4 weeks. MiR-17-3p mimics, inhibitors and their respective NC were synthesized (Life Technologies Corporation, MD, USA) and transfected into cells in the study. The highest transfection efficiency occurred at 48 h and thus 72 h post-transfection was considered as the harvest time in the subsequent experiments.
Reporter vectors constructs and luciferase reporter assay
The miR-17-3p binding site of HOTAIR was amplified and cloned into a pmiRGlO Dual-luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA) to form the reporter vector HOTAIR wild type (HOTAIR-Wt). The putative binding site sequence of miR-17-3p in the HOTAIR was replaced and being referred to as HOTAIR-mutated-type, HOTAIR-Mt. The vectors and miR-17-3p mimics were co-transfected into HEK 293T cells, and the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) was used to test the luciferase activity.
ELISA
Culture supernatants were collected from 24-well plates and the concentrations of inflammatory cytokines were measured by enzyme-linked immunosorbent assay (ELISA) as per manufacturer’s instructions (R&D Systems, Abingdon, UK).
Western blot
For Western blotting, the protein was extracted using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitors (Roche, Guangzhou, China). The BCA™ Protein Assay Kit (Pierce, Appleton, WI, USA) was used to quantify the proteins. Western blot system was established using a Bio-Rad Bis-Tris Gel system according to the manufacturer’s instructions. Briefly, in the analysis, the primary antibodies were prepared in 5% blocking buffer at a dilution of 1:1,000. Further, they were incubated with the membrane at 4°C overnight, followed by a wash and incubation with the secondary antibody linked to horseradish peroxidase for 1 hour at room temperature. After rinsing, the polyvinylidene difluoride (PVDF) membrane carried blots and antibodies were transferred into the Bio-Rad ChemiDoc™ XRS system. Around 200 μl of Immobilon Western Chemiluminescent HRP Substrate (Millipore, MA, USA) was added to cover the membrane surface. The signals were recorded and the intensity of the bands was quantified using Image Lab™ Software (Bio-Rad, Shanghai, China).
Statistical analysis
The results were replicated thrice to ensure consistency and are represented as the mean ± SD values. All the statistical analyses were conducted using Graphpad 6.0 statistical software. A P-value of <0.05 was considered for a statistically significant result.
Results
LPS induced cell injury and expression of inflammatory cytokines in human articular chondrocyte C28/I2 cells
Human articular chondrocyte C28/I2 cells were incubated with different concentrations of LPS (1 µg/ml, 5 µg/ml, and 10 µg/ml) and were assessed for cell viability, apoptotic cells (estimation of apoptosis related factors Bax and Bcl-1) and presence of inflammatory cytokines (interleukins and tumour necrosis factor-alpha). LPS reduced cell viability by inducing cell injury in a concentration-dependent manner (Figure 1A). The apoptotic cells were higher in number at 10 µg/ml concentration of LPS (Figure 1B). LPS induced the expression of apoptotic genes (Bax) in human articular chondrocyte C28/I2 cells in a concentration-dependent manner. The expression of anti-apoptotic factor Bcl-2 was decreased while that of pro-apoptotic factor (Bax) (Figure 1B). In human articular chondrocyte C28/I2 cells treated with LPS, both the mRNA expressed and the amount of inflammatory cytokines, including IL-1β, IL-6, IL-8 and tumour necrosis factor-alpha (TNF-α) were increased when compared to the corresponding control group (P<0.01, Figure 1C and 1D).
Figure 1.

LPS induced cell injury and promoted expression of inflammatory cytokines in human articular chondrocyte C28/I2 cells. A: LPS inhibited cell viability. B: LPS promoted cell apoptosis. C and D: LPS promoted expression of inflammatory cytokines (mRNA and ELISA). Data were expressed as mean ± SD. **P<0.01, ***P<0.001, ****P<0.0001.
LPS induces the expression of HOTAIR in human articular chondrocyte C28/I2 cells
HOTAIR expression was significantly increased (P<0.01) in human articular chondrocyte c28/I2 cells following treatment with LPS compared to the control group cells as shown in Figure 2.
Figure 2.
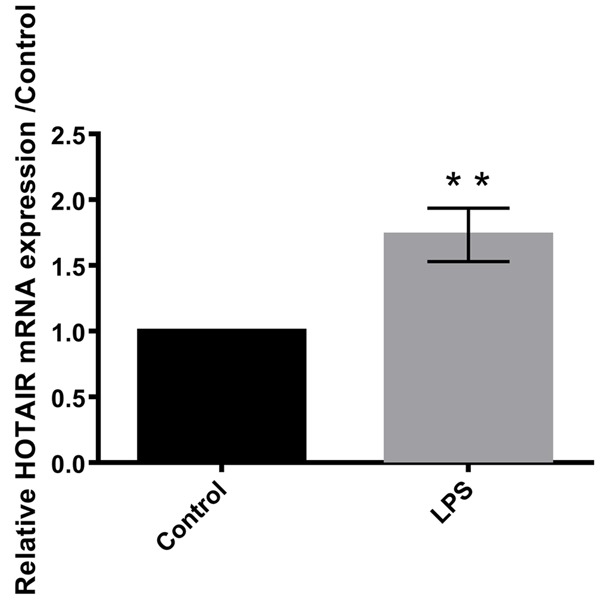
LPS induced the expression of HOTAIR in C28/I2 cells. Data were expressed as mean ± SD. **P<0.01.
Overexpression of HOTAIR further aggravates LPS-induced cell injury and expression of inflammatory cytokines, and while suppression of HOTAIR alleviates the injury
As shown in Figure 3A, the relative mRNA expression of HOTAIR was significantly higher in cells transfected with pEx-HOTAIR compared to control group of cells (P<0.01). The cell viability was found to be aggravated in shHOTAIR cells (P<0.05) with overexpression of HOTAIR (Figure 3B). The apoptotic cells were higher in transfected cells with pEx-HOTAIR when compared to the control group (P<0.05) as shown in Figure 3C. The inflammatory cytokines were highest in pEx-HOTAIR group (P<0.05) when compared to the control group as shown in Figure 3D. Further, suppression of HOTAIR seems to stop the injury in the human articular chondrocyte C28/I2 cells.
Figure 3.
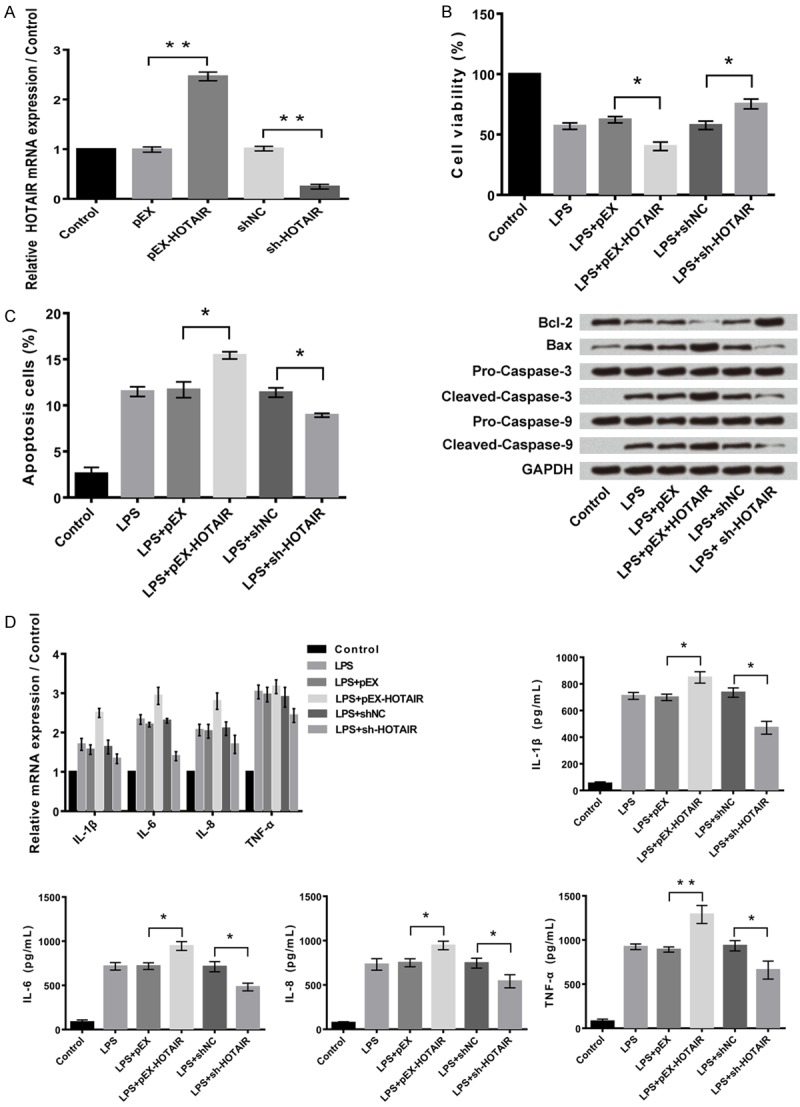
Overexpression of HOTAIR further aggravates LPS-induced cell injury and expression of inflammatory cytokines. A: Overexpression and suppression of HOTAIR in C28/I2 cells. B: Overexpression of HOTAIR aggravates LPS-induced cell viability inhibition, and while suppression of HOTAIR alleviates the injury. C: Overexpression of HOTAIR further aggravates LPS-induced cell apoptosis, and while suppression of HOTAIR alleviates the injury. D: Overexpression of HOTAIR further aggravates LPS-induced expression of inflammatory cytokines, and while suppression of HOTAIR alleviates the injury. Data were expressed as mean ± SD. *P<0.05, **P<0.01.
HOTAIR negatively regulates miR-17-3p, miR-17-3p is a target of HOTAIR
Relative mRNA expression of miR-17-3p was significantly suppressed (P<0.05) in cells transfected with pEx-HOTAIR compared to control group of cells and vice versa (P<0.05, Figure 4A). MiR-17-3p was found to be a target for HOTAIR in the relative luciferase activity. The relative luciferase activity percentage was found to be higher in the mutated HOTAIR than in the wild type (Figure 4B).
Figure 4.
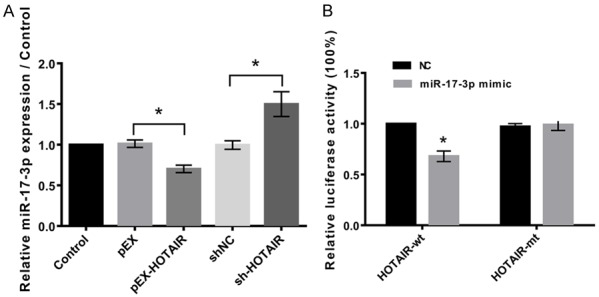
MiR-17-3p is a target of HOTAIR. A: HOTAIR negatively regulates miR-17-3p. B: Relative luciferase activity showed that miR-17-3p is a target of HOTAIR. Data were expressed as mean ± SD. *P<0.05.
Suppression of HOTAIR reduces cell injury by overexpression of miR-17-3p
Figure 5A shows that expression of miR-17-3p was significantly increased (P<0.01) in cells after transfection with miR-17-3p mimic and it was significantly decreased (P<0.01) in presence of miR-17-3P inhibitor. As shown in Figure 5B, the cell viability percentage was higher in transfected cells with sh-HOTAIR (P<0.01). Even the apoptotic cells were found to be higher in sh-HOTAIR cells in Figure 5C (P<0.01). As per Figure 5D, the inflammatory cytokines were higher in transfected cells sh-HOTAIR in the presence of miR-17-3P inhibitor (P<0.001).
Figure 5.
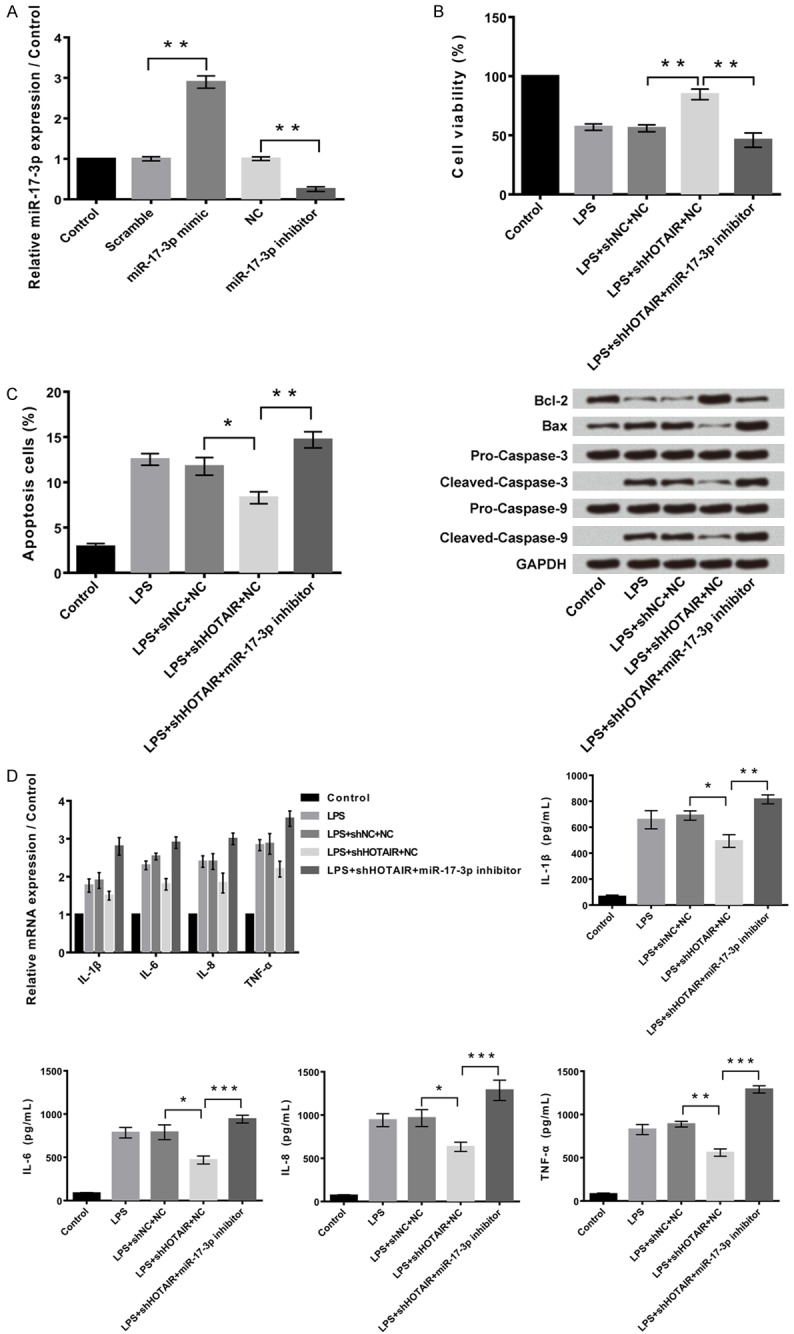
Suppression of HOTAIR reduces cell injury by overexpression of miR-17-3p. A: Overexpression and suppression of miR-17-3p in C28/I2 cells. B: Suppression of HOTAIR reduced LPS-induced cell proliferation inhibition by overexpression of miR-17-3p, and while suppression of miR-17-3p alleviates the injury. C: Suppression of HOTAIR reduced LPS-induced cell apoptosis by overexpression of miR-17-3p, and while suppression of miR-17-3p alleviates the injury. D: Suppression of HOTAIR reduced LPS-induced cell inflammatory cytokines expression by overexpression of miR-17-3p, and while suppression of miR-17-3p alleviates the injury. Data were expressed as mean ± SD. **P<0.05, **P<0.01, ***P<0.001.
MiR-17-3p negatively regulates ETV1, and ETV1 is a target of miR-17-3p
MiR-17-3p suppressed the expression of erythroblast transformation-specific translocation variant 1 (ETV1) when compared to the control group (P<0.05, Figure 6A). The relative mRNA expression of ETV1 was found to be the lowest in miR-17-3p mimic cells as per the western blot analysis. ETV1 was found to be a target for HOTAIR in the relative luciferase activity. The relative luciferase activity percentage was found to be higher in the miR-17-3p mimic group (P<0.05, Figure 6B).
Figure 6.
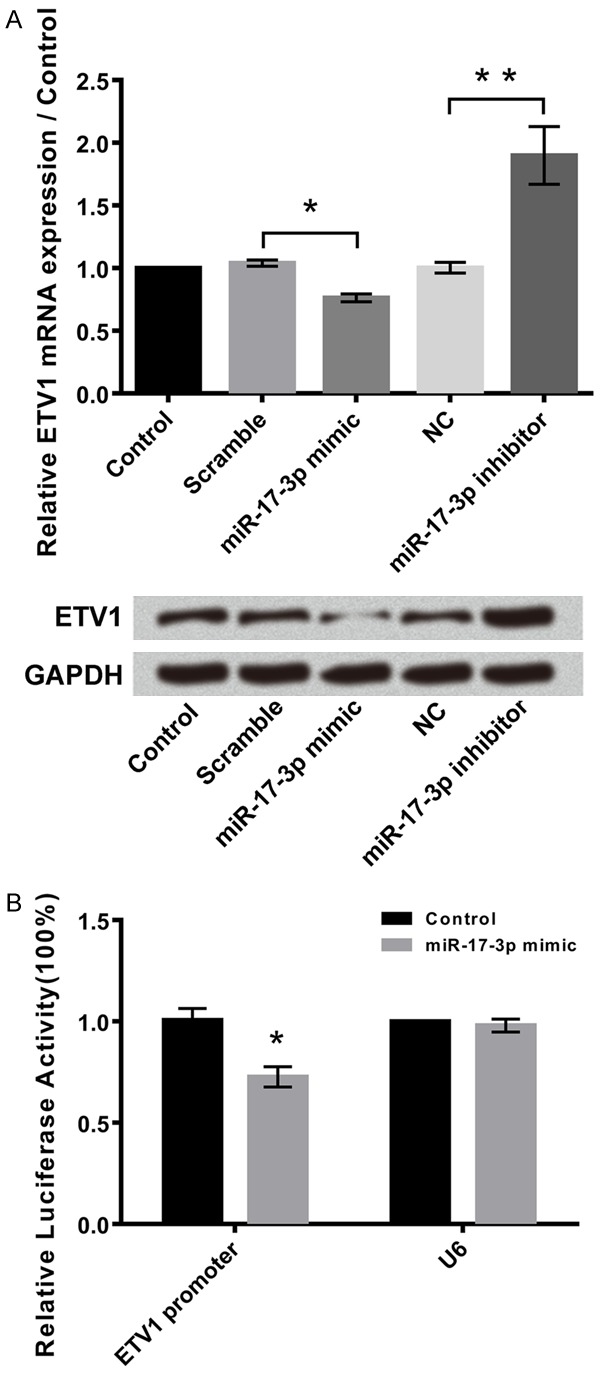
MiR-17-3p negatively regulates ETV1, and ETV1 is a target of miR-17-3p. A: miR-17-3p negatively regulated ETV1. B: ETV1 was a target of miR-17-3p. Data were expressed as mean ± SD. **P<0.05, **P<0.01.
Overexpression of ETV1 promotes LPS-induced cell injury, and while suppression of ETV1 alleviates cell injury
Figure 7A shows that the expression of ETV1 was significantly (P<0.01) increased following transfection with pEx-ETV1. The relative mRNA expression of ETV1 was found to be the lowest in sh-ETV1 cells as per the western blot analysis. As shown in Figure 7B, the cell viability percentage was higher in transfected cells with sh-ETV1 (P<0.05). Even the apoptotic cells were found to be higher in sh-ETV1 cells in Figure 7C when compared to the control group (P<0.05). As per Figure 7D, the inflammatory cytokines were higher in transfected cells pEx-ETV1 when compared to the control group (P<0.05).
Figure 7.
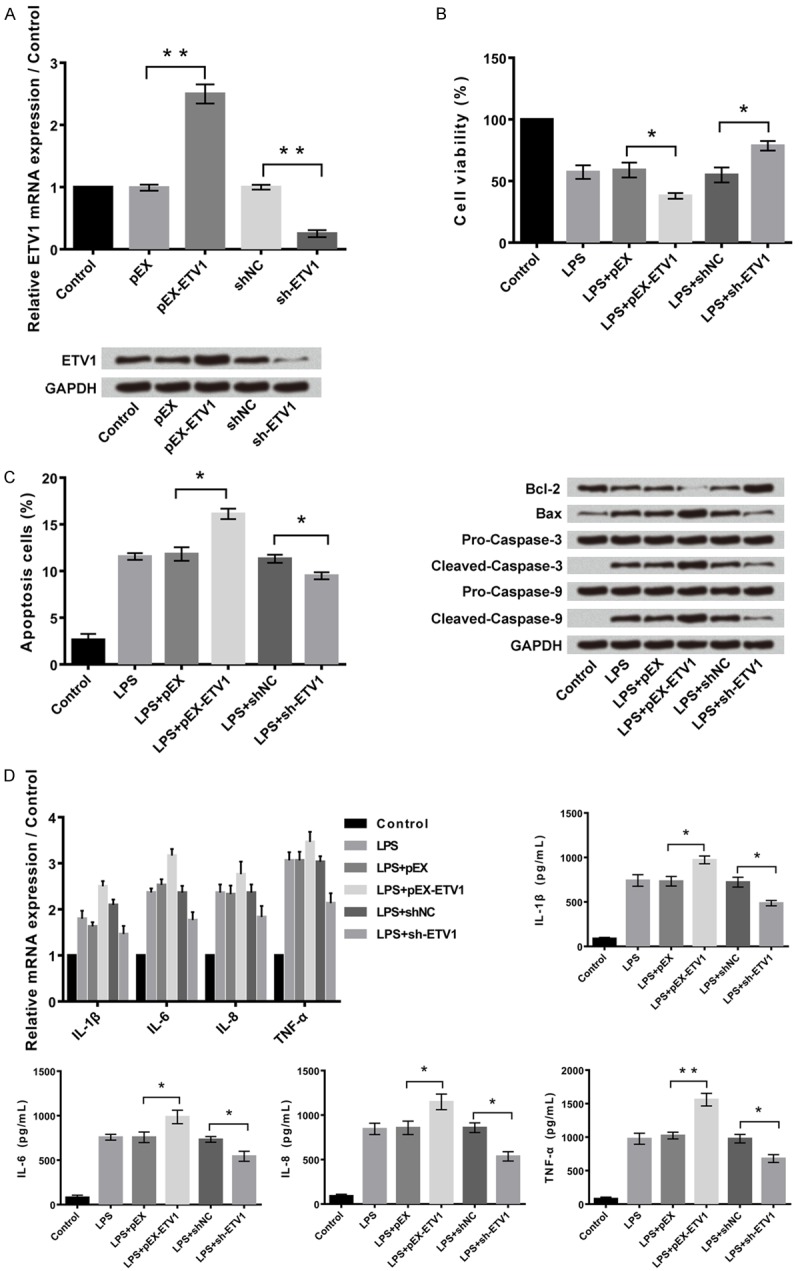
Overexpression of ETV1 promotes LPS-induced cell injury, and while suppression of ETV1 alleviates cell injury. A: Overexpression and suppression of ETV1 in C28/I2 cells. B: Overexpression of ETV1 promoted LPS-induced cell proliferation inhibition, and while suppression of ETV1 alleviated the injury. C: Overexpression of ETV1 promoted LPS-induced cell apoptosis, and while suppression of ETV1 alleviated the injury. D: Overexpression of ETV1 promoted LPS-induced cell inflammatory cytokines expression, and while suppression of ETV1 alleviated the injury. **P<0.05, **P<0.01.
Signaling of LPS-induced cell injury via ETV1
ETV1 promoted LPS-induced cell injury by activation of MAPK/c-Jun and NF-κB pathways as shown in Figure 8.
Figure 8.
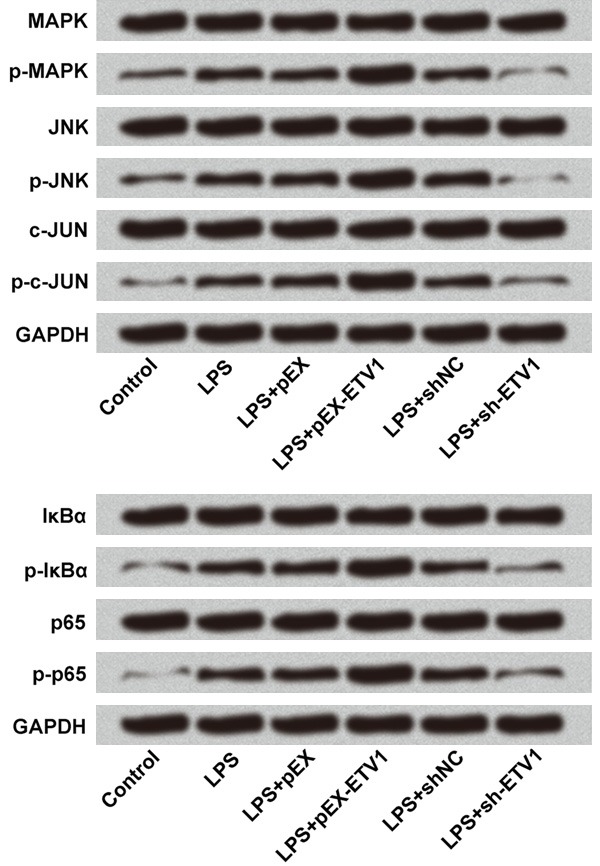
Signaling of LPS-induced cell injury via ETV1. ETV1 promoted LPS-induced cell injury by activation of MAPK/c-Jun and NF-κB pathways.
Discussion
Osteoarthritis (OA) is a widespread joint disorder resulting from chronic inflammatory responses owing to joint instability, aging, and obesity. Genetic studies involving several family groups and twin studies indicate the association of epigenetic effects such as histone modifications, DNA methylations and noncoding RNAs with OA susceptibility [4,5].
MicroRNAs (miRs) are the most well studied short noncoding RNA observed to be differentially expressed in osteoarthritic and normal cartilages. miR-9 is known to be upregulated [7,11] in osteoarthritic cartilage and expression of miR-27a, miR-140 and miR-146 is decreased [11-14]. Specific expression of miRs such as miR-455, miR-381, miR-193b and miR-92b [15] during chondrogenesis have been shown in several studies Potential role of miRs have been demonstrated in vitro and in vivo in the genesis and development of OA [16,17].
In recent times, a lot of attention is being grabbed by long non coding RNAs (lncRNA), which are known to be pervasively transcribed in mammalian genome [18]. The lncRNAs are implicated in a large number of regulatory processes, where they have multiple functions to perform. The first discovered lncRNA, HOTAIR has been largely studied in the context of tumor biology and has found to be associated with spermiogenesis [16], aortic valve calcification [17], neurodegeneration diseases [19] and hepatitis B virus-induced liver carcinogenesis [20]. The genetic variants of HOTAIR were observed to have led to the risk of osteosarcoma [19]. It was identified that HOTAIR was expressed in OA cartilage in higher levels when compared to the normal samples [14]. However, the expression profile and potential biological function of HOTAIR in the OA is less known.
In the present study, we compared the expression profile of lncRNAs, particularly HOTAIR in OA samples. We report that overexpression of HOTAIR aggravated LPS-induced cell injury, LPS-induced cell apoptosis and a spike in LPS-induced expressions of inflammatory cytokines in the human articular chondrocytes C28/I2 cells (Figure 3). This implicates the participation of HOTAIR in regulating osteogenic differentiation during OA.
Bioinformatics software reports the complementarity in the sequence of HOTAIR and miR-17-5p, suggesting some role of HOTAIR in the regulation of this miR-17/92 cluster [21]. The upregulation/downregulation of HOTAIR in osteogenic differentiation related parameters were observed to be reversed by upregulation/downregulation+ of miR-17-5p target gene SMAD7, strengthening the role of HOTAIR in osteogenesis through modulating the expression of miR-17-5p and its target gene SMAD7 [22]. Independent prediction analysis tools identified a conserved binding site of ETV1 as a possible target of miR-17. Knockdown experiments of ETV1 and ETV1-specific siRNA affects the overexpression of miR-17 [23].
The effects and mechanisms of lncRNA HOTAIR on the LPS-induced C28/I2 cell injury in the present study revealed very promising results it showed that LPS induced the expression of HOTAIR. Overexpression of HOTAIR further aggravated cell injury in C28/I2 cells, and while suppression of HOTAIR inhibited the cell injury. Further studies showed that miR-17-3p was a target of HOTAIR, and HOTAIR negatively regulated the expression of miR-17-3p (Figures 4 and 5). In addition, we found that HOTAIR regulated the inflammatory injury by down-regulation of miR-17-3p. Moreover, our findings suggested that miR-17-3p negatively regulated ETV1, and ETV1 was a target of miR-17-3p (Figure 6A and 6B). Overexpression of ETV1 promotes LPS-induced cell injury, and while suppression of ETV1 alleviates cell injury (Figure 7A and 7B). Besides, the results showed that ETV1 could regulate the inflammatory damage of C28/I2 cells by activation of MAPK/c-Jun and NF-κB pathways (Figure 8).
We are aware of the limitations of the study where we are providing indirect experimental evidences to indicate the functional links between lncRNA (HOTAIR) and its predicted target in OA. But yet, it leaves scope for further research in confirming our predictions of these target genes and demonstrating their functional role in cartilage degeneration and osteogenesis.
In summary, the study provides the expression profile/levels of lncRNAs in chondrocytes and attempts to validate them by studying the possible underlying mechanisms of the target genes and their pathways. These findings suggests that lncRNA HOTAIR is a potential biomarker for OA diagnosis and therapeutics, However, this also warrants further exploratory studies to understand the molecular mechanisms and biological functions of lncRNAs in OA pathogenesis.
Acknowledgements
This study was supported by the Key Science and Technology Program of Yiwu City (No.2009-G3-02).
Disclosure of conflict of interest
None.
References
- 1.Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–113. doi: 10.1186/ar148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen WK, Yu XH, Yang W, Wang C, He WS, Yan YG, Zhang J, Wang WJ. lncRNAs: novel players in intervertebral disc degeneration and osteoarthritis. Cell Prolif. 2017;50 doi: 10.1111/cpr.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 4.Pottie P, Presle N, Terlain B, Netter P, Mainard D, Berenbaum F. Obesity and osteoarthritis: more complex than predicted! Ann Rheum Dis. 2006;65:1403–1405. doi: 10.1136/ard.2006.061994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Livshits G, Kato BS, Zhai G, Hart DJ, Hunter D, MacGregor AJ, Williams FM, Spector TD. Genomewide linkage scan of hand osteoarthritis in female twin pairs showing replication of quantitative trait loci on chromosomes 2 and 19. Ann Rheum Dis. 2007;66:623–627. doi: 10.1136/ard.2006.060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynard LN, Loughlin J. Genetics and epigenetics of osteoarthritis. Maturitas. 2012;71:200–204. doi: 10.1016/j.maturitas.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Zhang C, Wang P, Jiang P, Lv Y, Dong C, Dai X, Tan L, Wang Z. Upregulation of lncRNA HOTAIR contributes to IL-1beta-induced MMP overexpression and chondrocytes apoptosis in temporomandibular joint osteoarthritis. Gene. 2016;586:248–253. doi: 10.1016/j.gene.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Song J, Ahn C, Chun CH, Jin EJ. A long noncoding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J Orthop Res. 2014;32:1628–1635. doi: 10.1002/jor.22718. [DOI] [PubMed] [Google Scholar]
- 10.Suarez Y, Wang C, Manes TD, Pober JS. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J Immunol. 2010;184:21–25. doi: 10.4049/jimmunol.0902369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones SW, Watkins G, Le Good N, Roberts S, Murphy CL, Brockbank SM, Needham MR, Read SJ, Newham P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthritis Cartilage. 2009;17:464–472. doi: 10.1016/j.joca.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Akhtar N, Rasheed Z, Ramamurthy S, Anbazhagan AN, Voss FR, Haqqi TM. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010;62:1361–1371. doi: 10.1002/art.27329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyaki S, Nakasa T, Otsuki S, Grogan SP, Higashiyama R, Inoue A, Kato Y, Sato T, Lotz MK, Asahara H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009;60:2723–2730. doi: 10.1002/art.24745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamasaki K, Nakasa T, Miyaki S, Ishikawa M, Deie M, Adachi N, Yasunaga Y, Asahara H, Ochi M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009;60:1035–1041. doi: 10.1002/art.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Kang Y, Zhang Z, Zhang H, Duan X, Liu J, Li X, Liao W. Expression of microRNAs during chondrogenesis of human adipose-derived stem cells. Osteoarthritis Cartilage. 2012;20:1638–1646. doi: 10.1016/j.joca.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 16.Vonk LA, Kragten AH, Dhert WJ, Saris DB, Creemers LB. Overexpression of hsa-miR-148a promotes cartilage production and inhibits cartilage degradation by osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2014;22:145–153. doi: 10.1016/j.joca.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Park SJ, Cheon EJ, Lee MH, Kim HA. MicroRNA-127-5p regulates matrix metalloproteinase 13 expression and interleukin-1betainduced catabolic effects in human chondrocytes. Arthritis Rheum. 2013;65:3141–3152. doi: 10.1002/art.38188. [DOI] [PubMed] [Google Scholar]
- 18.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 20.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao CG, Yang YY, He X, Xu T, Huang C, Huang Y, Zhang L, Lv XW, Jin Y, Li J. New advances of microRNAs in the pathogenesis of rheumatoid arthritis, with a focus on the crosstalk between DNA methylation and the microRNA machinery. Cell Signal. 2013;25:1118–1125. doi: 10.1016/j.cellsig.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 22.Jia J, Feng X, Xu W, Yang S, Zhang Q, Liu X, Feng Y, Dai Z. MiR-17-5p modulates osteoblastic differentiation and cell proliferation by targeting SMAD7 in non-traumatic osteonecrosis. Exp Mol Med. 2014;46:e107. doi: 10.1038/emm.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen R, Greenberg E, Nemlich Y, Schachter J, Markel G. miR-17 regulates melanoma cell motility by inhibiting the translation of ETV1. Oncotarget. 2015;6:19006–19016. doi: 10.18632/oncotarget.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]