

Abstract
Background and aims: Hepatocellular carcinoma (HCC) is a common cancer worldwide. Researchers have found that UDCA can be used to inhibit the growth of tumors. Microtubule-associated protein light chain 3B (LC3B) is an important reglator of autophagosomes. No researches have been published on the relationship of UDCA and LC3B. Methods: A Cell Counting Kit-8 cell viability assay, cell migration assay, quantitative reverse transcription PCR (qRT-PCR) and western blot were conducted for the SMMC-7721 and HepG2 cell lines. Hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) were used to analyze nude mice with 7721 xenograftes. The expression of LC3B was evaluated both in vivo and in vitro. Results: Our studies demonstrated that UDCA reduced the viability of the primary HCC cell lines 7721 and HepG2 (Student’s t-test, P<0.05) and inhibited the migration of 7721 cells (Student’s t-test, P<0.05). UDCA also increased the expression of LC3B and p53 in vitro (Student’s t-test, P<0.05). Additionally, UDCA inhibited the growth of tumors (Student’s t-test, P<0.05) and promoted the expression of LC3B in nude mice. Conclusion: Our data showed that UDCA promoted the expression of LC3B, with suppressed HCC in vivo and in vitro.
Keywords: Ursodeoxycholic acid, hepatocellular carcinoma, LC3B, p53, authophagy
Introduction
Liver cancer is the sixth most common cancer (782,451 new cases), the second cause of cancer related death (745,517 cases), and accounts for 9.1% of all cancers [1]. Hepatocellular carcinoma (HCC) represents more than 90% of primary liver cancers and is a major global health problem. The incidence of HCC increases progressively with advancing age in all populations, and reaches a peak at 70 years of age. In Chinese and black African populations, the mean age of patients with HCC is appreciably younger [2].
Ursodeoxycholic acid (UDCA), as a hydrophilic bile acid, is widely used to treat hepatobiliary diseases ina clinical setting. UDCA can promote the secretion of bile and inhibit bile reabsorption from the intestines. Therefore, UDCA is used to treat cholestasis, and it is also known to exert direct cytoprotective effects on hepatocytes [3,4]. Researchers have found that UDCA can be used to inhibit the growth of tumors. Lim et al. [5] demonstrated that UDCA killed drug-resistant gastric cancer cells via the induction of autophagic death. Chung et al. [6] found that UDCA suppressed HCC cell growth through the inhibition of the degradation of DLC1, and Zhu et al. showed that UDCA induced apoptosis of HCC cells [7]. However, the mechanism by which UDCA inhibits tumors is not well defined.
Autophagy is an evolutionarily conserved processthat functions in the degradation of long-lived proteins, aggregated proteins, damaged organelles and certain pathogens [8]. However, the functions of autophagy with respect to tumor cells are still controversial. Guo et al. [9]showed that the autophagy process was required by activated Ras in order to maintain oxidative metabolism and tumorigenesis, while, Elgendy et al. [10] suggested that Ras-induced autophagy may kill tumor cells that are on the verge of oncogenic transformation. Lim and Han [5] demonstrated than ursodexocycholic acid effectively killed drug-resistant gastric cancer cells through induction of autophagic death. In our research, we showed an incremental increase in the expression level of microtubuleassociated protein light chain 3B (LC3B), which is an important regulator of autophagosomes. This demonstrates that UDCA may inhibit HCC through the induction of autophagy.
Methods and materials
Reagents
UDCA (from Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO) (from Amresco 0231) to achieve the final concentrations of 0.3, 0.6, 0.8, 1.0 and 1.2 mmol/l. UDCA at varied doses was used to treat the HCC cells for 24 h, 48 h or 72 h to identify its effect on cell viability. Cells treated with UDCA at 0.8 and 1.2 mmol/l were further investigated to determine the expression of autophagy associated factors.
UDCA was suspended in the drinking water of the mice at concentrations of 90 mg/kg/day and 270 mg/kg/day.
Cell lines and culture
The human hepatoma cell line HepG2 was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). SMMC-7221 (7721) cells were agenous gift from the Central Laboratory of Shandong Province Hospital (Shandong, China). Both cell lines were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin G and 100 µg/ml streptmycin at 37°C in a humidified atmosphere cotaining 5% CO2.
To passage the cells, the cell monolayer was rinsed briefly with 1×phosphate-buffered saline (PBS) twice, 1 ml pre-warmed (37°C) 0.05% Trypsin-EDTA solution was added and the cells were incubated with this solution for 3-7 minutes (different cells were incubated for different lengths of time). Once the cell layer was dipersed, Trypsin was deactivated by the addition of 2-3 ml complete growth medium. After all the cells detached, we centrifuged the cell’ suspension for 4 minutes (1000 rpm/min). The supernatant was discarded and the cell pellet was resupended in an appropriate volume of medium for cell counting by Trypan Blue. The cells were split according to their confluence rate. The culture medium was changed once every two days.
Treatment of nude mice
In all, 5×106 cells (resuspended in 100 μl of DMEM) were injected subcutaneously into the posterior axillaryregion of BALB/c (nu/nu) nude mice (Vital River, Beijing, China). Tumor tissues were excised from the nude mice after the tumors were of sufficient size. Tumor cells were obtained from tumor tissues by grinding. Approximately 2×106 cells (resuspended in 100 μl of DMEM) were injected subcutaneously into the posterior axillary of BALB/c (nu/nu) nude mice. Three to 4 weeks later, the mice were assigned to different treatment groups. The mice were used in experiments 7 to 8 weeks after the inoculation of the tumor cells.UDCA was dissolved in the drinking water of the mice. Nude mice bearing tumors derived from 7721 cells received the water ad libitum. At the end of the experiment, the mice were sacrificed by cervical dislocation, and the tumors were excised. All animal care and experimentation conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health, and were approved by the animal ethics committee of Shandong University.
Cell treatment for time-course and dose-response studies
7721 and HepG2 cells were grown in DMEM complete growth medium followed by a 24 h incubation to allow the cells to attach. Afterwards, the medium was withdrawn and replaced with fresh medium and UDCA (UDCA stock solution was dissolved in the medium). We detected the absorbance at 24 h, 48 h and 72 h after UDCA treatment using a Cell Counting Kit-8 (CCK-8) cell viability assay (Dojindo, Japan) according to the manufacturer’s instructions.
Cell migrationassays
The migration of HCC cells was assessed using the 24-well polycarbonate membrane cell migration assay kit (Corning Incorporated Costar, Shanghai, China) according to the manufacter’s instructions. Briefly, the HCC cell lines were incubated in serum-free medium for 24 h. The cells were then transferred to the upper chamber of a Transwell plate by seeding 5×105 cells per well in 200 μl serum-free medium with diferent concentrations UDCA. Next, 0.7 ml of 10% FBS containing medium was added to the lower chamber as a chemoattractant. The cells were then incubated for 24 h at 37°C. Non-migrating cells on the upper membrane surface were scraped off with cotton swabs. Cells that migrated to the bottom of the membrane were stained with 0.1% crystal violet for 30 min, which was, followed by washesin water for 30 s to remove residual dye. Fourviews were examined per Transwell plate and the cells/view were counted at 200×magnification. Each experiment was performed in triplicate.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was isolated using TRIzol® Reagent (TaKaRa Bio Inc., Dalian, China) according to manufacturers’ instructions. Subse-quently, cDNA was generated using a PrimeScriptTM RT Reagent Kit with gDNA eraser (Takara Bio Inc., Dalian, China). RNA samples were extracted from both 7721 and HepG2 cells. cDNA was synthesized using a Superscript III First-Strand cDNA Synthesis kit (Takara Bio Inc., Dalian, China). qRT-PCR was peformed using SYBR Green PCR Master Mix. The primers used in this experment are shown in Table 1. The relative genexpression quantification method was used to calculate the fold changesin mRNA expression.
Table 1.
Primers used for qRT-PCR
| Gene | Sequences |
|---|---|
| LC3B | Forward: 5’-AAACGCATTTGCCATCACA-3’ |
| Reverse: 5’-GGACCTTCAGCAGTTTACAGTCAG-3’ | |
| p53 | Forward: 5’-ACATGACGGAGGTTGTGAGG-3’ |
| Reverse: 5’-GTAGTGGATGGTGGTACAGTCAGAG-3’ | |
| β-actin | Forward: 5’-TGGCACCCAGCACAATGAA-3’ |
| Reverse: 5’-CTAAGTCATAGTCCGCCTAGAAGCA-3’ |
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting
This protocol was approved by the Institutional Review Board of the Shandong Province Hospital, Shandong University, Shandong, China. Total protein was extracted from cells in RIPA lysis buffer (Beyotime, Shanghai, China) and was quantified by Bicinchoninic acid assay. In total, 30 μg of protein was separated using 12% SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The membrane was blocked in a 5% powdered milk solution and incubated with the primary antibody overnight at 4°C. After washing, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (ZSGB-BIO, Beijing, China) at 37°C for 1 h. Protein bands were visualized using Western Bright ECL (Thermo, Shanghai, China) and were detected using an Amersham Imager 600 (General Electric, USA). Antibodies included those against p53 (Cell Signaling Technology, Boston, MA, USA), β-actin (ZSGB-BIO, Beijing, China), and LC3B (Abcam, Shanghai, China).
Immunohistochemistry (IHC)
This protocol was approved by the Institutional Review Board of Shandong Province Hospital, Shandong University, Shandong, China. The tumors were evaluated by immunohistochemical staining. Briefly, the tumor specimens were fixed in 4% paraformaldehyde for 24 h, after which they were embedded in paraffin for subsequent immunohistochemical examination of LC3B expression. After deparaffinization and rehydration inturpentine and alcohol, respectively, the slides were incubated with hydrogen peroxide quench endogenous peroxidase activity. The tumor sections were then blocked with normal serum, and were treated sequentially with normal goat serum, anti-rabbit LC3B antibody (1:200), and biotin-labeled goat anti-rabbit IgG (1:400). Finally, the sections were dehydrated in alcohol and mounted with neutral resins.
Statistical analysis
Data were collected in an MS-Excel spreasheet. Group comparisons for continuous data were made using Student’s t-test for indepdent means, which was performed by IBM SPSS Statistics 22. All experiments were performed in triplicate. Statistical signcance was set at P<0.05.
Results
UDCA reduces the viability of the primary HCC cell lines 7721 and HepG2
To investigate the effect of UDCA on cell proliferation, in vitro studies were performed in 7721 and HepG2 cell lines.
A CCK-8 assay is based on the measurement of activity of a marker associated with the number of viable cell. Our results showed different decreases in OD (450 nm) value in cells treated with UDCA (0.6, 0.8, 0.9, 1.0, 1.1, 1.2 mmol/l) for 24 h, 48 h and 72 h (Figure 1). A significant difference was observed between the control groups and the experimental groups of 7721 cells that were treated with different dosages UDCA at 24 h (Student’s t-test, P<0.05). A statistically signicant difference was also observed between the experimental groups and the control groups of 7721 cells 48 h/72 h after UDCA treatment (Student’s t-test, P<0.05). The viability of HepG2 cells was signicantly reduced aftertreatment with UDCA compared with cells that were not treated with UDCA (Student’s t-test, P<0.05).
Figure 1.

CCK-8 analysis of the reduction of cell viability after UDCA treatment at different time points and different concentrations of UDCA. A. UDCA treatment of 7721 cells. B. UDCA treatment of HepG2 cells. All results are expressed as the mean ± S.D. from three independent experiments. *P<0.05 indicates a statistically significant difference compared with the control group.
UDCA inhibits the migration of 7721 cells
According to the results of the CCK-8 cell viability assay, HepG2 cells treated with UDCA showed an obvious reduction in number at 24 h (Student’s t-test, P<0.05), which would affect their migration. Although a statistically significant difference was observed in 7721 cells treated with UDCA at 24 h (Student’s t-test, P<0.05), the change was very small. Therefore, 7721 cells were selected to test the effect of UDCA on cell migrations. UDCA significantly inhibited the migration of 7721 cells (Student’s t-test, P<0.05). A significant reductionin migration was found when 7721 and HepG2 cells were treaed with UDCA at 48 h and 72 h (Student’s t-test, P<0.05), but we treated only 7721 cells with UDCA and assessed cell migration after 24 h. We observed that UDCA could inhibit the migrtion of 7721 cells, as shown in Figure 2. Finally, this inhibition was intensified as the dosage increased.
Figure 2.
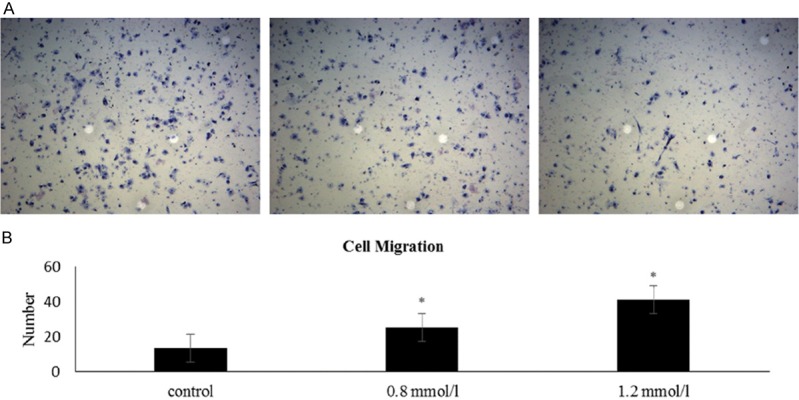
UDCA inhibits the migration of 7721. A. The control group without UDCA (left). The group treated with 0.8 mmol/l UDCA (middle). The group treated with 1.2 mmol/l UDCA (right). B. Histogram of cell migration. All results are expressed as the mean ± S.D. from three independent experiments. *P<0.05 indicates a statistically significant difference compared with the control group.
UDCA improves the level of LC3B and p53 in vitro
We used 0.8 and 1.2 mmol/l UDCA to treat 7721 and HepG2 cells. As revealed by qRT-PCR, LC3B mRNA levels were significantly up-regulated in the HCC cell lines 7721 and HepG2 (Student’s t-test, P<0.05), and themRNA level increased as the dosage increased. The western blot analysis showed the same changes. The expression of LC3B prtein increased as the dosage of UDCA increased, and this change in expression was also statistically significant (Student’s t-test, P<0.05). In order to investigate the reasons for the increase in LC3B protein, we examined whether other prteins were involved in UDCA-induced autophagy. We then found that the levels of p53 mRNA and protein were greatly increased in 7721 and HepG2 cells, upon treatment with UDCA. Additionally, these difrences in exprsion were statistically significant (Student’s t-test, P<0.05). The results are shown in Figure 3.
Figure 3.

UDCA improves the level of LC3B and p53 in 7721 and HepG2 cells. A. The mRNA expression levels of LC3B and p53 increased as the dosage of UDCA increased. The relative levels of LC3B and p53 were detected by qRT-PCR. B, C. The band and gray level analysis of protein expression of LC3B and p53. All results are expressed as the mean ± S.D. from three independent experiments. *P<0.05 indicates a statistically significant difference compared with the control group.
UDCA inhibits tumor growth and promotes the expression of LC3B in nude mice
To determine whether we were able to recapitulate the molecular events we observed in 7721 and HepG2 cells, an HCC xenograft model was established in BALB/c nude mice. BALB/c nude mice that were inoculated with 7721 cells were treated with UDCA suspended in the drinking water. We monitored the tumor growth overan 8-week period. As shown in Figure 4, 8 weeks after transplantation, tumor growth in nude mice that were treated with UDCA was significantly slower than that in control mice that were not treated with UDCA (Student’s t-test, P<0.05). Hematoxylin-eosin staining of tumors from nude mice showed that the percentage of cells that underwent cell death increased as the dosages of UDCA increased (Figure 5I). IHC staining (Figure 5II) of sections from paraffin-embedded xenograft tumors showed an increase in LC3B expression in tumors from mice treated with UDCA.
Figure 4.

UDCA inhibits in vivo growth of xenograft tumors. A. Images of xenografts from mice treated with UDCA (control group (left) n=2, 90 mg/kg/day (middle): n=6, 270 mg/kg/day (right): n=6). B. The tumor volumes were difference among the various groups. All results are expressed as the mean ± S.D. from three independent experiments. *P<0.05 indicates a statistically significant difference compared with the control group.
Figure 5.

I. Hematoxylin-eosin staining of tumor sections obtained from nude mice. A. Xenograft tumors from mice treated with 270 mg/kg/day UDCA. B. Xenograft tumors from mice treated with 90 mg/kg/day UDCA. C. Control group without UDCA. A-C. Low (×200) magnifications. a-c. High (×400) magnifications (a-c each show amplified areas indicated by the arrows in A-C). II. IHC staining of paraffin-embedded xenograft tumors. A. Control group without UDCA treatment. B. Xenograft tumors from mice that were treated with 90 mg/kg/day UDCA. C. Xenograft tumors from mice that were treated with 270 mg/kg/day UDCA. The arrow indicates LC3B staining.
Discussion
Autophagy is a type of self degradative and autophagosome lysosome fusion. Autophagy is characterized by a set of autophagy-related (ATG) proteins [13], such as, Beclin-1 [14] and a lipid-conjugated form of microtubule-associated prtein1 light chain 3 (LC3) [15]. This process primarily acts as a pro-survival mechanism to ovcome stressful conditions, but it ultimately commits the cell to death during prolonged states of stress or inseverely stresful environments [16]. LC3 has three isoforms (LC3A, LC3B and LC3C) [17]. LC3B is used as a marker of autophagy, because it is the key regulator of autophagosome fomation. Lim et al. [5] found that UDCA could increase the ATG5 level to induce autophagy. However, since no studies have been coducted on the relationship of UDCA and LC3B, we examined this relationship in current study.
Although UDCA is widely used to treat hepatobiliary diseaes, many studies have shown that it has antitumor effects. Tsagarakis et al. [18] found that UDCA exerted a concentration-dependent effect on apoptsis of HepG2 cells. Tarao K et al. [19] considered that UDCA may prevent the development of HCC in individuals with heatitis C virus-associated liver cirrhosis.
In the current study, we demonstrated that UDCA reduced the viability of HCC cells in both a dose-dependent and a time-dependent manner. In addition, UDCA could suppress cell migration. Both of these results demonstrate that UDCA can inhibit HCC.
To test the mechanism through which UDCA inhibits HCC, we examined the relationship between UDCA and LC3B. Our research showed that UDCA could increase the levels of LC3B mRNA and protein both in vivo and in vitro.
Rosenfeldt et al. [20] demonstrated that p53 status determined the role of autophagy in the development of pancreatic tumors. They found that p53 played an important role in autophagy. Therefore, we studied the changes in p53 expression in 7721 and HepG2 cells upon treatment with UDCA. We found that the expression of p53 increased with UDCA treatment, which suggests that UDCA promotes the expression of LC3B via an increase in the expression of p53.
All of the results discussed above indicated that UDCA plays an important role in autophagy in HCC cell lines and that UDCA may be used to treat or prevent HCC.
Future studies will involve verification of the relationship of LC3B and p53. We also plan to test other mechanisms of autophagy in the UDCA-mediated inhibition of HCC. For example, Du et al. [21] demonstrated that microRNA-182 was up-regulated in HCC tissues. We could therefore study the relationship between microRNA-182 and UDCA.
Conclusion
In conclusion, our results showed that LC3B is frequently up-regulated in HCC cell lines and tumor xenografts of nude mice treated with UDCA. This may provide a direction for future studies on the molecular mechanisms of UDCA on HCC. Finally, UDCA is a potential drug candidate for HCC.
Acknowledgements
This work was supported in part by the following: the National Natural Science Foundation of China, grant numbers 81472685 and 81600469, the Science and Technology Development Projects of Shandong Province, grant number 2013GSF11852 and 2016GSF201126, a project funded by China Postdoctoral Science Foundation, grant number 2013M541926, the Postdoctoral Innovation Project Special Foundation of Shandong Province, grant number 201302031, the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province, grant number BS2014YY37, the Major Special Plan of Science and Technology of Shandong Province, grant number 2015ZDXX0802A01, the Shandong Provincial Natural Science Foundation, grant number ZR2013HL038.
Disclosure of conflict of interest
None.
References
- 1.Baselga J, Bhardwaj N, Cantley LC, DeMatteo R, DuBois RN, Foti M, Gapstur SM, Hahn WC, Helman LJ, Jensen RA, Paskett ED, Lawrence TS, Lutzker SG, Szabo E. AACR cancer progress report 2015. Clin Cancer Res. 2015;21(Suppl):S1–128. doi: 10.1158/1078-0432.CCR-15-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Association for The Study of The Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–43. doi: 10.1016/j.jhep.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol. 2001;35:134–46. doi: 10.1016/s0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 4.Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, Poelstra K, Haisma HJ, Jansen PL, Moshage H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology (Baltimore, Md) 2004;39:1563–73. doi: 10.1002/hep.20246. [DOI] [PubMed] [Google Scholar]
- 5.Lim SC, Han SI. Ursodeoxycholic acid effectively kills drug-resistant gastric cancer cells through induction of autophagic death. Oncol Rep. 2015;34:1261–8. doi: 10.3892/or.2015.4076. [DOI] [PubMed] [Google Scholar]
- 6.Chung GE, Yoon JH, Lee JH, Kim HY, Myung SJ, Yu SJ, Lee SH, Lee SM, Kim YJ, Lee HS. Ursodeoxycholic acid-induced inhibition of DLC1 protein degradation leads to suppression of hepatocellular carcinoma cell growth. Oncol Rep. 2011;25:1739–46. doi: 10.3892/or.2011.1239. [DOI] [PubMed] [Google Scholar]
- 7.Zhu L, Shan LJ, Liu YJ, Chen D, Xiao XG, Li Y. Ursodeoxycholic acid induces apoptosis of hepatocellular carcinoma cells in vitro. J Dig Dis. 2014;15:684–93. doi: 10.1111/1751-2980.12191. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 9.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science (New York, NY) 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–62. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 15.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 16.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koukourakis MI, Kalamida D, Giatromanolaki A, Zois CE, Sivridis E, Pouliliou S, Mitrakas A, Gatter KC, Harris AL. Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One. 2015;10:e0137675. doi: 10.1371/journal.pone.0137675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsagarakis NJ, Drygiannakis I, Batistakis AG, Kolios G, Kouroumalis EA. A concentration-dependent effect of ursodeoxycholate on apoptosis and caspases activities of HepG2 hepatocellular carcinoma cells. Eur J Pharmacol. 2010;640:1–7. doi: 10.1016/j.ejphar.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 19.Tarao K, Fujiyama S, Ohkawa S, Miyakawa K, Tamai S, Hirokawa S, Masaki T, Tanaka K. Ursodiol use is possibly associated with lower incidence of hepatocellular carcinoma in hepatitis C virus-associated liver cirrhosis. Cancer Epidemiol Biomarkers Prev. 2005;14:164–9. [PubMed] [Google Scholar]
- 20.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 21.Du C, Weng X, Hu W, Lv Z, Xiao H, Ding C, Gyabaah OA, Xie H, Zhou L, Wu J, Zheng S. Hypoxia-inducible MiR-182 promotes angiogenesis by targeting RASA1 in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:67. doi: 10.1186/s13046-015-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]