

Abstract
Segmental progeroid syndromes are commonly represented by genetic conditions which recapitulate aspects of physiological aging by similar, disparate, or unknown mechanisms. Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease caused by mutations in the gene for ACVR1/ALK2 encoding Activin A receptor type I/Activin-like kinase 2, a bone morphogenetic protein (BMP) type I receptor, and results in the formation of extra-skeletal ossification and a constellation of others features, many of which resemble accelerated aging. The median estimated lifespan of individuals with FOP is approximately 56 years of age. Characteristics of precocious aging in FOP include both those that are related to dysregulated BMP signaling as well as those secondary to early immobilization. Progeroid features that may primarily be associated with mutations in ACVR1 include osteoarthritis, hearing loss, alopecia, subcutaneous lipodystrophy, myelination defects, heightened inflammation, menstrual abnormalities, and perhaps nephrolithiasis. Progeroid features that may secondarily be related to immobilization from progressive heterotopic ossification include decreased vital capacity, osteoporosis, fractures, sarcopenia, and predisposition to respiratory infections. Some manifestations of precocious aging may be attributed to both primary and secondary effects of FOP. At the level of lesion formation in FOP, soft tissue injury resulting in hypoxia, cell damage, and inflammation may lead to the accumulation of senescent cells as in aged tissue. Production of Activin A, platelet-derived growth factor, metalloproteinases, interleukin 6, and other inflammatory cytokines as part of the senescence-associated secretory phenotype could conceivably mediate the initial signaling cascade that results in the intense fibroproliferative response as well as the tissue-resident stem cell reprogramming leading up to ectopic endochondral bone formation. Consideration of FOP as a segmental progeroid syndrome offers a unique perspective into potential mechanisms of normal aging and may also provide insight for identification of new targets for therapeutic interventions in FOP.
Keywords: progeroid syndrome, fibrodysplasia ossificans progressiva, activin A, ACVR1, cell senescence
Introduction
Aging may be a unique biological process, since evolutionarily there appears to be an absence of genes specifically selected to cause it (1, 2). Rather, age-related changes may be the unprogrammed results of optimization for early reproductive success. Thus, senescence at the organismal level represents a phenomenon with low mechanistic conservation among disparate metazoans and so mechanisms of human aging do not necessarily have metazoan counterparts in every situation. For example, replicative senescence or the loss of proliferative capacity in replication-competent somatic tissues is not a potential mechanism for aging in organisms where soma compartments are post-mitotic, such as C. elegans (3).
A complementary approach to studying a model system for aging in a lower organism is to directly study human aging. Although this would closely capture aspects of aging that are relevant to humans, it does not obviate consideration for the highly polygenic nature of age-related pathologies (4), the confounding effects of outbreeding, or environmental effects based on where and how individuals live. An approach to providing a scientifically tractable system, at least with respect to the former, is to study genetic diseases whose phenotypes mimic at least some (i.e., “segmental”) features of the usual human aging process (4, 5). Such segmental progeroid (i.e., premature or accelerated aging-like) syndromes are usually monogenic and may thus be simple enough to provide insights into the causes of their pathology. Studied within the context of theories for physiological aging, observations made in segmental progeroid syndromes may also explain certain aspects of normal aging. Despite being only partial phenocopies of normal aging (i.e., some tissues show aging features and other not), these segmental progeroid syndromes provide experimental tractability, with varying fidelity, that is the rationale for their use as paradigm for natural deteriorative changes that occur over time. Single-gene mutations that impact multiple aspects of the physiological aging phenotype may exert their action through developmental alterations that have consequences for post-maturational aging, and importantly, for regulation of the rates of post-maturational aging after normal development.
Here we propose that consideration of fibrodysplasia ossificans progressiva (FOP) as a segmental progeroid syndrome offers a unique perspective into potential mechanisms of normal aging and may also provide insight for identification of new targets for therapeutic interventions in FOP.
Segmental Progeroid Syndromes as a Model to Investigate Human Aging
Representative segmental progeroid syndromes are shown in Table 1. Several are monogenic or at least affect the same or similar pathways when more than one gene causes the same phenotype within the same syndrome. The putative mechanism(s) by which aging phenotypes are manifested are similar in several syndromes, including decreased genome maintenance and accelerated cellular senescence. All of the syndromes reduce mean lifespan or life expectancy.
Table 1.
| Syndrome | Inheritance | Approximate mean life-span (years) | Causative mutation | Possible mechanistic relevance to natural aging |
|---|---|---|---|---|
| Down | De novo trisomy | 60 | Many genes involved in phenotypea | Decreased genome maintenance |
| Werner | Autosomal recessive | 47 | WRN | Decreased genome maintenance; altered DNA damage responses; accelerated cell senescence |
| Dyskeratosis congenitab | X-linked; autosomal dominant | Variablec | DKC1; TERC | Accelerated cell senescence |
| Cockayne | Autosomal recessive | 20 | CS-A (ERCC8); CS-8 (ERCC6) | Decreased genome maintenance |
| Hutchinson-Gilford | Dominant negative | 12 | LMNA | Altered DNA damage responses; accelerated cell senescence |
| Ataxia telangiectasia | Autosomal recessive | 20 | ATM | Decreased genome maintenance; Accelerated neurodegeneration; Reduced immune diversity |
| Berardinelli-Seipd | Autosomal recessive | 40 | AGPAT2; BSCL2 | Altered insulin signaling; decreased membrane integrity; increased glycation damage |
| Fibrodysplasia ossifcans progressiva | Sporadic; autosomal dominant | 56e | ACVR1 (ALK2) | Injury-induced senescence; overactive activin A-BMP pathway signaling |
For examples, GATA1, JAK2, DSCR1, DYRK1A.
Information shown for the two most common forms.
Life expectancy ranges from infancy to 60s.
Congenital generalized lipodystrophy type 1 and 2.
Estimated median life expectation.
In the case of FOP, possible mechanisms for generation of an accelerated aging phenotype include injury-induced senescence and overactive activin A signaling. In comparison to other segmental progeroid syndromes, FOP represents an opportunity to study two different mechanisms by which aging phenotypes may be produced. Injury-induced senescence, especially in soft tissue such as muscle, has recently been described (8, 9) and muscle injury is a known cause of episodic inflammatory exacerbations or flare-ups in FOP (10, 11). Increased signaling through the bone morphogenetic protein (BMP) pathway, especially by activin A, has been implicated in osteoarthritis, sarcopenia, neurodegeneration, and other features associated with aging and FOP (discussed below under section Segmental Progeroid Features of FOP). Furthermore, activin A is a component of the senescence-associated secretory phenotype (SASP) (12, 13) and in FOP the mutated ACVR1/ALK2 encoding Activin A receptor type I/Activin-like kinase 2 (ACVR1/ALK2), a BMP type I receptor, is exquisitely sensitive to increased levels (14). Thus, injury-induced senescence leading to increased production of activin A may precipitate flare-ups in FOP and increased BMP signaling through activating mutations in ACVR1 may contribute to accelerated age-related changes in certain tissues. To test this hypothesis it will be necessary to examine the senescent cell burden in FOP lesion formation using markers of senescence in both patient samples and mouse models of FOP as well as analysis of the SASP in mouse models of FOP.
Fibrodysplasia Ossificans Progressiva (FOP)
FOP is a strongly debilitating genetic disorder with hallmark features of congenital first toe malformations, progressive heterotopic ossification (HO) that produces normal bone at extra-skeletal locations, and accelerated features of aging (10, 11). The worldwide prevalence is 1/1,300,000–1/2,000,000 (15, 16). There is no ethnic, racial, gender, or geographic predilection to FOP. Early in life, episodic bouts of inflammatory soft tissue protuberances (i.e., exacerbations or flare-ups) develop which are often caused by injury, intramuscular injections, viral infections, muscular overuse, or fatigue (17, 18). These exacerbations convert connective tissues, including skeletal muscle, into HO. Tendons, ligaments, fascia, and aponeuroses are also affected, and together with transformed muscle, result in joint ankyloses and immobility. Atypical forms of FOP have been reported (19). Approximately 97% of patients with FOP harbor an activating mutation (617G > A; R206H) in ACVR1/ALK2 (6). Individuals with FOP variants also have heterozygous ACVR1 missense mutations in conserved amino acids. FOP is diagnosed clinically, with confirmation by genetic testing if available. The majority of FOP cases are sporadic (i.e., non-inherited mutations), but a small number of cases demonstrate germline transmission with inheritance in an autosomal dominant fashion (6). Although progressive HO is a hallmark feature, changes in early adulthood reminiscent of premature aging are evident.
Currently, there are no curative interventions, and the mainstay of treatment is focused on symptomatic relief using brief courses of high-dose corticosteroids for flare-ups, which may help to reduce the intense pain and edema associated with the early stages of ectopic bony lesion formation (10, 20). Steps to mitigate the likelihood of falls, decline in pulmonary function, and acquisition of viral infections are important prophylactic measures. The median life expectancy is about 56 years of age (7). Most patients require partial or complete assistance for ambulation by age 30, and common proximal causes of death include thoracic insufficiency syndrome and pneumonia (7). Factors contributing to the accelerated aging phenotype of FOP may be primarily related to ACVR1/ALK2 mutation, secondarily related to immobilization and disuse due to HO-associated joint ankyloses, or a combination of the two. Endpoints of current clinical trials focus on reducing heterotopic bone formation (20), but it is unclear if those therapies targeting mutant ACVR1/ALK2 signaling will also delay, prevent, or ameliorate the progeroid features of FOP. Furthermore, it is unclear if targeting the activin-A ligand, vs. the receptor or post-receptor pathways, will be sufficient to mitigate all aspects of the condition. Also, it is unknown if or how targeting activin-A and its signaling networks will impact its role in hypothalamic-pituitary-gonadal feedback.
Segmental Progeroid Features of FOP
Progeroid features in FOP that may primarily be associated with mutations in ACVR1 include alopecia, subcutaneous lipodystrophy, hearing loss, myelination defects, osteoarthritis, heightened inflammation, menstrual abnormalities, and perhaps nephrolithiasis (Table 2).
Table 2.
Progeroid features in FOP.
| System or tissue | Aging features in FOP | Characteristic(s) in FOP |
|---|---|---|
| Skin | Alopecia; Subcutaneous lipodystrophy (21–24) |
Alopecia seen in both sexes (19, 25–27); lipodystrophy may be associated with jaw ankylosis or recurrent flare-ups |
| Central nervous | Hearing loss; Myelination defects |
Conductive and sensorineural hearing loss (19, 28–30); re-myelination deficits (31–33) |
| Respiratory | Decreased vital capacity; Pulmonary hypertension |
Restrictive pulmonary function (7, 34) |
| Bone | Osteoporosis; Fractures |
Osteoporosis (secondary) (35) |
| Muscle | Sarcopenia (36–38) | Sarcopenia of disuse is prominent |
| Joint | Osteoarthritis (19, 39, 40) | Often symmetrical |
| Immune | Inflammation; Predisposition to respiratory infections (7) |
Acute inflammatory episodes (flare-ups) (10); chronic inflammatory state (41–45) |
| Reproductive | Menstrual abnormalities | Amenorrhea (19, 46, 47) |
| Renal | Nephrolithiasis (48) | Three times more likely compared to general population (49) |
Alopecia is frequently observed in individuals with FOP and clinically is seen in both males and females. Evidence suggests that BMP signaling is involved in the control of the hair cycle (25). Increased BMP signaling through expression of BMP4, or its inhibition by the antagonist Noggin, causes progressive alopecia (26). In androgen-dependent alopecia, elevated BMP signaling in early (refractory) telogen likely mediates the retention of quiescent bulge stem cells (27). The case for elevated BMP signaling in lipodystrophy is less direct. Increased Fra-1 causes severe lipodystrophy (21) and both BMP-2 and TGF-β stimulate AP-1 activities, including the DNA binding activity of Fra-1 (22). Alternative explanations for subcutaneous lipodystrophy include decreased caloric intake after jaw ankylosis and the effects of recurrent inflammatory flare-ups. With respect to the latter, activation of the NF-κβ pathway during periods of acute or chronic inflammation may contribute to loss of subcutaneous fat. For example, activation of the NF-κβ pathway due to ubiquitination defects has been associated with lipodystrophy (23, 24).
Conductive and sensorineural hearing loss are common in FOP (28) and with prebycusis. Conductive hearing loss occurs when sound waves are not relayed efficiently to the inner ear, while sensorineural hearing loss is related to sensory organ (cochlea and associated structures) dysfunction or damage to the vestibulocochlear nerve (cranial nerve VIII). In humans, NOGGIN (NOG) gene mutations are associated with a few autosomal dominant conditions like proximal symphalangism and multiple synostoses which are characterized by skeletal defects and fusion of adjacent bones. Synostosis of one or more ossicles in the ear promotes conductive hearing loss. Proper formation of the skeleton requires balanced levels of BMPs and Noggin and the conductive hearing loss in Nog+/−mice results from an ectopic bridge of bone between the stapes and the tympanum, interfering with the normal mobility of the ossicle (29). BMP signaling is also required for inner ear development, including patterning of sensory regions in the cochlea that process sound (30). It is likely that hearing loss in FOP is due to increased BMP signaling very early in life affecting both/either the cochlear sensory regions and/or motion of ossicles. Later in life, synostosis of the ossicles due to HO may be the predominant cause of progressive hearing loss.
Demyelinated lesions and focal inflammatory changes of the CNS are seen in both mouse models of FOP and in CNS white matter lesions in FOP patients (31). BMP signaling is a potent inhibitor of oligodendroglial differentiation and remyelination (32), and gain-of-function mutations in ACVR1/ALK2 predictably enhance this potent inhibition. Dysregulated BMP signaling causes CNS demyelination, and CNS demyelination is one of the underlying mechanisms for the observed atypical neurologic phenotypes in FOP patients. With normal aging, decreased CNS remyelination becomes more prominent over time (33, 50). In addition, aging is associated with decreased hippocampal neurogenesis and concomitant hippocampus-dependent cognitive functions (51). There is an inverse relationship between CNS levels of BMP4 expression and noggin with age, with the former increasing substantially in the mouse dentate gyrus and the latter decreasing. This results in a profound elevation of phosphorylated-SMAD1/5/8, a key effector of BMP signaling. As with aging in mice, a large increase in BMP4 expression is seen in the dentate gyrus of older humans without known cognitive dysfunction (51). Increased BMP signaling is related to impairments in neurogenesis and to age-related cognitive changes (51) and aspects of these processes may be phenocopied in FOP.
Accelerated osteoarthritis is commonly found in FOP. Terminal differentiation of chondrocytes may be delayed or prevented by abrogation of BMP signaling in articular cartilage, and mitigation of this blockage or increased BMP signaling may then contribute to endochondral ossification and breakdown of cartilage matrix (39). In cartilage, TGFß and BMP are necessary for normal joint development and maintenance and their dysregulation has been associated with the pathogenesis of osteoarthritis. Interestingly, osteoarthritic patients have significantly higher serum levels of BMP-2 and BMP-4 compared to non-diseased humans and appear to characterize patients who have degenerative joint disease severe enough to require total joint replacement (40).
Heightened inflammation in FOP can be acute (as in episodic flare-ups) as well as chronic (as in an elevated pro-inflammatory state). The inflammatory nature of flare-ups in FOP is clinically obvious and well-described (41). In FOP patients without clinically evident HO, increased serum levels of cytokines, including IL3-, IL-7, IL-8, and IL-10, suggest a persistent pro-inflammatory state (42). So-called “inflammaging” refers to the chronic, sterile, low-grade inflammation which develops as part of normal aging, and is thought to contribute to the pathogenesis of multiple age-related diseases (43). In FOP, both acute and chronic inflammation may be related to the role of activin A in the initiation and persistence of the inflammatory response (44, 45).
Early menstrual abnormalities in FOP, including amenorrhea, are clinically recognized but have not been objectively studied or described. Roles for activin A in the ovulation cycle as well as in endometrial repair after menses have been reported and are perhaps causally related (46, 47).
Progeroid features in FOP that may secondarily be related to immobilization from progressive HO include decreased vital capacity, osteoporosis, fractures, sarcopenia, and predisposition to respiratory infections (Table 2). These manifestations represent an opportunity to study the contribution of disuse to the normal aging phenotype typified by the decreased physical activity, sedentary predilection, and increased likelihood of prolonged bed rest in older adults. If physiological aging is the result of primary aging processes interacting with or superimposed upon the pathophysiological consequences of inactivity (36), then specific characteristics of precocious aging in FOP due to disuse would be amenable to study in isolation. As an illustration, unloading of the normotopic skeleton due to bridging heterotopic bone results in osteoporosis. Another example is the increase in chest wall rigidity and decreases in elastic recoil and force-generating capacity of respiratory muscles that contribute to diminished vital capacity and predisposition to respiratory infection.
Some manifestations of precocious aging may be attributed to both primary and secondary effects of FOP. Sarcopenia in FOP likely represents the effects of both disuse atrophy due to joint ankyloses as well as increased activin A signaling causing both increased muscle catabolism and inhibition of myoblast differentiation (37, 38). Nephrolithiasis in FOP could be related to inadequate fluid intake due to functional difficulties in voiding, immobilization itself, and the effects of activin A on kidney function (48, 49).
Injury, Reprogramming in vivo, and Cellular Senescence
Injury, in general, is associated with accumulation of senescent cells [see (8) and Figure 1]. Growing evidence suggests that injury-induced reprogramming in skeletal muscle is facilitated by the accumulation of senescent cells at or near the site of damaged tissue (8). Bothe acute and chronic injury enables transcription-factor-mediated reprogramming in damaged muscle (8). The reprogramming effect of senescence appears to be due to the release of interleukin 6 (IL-6) and perhaps other components of the senescence-associated secretory phenotype (SASP) (8, 52). Senescence and the SASP facilitate the reprogramming of neighboring non-senescent cells but also recruit macrophages for the removal of necrotic tissue (8).
Figure 1.
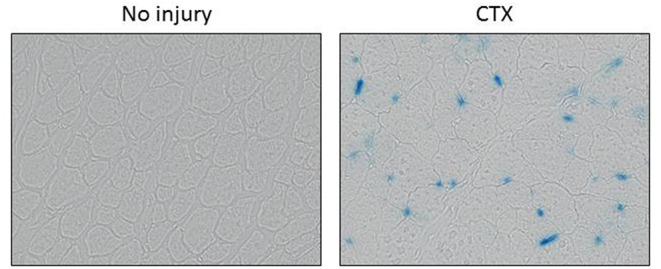
Muscle injury-induced senescence. Senescence-associated β-galactosidase (SAβ-gal) staining of the tibialis anterior muscle of a wild-type mouse is shown without injury (left) and 5 days after injury via cardiotoxin (CTX) injection. SAβ-gal-stained cells appear blue. Images are courtesy of Haitao Wang, Ph.D., Mayo Clinic, Rochester, MN, USA).
Paracrine release of IL-6 and other factors secreted by senescent cells promote reprogramming by Oct4, Sox2, Klf4, and c-Myc (OSKM) in non-senescent cells (53) into pluripotent cells (also called induced pluripotent stem cells or iPSCs). A direct relationship has been demonstrated between senescence and OSKM-driven reprogramming. In cells lacking p16INK4a/ARF (i.e., cells that do not undergo senescence), their ability to reprogram is severely compromised (9, 53). Furthermore, pharmacological inhibition of NFkB, a major driver of cytokine production and the SASP, reduces in vivo reprogramming (9). Aging, which is associated with higher levels of cellular senescence, also favors OSKM-driven reprogramming. Similarly, in physiological conditions of wound healing, senescence triggered by injury could promote cell dedifferentiation to mediate repair of damaged tissue (9, 53).
Roles of Cellular Senescence in FOP Lesion Formation
In FOP, injuries due to soft tissue trauma, viral infection, muscular stretching, and even fatigue due to overuse can precipitate a flare-up. Tissue damage causes pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in response to microbial and endogenous injury in the setting of a hypoxic microenvironment (54, 55). As the result of tissue injury, senescent cells accumulate and potentially contribute to early events that enhance BMP signaling and facilitate the reprogramming of tissue-resident stem cells (Figure 2).
Figure 2.

Potential roles for cellular senescence in FOP lesion formation. The major hypothesized contributions of senescence are through the production of activin A, IL-6, and other components of the SASP. T, inhibitory pathways;, inhibitory action of senotherapeutic drugs.
Senescence is a cellular response to damage characterized by an irreversible cell cycle arrest and then by the SASP (56, 57). The SASP produces at least two factors that can directly promote increased BMP signaling and stem cells reprogramming—activin A and IL-6, respectively (Figure 2). It is well-established that activin A stimulates BMP signaling in FOP cells, owing to the causative mutations in the ACVR1 gene. In addition to the permissive effects of IL-6 in reprogramming, FOP cells show an increased efficiency of iPSC generation (58). In normal cells, the generation of iPSCs is facilitated by transduction of mutant ACVR1 or SMAD1 or by the early addition of BMP4 during the reprogramming. ID genes, downstream targets of BMP-SMAD signaling, are important for iPSC generation and their signaling through this pathway can inhibit cell senescence due to p16/INK4A, which otherwise serves to prevent reprogramming (58). Thus, ID1 and other ID genes may serve to both enhance expansion of the FOP early lesion as well as stimulate production of osteochondral progenitor cells. Enhanced BMP signaling promotes a tremendous fibroproliferative response, perhaps further accelerated by secretion of platelet-derived growth factor (PDGF) and matrix metalloproteinases (MMPs) by the SASP (Figure 2). Osteochondral progenitor cells derived from reprogrammed stem cells ultimately contribute to the endochondral bone formation which is the hallmark of mature FOP lesions. Other events that may contribute to formation of heterotopic bone cannot be excluded (59). However, in mouse models it will be possible to demonstrate if senescence-mediated tissue reprogramming in FOP lesions shifts lineage determination from a myogenic to a chondrogenic fate after injury.
Senescent cells may play multiple roles in the formation of HO in FOP and drugs which target senescent cells and/or the SASP may be candidates for therapeutic interventions. Compounds which selectively clear senescent cells (so-called senolytics) were first described on the basis of targeting pro-survival networks in senescent cells (60). Compounds that reduce the SASP (i.e., senomodulators), including inhibitors of the JAK/STAT pathway that plays an important role in regulating cytokine production, reduce systemic and adipose tissue inflammation in old mice (61). Rapamycin, a senomodulator, may be of particular benefit in FOP, since it also reduces activin-A mediated mTOR signaling (62). Many senotherapeutic agents have been reported, are effective in delaying or alleviating multiple age-related conditions in pre-clinical models, and are now being evaluated in clinical trials (63, 64). Their potential use in FOP offers a novel therapeutic approach to injury-induced flare-ups in FOP which should be further explored. We propose that senescent cell clearance and/or reduction in the SASP will ameliorate HO formation in mouse models of FOP and can be translated for use in patients with FOP.
Conclusions
Monogenic segmental progeroid syndromes are important models for studying aspects of physiological aging. Features of precocious aging in FOP include both those that are related to dysregulated BMP signaling as well as those secondary to early immobilization and disuse. At the level of lesion formation in FOP, soft tissue injury resulting in hypoxia, cell damage, and inflammation may result in the accumulation of senescent cells as in aged tissue. Production of Activin A, interleukin 6, and other inflammatory cytokines as part of the SASP could mediate the initial signaling cascade that results in intense fibrosis as well as tissue-resident stem cell reprogramming prior to ectopic endochondral bone formation. This proposal requires experimental validation, but is amendable to testing in animal models. Consideration of FOP as a segmental progeroid syndrome may offer a unique perspective into potential mechanisms of normal aging, may increase understanding of BMP signaling as related to bone homeostasis and repair, and may also provide insight for identification of new targets for therapeutic interventions in FOP such as use of senotherapeutic drugs now in phase 1 and phase 2 clinical trials for aging-related conditions.
Author Contributions
RP conceived the work, with substantial contributions from HW and FK. RP drafted the initial manuscript with HW and FK revising it critically. RP, HW, and FK gave final approval to the work and agreed to be accountable for all aspects.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was enabled by support by the Radiant Hope Foundation (to RP, HW), the Robert and Arlene Kogod Professorship in Geriatric Medicine (to RP), and the Isaac and Rose Nassau Professorship of Orthopedic Molecular Medicine (to FK).
References
- 1.Kirkwood TB. Human senescence. Bioessays. (1996) 18:1009–16. 10.1002/bies.950181211 [DOI] [PubMed] [Google Scholar]
- 2.Partridge L, Gems D. The evolution of longevity. Curr Biol. (2002) 12:R544–6. 10.1016/S0960-9822(02)01048-5 [DOI] [PubMed] [Google Scholar]
- 3.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. (2002) 419:808–14. 10.1038/nature01135 [DOI] [PubMed] [Google Scholar]
- 4.Martin GM. Genetics and aging; the Werner syndrome as a segmental progeroid syndrome. Adv Exp Med Biol. (1985) 190:161–70. 10.1007/978-1-4684-7853-2_5 [DOI] [PubMed] [Google Scholar]
- 5.Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. (1978) 14:5–39. [PubMed] [Google Scholar]
- 6.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. (2006) 38:525–7. 10.1038/ng1783 [DOI] [PubMed] [Google Scholar]
- 7.Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. (2010) 92:686–91. 10.2106/JBJS.I.00705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, Cazin C, et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. (2017) 20:407–14.e4. 10.1016/j.stem.2016.11.020 [DOI] [PubMed] [Google Scholar]
- 9.Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. (2016) 354:aaf4445. 10.1126/science.aaf4445 [DOI] [PubMed] [Google Scholar]
- 10.Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis. (2011) 6:80. 10.1186/1750-1172-6-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatr Endocrinol Rev. (2013) 10(Suppl 2):437–48. [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. (2017) 21:21–8. 10.1016/j.ebiom.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. (2015) 4:e12997. 10.7554/eLife.12997.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Shore EM, Pignolo RJ, Kaplan FS. Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone. (2018) 109:218–24. 10.1016/j.bone.2017.11.014 [DOI] [PubMed] [Google Scholar]
- 15.Baujat G, Choquet R, Bouee S, Jeanbat V, Courouve L, Ruel A, et al. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: an estimate based on a record linkage of two national databases. Orphanet J Rare Dis. (2017) 12:123. 10.1186/s13023-017-0674-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connor JM, Evans DA. Genetic aspects of fibrodysplasia ossificans progressiva. J Med Genet. (1982) 19:35–9. 10.1136/jmg.19.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, et al. The natural history of flare-ups in fibrodysplasia ossificans progressiva (FOP): a comprehensive global assessment. J Bone Miner Res. (2016) 31:650–6. 10.1002/jbmr.2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pignolo RJ, Kaplan FS. Clinical staging of Fibrodysplasia Ossificans Progressiva (FOP). Bone. (2018) 109:111–4. 10.1016/j.bone.2017.09.014 [DOI] [PubMed] [Google Scholar]
- 19.Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. (2009) 30:379–90. 10.1002/humu.20868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan FS, Pignolo RJ, Al Mukaddam MM, Shore EM. Hard targets for a second skeleton: therapeutic horizons for fibrodysplasia ossificans progressiva (FOP). Expert Opin Orphan Drugs. (2017) 5:291–4. 10.1080/21678707.2017.1304211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luther J, Driessler F, Megges M, Hess A, Herbort B, Mandic V, et al. Elevated Fra-1 expression causes severe lipodystrophy. J Cell Sci. (2011) 124(Pt 9):1465–76. 10.1242/jcs.079855 [DOI] [PubMed] [Google Scholar]
- 22.Lai CF, Cheng SL. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J Biol Chem. (2002) 277:15514–22. 10.1074/jbc.M200794200 [DOI] [PubMed] [Google Scholar]
- 23.Aksentijevich I, Zhou Q. NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. 10.3389/fimmu.2017.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rishikaysh P, Dev K, Diaz D, Qureshi WM, Filip S, Mokry J. Signaling involved in hair follicle morphogenesis and development. Int J Mol Sci. (2014) 15:1647–70. 10.3390/ijms15011647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botchkarev VA, Botchkareva NV, Roth W, Nakamura M, Chen LH, Herzog W, et al. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nat Cell Biol. (1999) 1:158–64. 10.1038/11078 [DOI] [PubMed] [Google Scholar]
- 27.Nesterova A, Yuryev A. Androgenic alopecia: cross-talk between cell signal transduction pathways. In: Hair and Scalp Disorders. Intech (2017), p. 141–74. 10.5772/67845 [DOI] [Google Scholar]
- 28.Levy CE, Lash AT, Janoff HB, Kaplan FS. Conductive hearing loss in individuals with fibrodysplasia ossificans progressiva. Am J Audiol. (1999) 8:29–33. 10.1044/1059-0889(1999/011) [DOI] [PubMed] [Google Scholar]
- 29.Hwang CH, Wu DK. Noggin heterozygous mice: an animal model for congenital conductive hearing loss in humans. Hum Mol Genet. (2008) 17:844–53. 10.1093/hmg/ddm356 [DOI] [PubMed] [Google Scholar]
- 30.Ohyama T, Basch ML, Mishina Y, Lyons KM, Segil N, Groves AK. BMP signaling is necessary for patterning the sensory and nonsensory regions of the developing mammalian cochlea. J Neurosci. (2010) 30:15044–51. 10.1523/JNEUROSCI.3547-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kan L, Kitterman JA, Procissi D, Chakkalakal S, Peng CY, McGuire TL, et al. CNS demyelination in fibrodysplasia ossificans progressiva. J Neurol. (2012) 259:2644–55. 10.1007/s00415-012-6563-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabo JK, Aumann TD, Merlo D, Kilpatrick TJ, Cate HS. Remyelination is altered by bone morphogenic protein signaling in demyelinated lesions. J Neurosci. (2011) 31:4504–10. 10.1523/JNEUROSCI.5859-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shields SA, Gilson JM, Blakemore WF, Franklin RJ. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia. (1999) 28:77–83. [DOI] [PubMed] [Google Scholar]
- 34.Kussmaul WG, Esmail AN, Sagar Y, Ross J, Gregory S, Kaplan FS. Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. (1998) 104–9. 10.1097/00003086-199801000-00015 [DOI] [PubMed] [Google Scholar]
- 35.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1994:238–47. 10.1097/00003086-199407000-00037 [DOI] [PubMed] [Google Scholar]
- 36.Harridge SD, Lazarus NR. Physical activity, aging, and physiological function. Physiology. (2017) 32:152–61. 10.1152/physiol.00029.2016 [DOI] [PubMed] [Google Scholar]
- 37.Ding H, Zhang G, Sin KW, Liu Z, Lin RK, Li M, et al. Activin A induces skeletal muscle catabolism via p38beta mitogen-activated protein kinase. J Cachexia Sarcopenia Muscle. (2017) 8:202–12. 10.1002/jcsm.12145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trendelenburg AU, Meyer A, Jacobi C, Feige JN, Glass DJ. TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet Muscle. (2012) 2:3. 10.1186/2044-5040-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauge C, Girard N, Lhuissier E, Bazille C, Boumediene K. Regulation and role of TGFβ signaling pathway in aging and osteoarthritis joints. Aging Dis. (2014) 5:394–405. 10.14336/AD.2014.0500394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albilia JB, Tenenbaum HC, Clokie CM, Walt DR, Baker GI, Psutka DJ, et al. Serum levels of BMP-2, 4, 7 and AHSG in patients with degenerative joint disease requiring total arthroplasty of the hip and temporomandibular joints. J Orthop Res. (2013) 31:44–52. 10.1002/jor.22182 [DOI] [PubMed] [Google Scholar]
- 41.Convente MR, Wang H, Pignolo RJ, Kaplan FS, Shore EM. The immunological contribution to heterotopic ossification disorders. Curr Osteoporos Rep. (2015) 13:116–24. 10.1007/s11914-015-0258-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barruet E, Morales BM, Cain CJ, Ton AN, Wentworth KL, Chan TV, et al. NF-κB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight. (2018) 3:122958. 10.1172/jci.insight.122958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. (2018) 14:576–90. 10.1038/s41574-018-0059-4 [DOI] [PubMed] [Google Scholar]
- 44.Hedger MP, Winnall WR, Phillips DJ, de Kretser DM. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam Horm. (2011) 85:255–97. 10.1016/B978-0-12-385961-7.00013-5 [DOI] [PubMed] [Google Scholar]
- 45.Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci USA. (2007) 104:16239–44. 10.1073/pnas.0705971104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaitu'u-Lino TJ, Phillips DJ, Morison NB, Salamonsen LA. A new role for activin in endometrial repair after menses. Endocrinology. (2009) 150:1904–11. 10.1210/en.2008-0738 [DOI] [PubMed] [Google Scholar]
- 47.Lockwood GM, Muttukrishna S, Ledger WL. Inhibins and activins in human ovulation, conception and pregnancy. Hum Reprod Update. (1998) 4:284–95. 10.1093/humupd/4.3.284 [DOI] [PubMed] [Google Scholar]
- 48.Maeshima A, Miya M, Mishima K, Yamashita S, Kojima I, Nojima Y. Activin A: autocrine regulator of kidney development and repair. Endocr J. (2008) 55:1–9. 10.1507/endocrj.KR-113 [DOI] [PubMed] [Google Scholar]
- 49.Gupta RR, Delai PLR, Glaser DL, Rocke DM, Al Mukaddam M, Pignolo RJ, et al. Prevalence and risk factors for kidney stones in fibrodysplasia ossificans progressiva. Bone. (2018) 109:120–3. 10.1016/j.bone.2017.12.010 [DOI] [PubMed] [Google Scholar]
- 50.Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. (2002) 22:2451–9. 10.1523/JNEUROSCI.22-07-02451.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyers EA, Gobeske KT, Bond AM, Jarrett JC, Peng CY, Kessler JA. Increased bone morphogenetic protein signaling contributes to age-related declines in neurogenesis and cognition. Neurobiol Aging. (2016) 38:164–75. 10.1016/j.neurobiolaging.2015.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brady JJ, Li M, Suthram S, Jiang H, Wong WH, Blau HM. Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nat Cell Biol. (2013) 15:1244–52. 10.1038/ncb2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mosteiro L, Pantoja C, de Martino A, Serrano M. Senescence promotes in vivo reprogramming through p16(INK)(4a) and IL-6. Aging Cell. (2018) 17:e12711 10.1111/acel.12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Behrens EM, Pignolo RJ, Kaplan FS. ECSIT links TLR and BMP signaling in FOP connective tissue progenitor cells. Bone. (2018) 109:201–9. 10.1016/j.bone.2017.12.024 [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, et al. Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J Bone Miner Res. (2016) 31:1652–65. 10.1002/jbmr.2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. (2010) 16:238–46. 10.1016/j.molmed.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 58.Hayashi Y, Hsiao EC, Sami S, Lancero M, Schlieve CR, Nguyen T, et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc Natl Acad Sci USA. (2016) 113:13057–62. 10.1073/pnas.1603668113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez DM, Ramirez MR, Reginato AM, Medici D. Molecular and cellular mechanisms of heterotopic ossification. Histol Histopathol. (2014) 29:1281–5. 10.14670/HH-29.1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14:644–58. 10.1111/acel.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. (2015) 112:E6301–10. 10.1073/pnas.1515386112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest. (2017) 127:3339–52. 10.1172/JCI93521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc. (2017) 65:2297–301. 10.1111/jgs.14969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tchkonia T, Kirkland JL. Aging, cell senescence, and chronic disease: emerging therapeutic strategies. JAMA. (2018) 320:1319–20. 10.1001/jama.2018.12440 [DOI] [PubMed] [Google Scholar]