

Abstract
Whilst adequate for most existing pathological tests, formalin is generally considered a poor DNA preservative and use of alternative fixatives may prove advantageous for molecular testing of tumour material; an increasingly common approach to identify targetable driver mutations in lung cancer patients. We collected paired PAXgene® tissue‐fixed and formalin‐fixed samples of block‐sized tumour and lung parenchyma, Temno‐needle core tumour biopsies and fine needle tumour aspirates (FNAs) from non‐small cell lung cancer resection specimens. Traditionally processed formalin fixed paraffin wax embedded (FFPE) samples were compared to paired PAXgene® tissue fixed paraffin‐embedded (PFPE) samples. We evaluated suitability for common laboratory tests (H&E staining and immunohistochemistry) and performance for downstream molecular investigations relevant to lung cancer, including RT‐PCR and next generation DNA sequencing (NGS). Adequate and comparable H&E staining was seen in all sample types and nuclear staining was preferable in PAXgene® fixed Temno tumour biopsies and tumour FNA samples. Immunohistochemical staining was broadly comparable. PFPE samples enabled greater yields of less‐fragmented DNA than FFPE comparators. PFPE samples were also superior for PCR and NGS performance, both in terms of quality control metrics and for variant calling. Critically we identified a greater number of genetic variants in the epidermal growth factor receptor gene when using PFPE samples and the Ingenuity® Variant Analysis pipeline. In summary, PFPE samples are adequate for histopathological diagnosis and suitable for the majority of existing laboratory tests. PAXgene® fixation is superior for DNA and RNA integrity, particularly in low‐yield samples and facilitates improved NGS performance, including the detection of actionable lung cancer mutations for precision medicine in lung cancer samples.
Keywords: Histopathology, Lung Cancer, Immunohistochemistry, DNA sequencing
Introduction
Lung cancer is a leading cause of cancer mortality in the UK 1. Overall median survival is approximately 17 months, with fewer than 25% of cases suitable for surgical resection 2, 3. Moreover, most patients present with metastatic disease in which median survival falls to 6–9 months 4, 5. Multiple treatment strategies are employed, including ‘personalised’ or ‘stratified’ approaches, particularly for patients with non‐small cell lung cancer (NSCLC), whereby identification of driver genetic mutations to guide efficacious treatment is beneficial (for recent reviews see 6, 7, 8). To date, efforts have largely focussed upon targeting consequences of mutations in the epidermal growth factor receptor (EGFR) gene 9, 10, 11, 12, 13, 14, as well as ALK and ROS1 translocations 15, 16, 17, although other genes, including MET gene amplification, may be of future benefit 18.
Technological innovations, including next generation DNA sequencing (NGS), to target‐sequence panels of causal cancer‐genes, now represent a practical modality for developed healthcare systems, including the UK National Health Service (NHS) 19, 20; however, current fixation protocols are often a bottle‐neck for molecular analysis and over/under‐fixation of samples is highly detrimental 21, 22, 23, 24. As many samples from lung cancer patients are physically small, low‐ and/or poor quality‐DNA yields can be hindered by fixation artefacts 25. Furthermore, health risks are associated with occupational exposure to formaldehyde 26, 27, 28.
These data are the findings of an Innovate UK‐funded study delivered by a partnership of NHS laboratories, academic institutions together with an industrial partner working to align advances in pre‐analytical processing with established workflows for handling pathological samples. Here we compared paired PAXgene® and formalin‐fixed samples from resection specimens and evaluated technical performance for histology, immunohistochemistry, DNA/RNA preservation and performance in molecular testing pertinent to the study of lung cancer.
Methods
Histopathological tumour sampling, fixation and processing schedules
This study was approved by the research ethics committee via the Royal Papworth Hospital Research Tissue Bank (08/H0304/56+5 and 18/EE/0269). Unfixed lung resection specimens from informed and consenting patients were sampled and paired block‐sized pieces of; (1) lung tumour and (2) background lung parenchyma were placed into buffered neutral formalin (Genta Medical, NewYork, UK) or PAXgene®‐tissue fixative (QIAGEN®, Manchester, UK). Similarly, paired PAXgene® and formalin fixed 16G Temno needle core biopsies (Careusion, BD, Wokingham, UK) of tumour material were collected from neighbouring tumour regions and fixed for 24–72 h.
Endobronchial or endoscopic ultrasound guided FNA samples are now accepted to be the preferred technique for the diagnosis and staging of advanced stage lung tumours 29, 30, 31, 32. To technically replicate these samples we aspirated tumour material from unfixed specimens using a wide‐bore needle and syringe, placed into either ThinPrep Cytolyt fixative (Hologic Inc., Manchester, UK) or PAXgene‐FNA® fixative (QIAGEN®, Manchester, UK) for >1 h. The plasma/thrombin clot preparation method is detailed in supplementary material, Supplementary materials and methods. Following fixation, all PAXgene® samples were transferred to PAXgene® stabiliser solution (QIAGEN®, Manchester, UK) and stored at −20 °C for batched formalin‐free processing. For complete processing schedules see supplementary material, Tables S1 and S2.
H&E staining, histomorphological assessment and scoring
All samples were embedded in paraffin wax (60 °C) and blocks stored at −20 °C. Immediately prior to sectioning, blocks were removed from the freezer and 4 μm sections cut, stained with H&E and coverslipped (Multistainer, Leica, Milton Keynes, UK). Using a published scoring system, nuclear, cytoplasmic and cell membrane features were each assigned a score of 0–4 by two blinded observers (see 33 for details).
Antigen retrieval and immunohistochemistry
Antigen retrieval is widely performed to counteract the effects of cross‐linking caused by formalin fixation. Despite PAXgene® being a non‐cross‐linking fixative, we opted to carry out antigen retrieval for both PFPE and FFPE material. Our reasoning for this was two‐fold; (1) to ensure a fair comparison between FFPE and PFPE samples and (2) laboratories opting to use PAXgene® technology might wish to harmonise FFPE and PFPE IHC protocols where possible. Antigen retrieval (20 min/96 °C) was performed with high pH antigen retrieval solution (PT module, DakoCytomation, Ely, UK) following the manufacturer's protocol. Immunohistochemistry was performed using batches of freshly prepared monoclonal anti‐human ‐Ki67, ‐MNF116, ‐p63, ‐cytokeratin 7 (CK‐7), ‐CK5/6, and ‐thyroid transcription factor‐1 (TTF‐1) antibodies. Negative controls consisted of slides undergoing pre‐treatment and incubation with antibody diluent followed by detection system reagents. Immunostaining was performed using the EnVision FLEX, high‐pH detection system and Autostainer Link48 Immunostainer, visualised using 3,3′‐diaminobenzidine tetrahydrochloride and counterstained using haematoxylin (all DakoCytomation, Ely, UK). Two independent observers scored the immunostaining, marking the sections out of 5 for intensity/specificity of staining; where 1 or 2 = unacceptable, 3 = borderline acceptable and 4 or 5 = acceptable. For the assessment of histomorphology, paired PFPE and FFPE pieces of block‐sized tumour samples, Temno biopsies and tumour FNA preparations were scored from a total of n = 23 patients and immunohistochemistry performed on all samples from n = 10 patients.
Molecular testing of samples
Paired PFPE and FFPE samples from n = 8 subjects were referred to QIAGEN Ltd., Manchester, UK; QIAGEN GmbH, Hilden, Germany, and paired PFPE and FFPE samples from n = 23 patients (including the n = 8 also sent to QIAGEN) sent to Queens University Belfast (QUB). The samples comprised n = 10, 5 μm tissue sections on slides (for DNA) and n = 5, 10 μm tissue sections/curls (for RNA) in sterile 1.5 ml Eppendorf tubes (Eppendorf, Stevenage, UK) and shipped using standard UK Royal Mail 48 h delivery.
QIAGEN analysis of DNA and RNA quality and integrity
Nucleic acids from all paired samples were extracted from FFPE samples using the QIAamp® DNA FFPE Kit and RNeasy® FFPE Kit (QIAGEN®, Manchester, UK) and from PFPE samples using the PAXgene® Tissue DNA or RNA kits (PreAnalytiX® GmbH, Hombrechtikon, Switzerland) following the manufacturers' protocols. DNA yield and purity (A 260 nm/A 280 nm) was measured using the NanoDrop 2000 Spectrophotometer and by QUBIT dsDNA Broad Range Fluorimetry (ThermoFisher Scientific, Hemel Hempstead, UK). DNA fragmentation was evaluated by Tapestation 2200 (Agilent, Stockport, UK).
QIAGEN β‐actin RT‐PCR and EGFR therascreen® assays
Five microliter from the extracted RNA eluate was used in a 294 bp β‐actin real‐time RT‐PCR assay with reactions performed in duplicates. For primer sequences, see supplementary material, Supplementary materials and methods. DNA mutation analysis was performed using the therascreen® EGFR RGQ PCR Kit (QIAGEN, Manchester, UK) for detection of common exon 19–21 EGFR mutations using RT‐PCR, according to the manufacturer's instructions.
QIAGEN Gene Reader™ NGS workflow including unique molecular indexing
DNA extracts from paired PFPE and FFPE samples were sequenced with the QIAGEN GeneReader™ NGS workflow. The GeneReader™ QIAact Lung DNA Panel (QIAGEN, Manchester, UK) was used for target enrichment PCR and library preparation from 40 ng of DNA following the manufacturer's protocol. The GeneReader™ workflow was used to compare quality control metrics for NGS performance. Unique molecular indexing (UMI) technology allows ligation of each original DNA molecule with a QIAact adapter containing a 12‐random base UMI prior to PCR amplification. Although developed to identify consequences of PCR or sequencing errors in NGS and to distinguish low frequency genetic variants from artefactual data it can also serve as a proxy measurement to compare how DNA preservation can affect sequencing coverage and fidelity between paired PFPE and FFPE samples.
QUB analysis of DNA and RNA quality and integrity
Nucleic acids were extracted from FFPE specimens using the Maxwell 16 DNA/RNA Purification kit (Promega, Southampton, UK) and from PFPE specimens using the PAXgene® Tissue DNA/RNA kit (QIAGEN, Manchester, UK). Nucleic acid yield/purity was measured using the Qubit 2.0 Fluorometer and the Nanodrop 2000 Spectrophotometer (ThermoFisher, Hemel Hempstead, UK). Fragmentation of extracted DNA was measured using the Fragment Analyser Automated system for High Sensitivity Genomic DNA (DNF‐488 high sensitivity Genomic DNA kit) from Advanced Analytical (Ankeny, USA). 40–250 ng of extracted DNA was prepared for enrichment and library construction for the QIAseq Targeted DNA Panel assay protocol (QIAGEN, Manchester, UK). Libraries were prepared using the KAPA Biosystems quantification kit (Sigma‐Aldrich/Merck KGaA, Darmstadt, Germany) and NGS performed on Illumina MiSeq or Illumina NextSeq 500 sequencing platforms (Illumina, San Diego, CA, USA). This panel allows for sequencing of 72 genes commonly mutated in human lung cancer samples (see supplementary material, Table S3).
Data summary and bioinformatics
Paired Student's t‐tests were used for comparisons between two groups. Multiple comparisons were assessed by one‐way ANOVA, followed by the appropriate post‐hoc test for significance. Data were analysed using the QIAGEN Targeted Sequencing Data Analysis Portal/GeneGlobe® Data Analysis Centre. Variants were called and annotated using the QIAGEN Ingenuity Variant Analysis software, and filtered for the following calling criteria: call quality of at least 30, read depth of at least 50 and a mutant allele fraction of at least 10%. FASTQ files were downloaded from BaseSpace and uploaded to the QIAGEN Data Analysis Centre and variants were called using QIAGEN Ingenuity® Variant Analysis software. The QIAGEN Clinical Insights Analyze (QCI®‐A) tool was applied for analysis of NGS results including QC parameters and variant calls. The DNA sequencing and bioinformatic pipeline is summarised in supplementary material, Figure S1.
Results
H&E and immunohistochemical staining of PFPE and FFPE samples
H&E staining of PFPE and FFPE samples was of high quality and blind scores generally comparable (Figure 1A–F). Interestingly, blinded observers preferred nuclear H&E staining of PFPE biopsies and tumour FNA preparations. Representative histomicrographs of PFPE and FFPE adenocarcinoma or squamous cell carcinoma sections are shown in Figure 1G–J. Although no efforts were made to optimise immunohistochemical protocols for PFPE samples, and antigen retrieval was performed on all samples, immunohistochemistry was successful and mean scores were statistically similar. Representative immunostaining is demonstrated in Figure 2.
Figure 1.
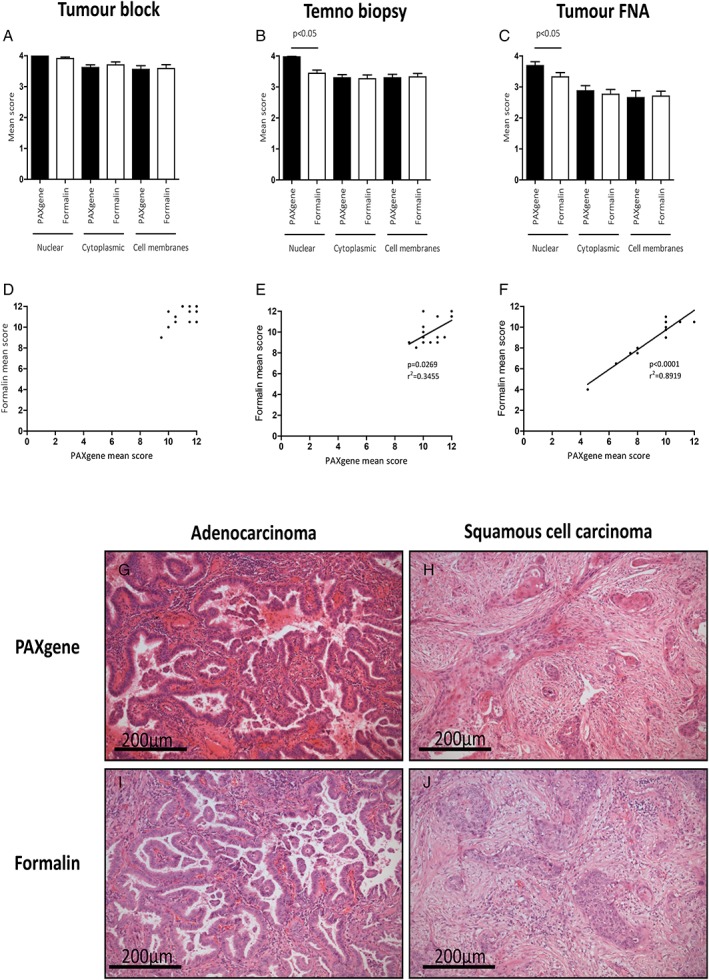
H&E staining and histomorphology. Histomorphology of PFPE and FFPE H&E stained sections of tumour block (A), Temno biopsy (B) and tumour FNA clot preparations (C). Nuclear staining of Temno biopsies and tumour FNA specimens was preferable in PFPE samples (both p < 0.05). Scores for nuclear, cytoplasmic and cell membranous staining were combined for tumour blocks (D), Temno biopsies (E) and for tumour FNA samples (F) and correlations between FFPE and PFPE scores were observed for Temno biopsies (r 2 = 0.3455, p = 0.0269) and tumour FNA samples (r 2 = 0.8919, p < 0.0001). Representative H&E staining of PFPE adenocarcinoma (G) and PFPE squamous cell carcinoma (H) and FFPE adenocarcinoma (I) and FFPE squamous cell carcinoma (J).
Figure 2.
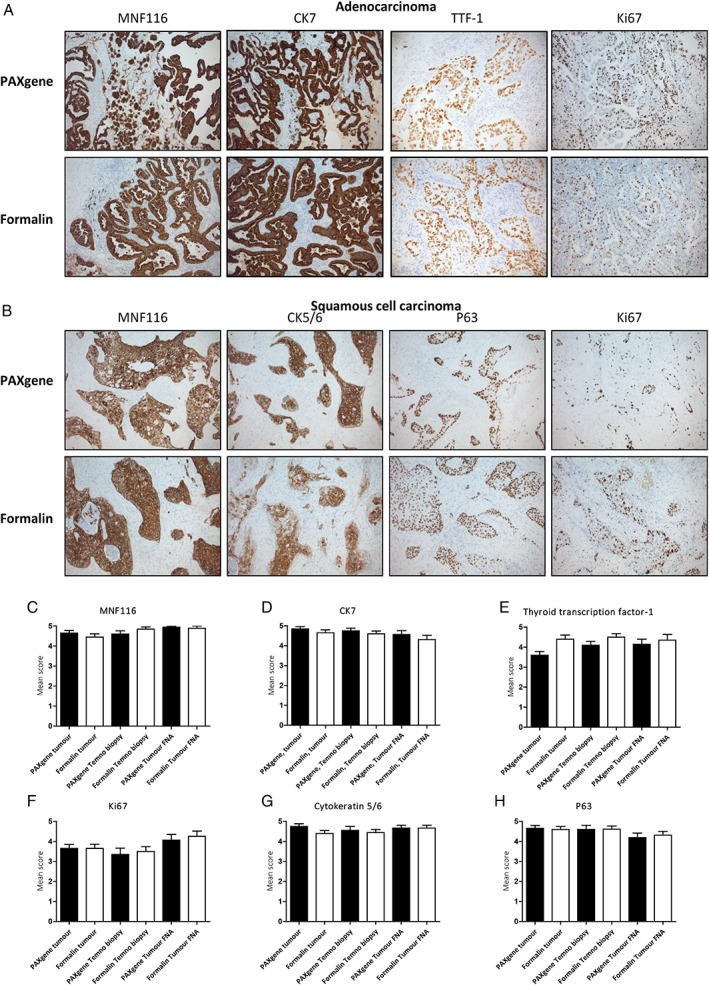
IHC staining. Representative images of PFPE and FFPE adenocarcinoma immunostained with MNF116, CK7, TTF‐1 and Ki67 (A) and squamous cell carcinoma immunostained with MNF116, CK5/6, p63 and Ki67 (B). IHC scores of MNF116 (C), CK7 (D), TTF‐1 (E), Ki67 (F), CK5/6 (G) and p63 (H) were comparable for PFPE and FFPE samples (n = 10).
QIAGEN analysis of DNA quality and integrity
Mean DNA yields from all PFPE sample types were greater than FFPE comparators, with the larger block‐sized samples yielding more DNA for both parenchyma (Figure 3A, 4714 ± 839.3 ng versus 583.4 ± 125.9 ng, p = 0.0011) and for block‐sized pieces of lung tumour (Figure 3B, 9476 ± 1371 ng versus 1955 ± 304.6 ng, p = 0.008). Similarly, DNA yields were greater from PFPE Temno tumour biopsies compared to FFPE Temno biopsies (Figure 3C, 770.1 ± 181.7 ng versus 42.5 ± 7.44 ng, p = 0.0053). Mean cycle threshold C T values for β‐actin RT‐PCR amplification were lower in PFPE lung parenchyma (31.48 ± 0.8484 versus 38.86 ± 0.6143, p = 0.002), block‐sized tumour samples (26.69 ± 0.2638 versus 38.01 ± 0.8508, p < 0.0001) and Temno tumour biopsies (31.61 ± 0.8326 versus 39.24 ± 0.6544, p = 0.0002) (Figure 3D).
Figure 3.
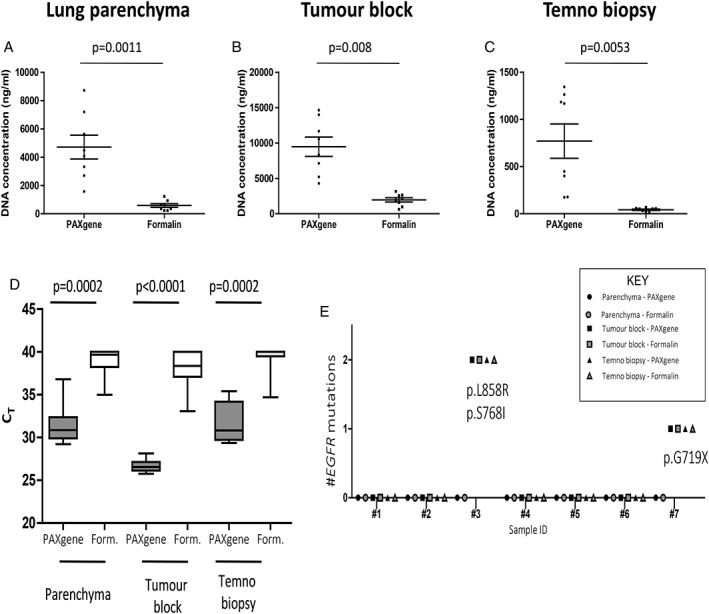
DNA analysis. DNA concentrations were greater in (A) PFPE lung parenchyma (4714 ± 839.3 ng versus 583.4 ± 125.9 ng, p = 0.0011), (B) PFPE tumour blocks, (9476 ng ±1371 ng versus 1955 ng ± 304.6 ng, p = 0.008) and (C) PFPE Temno biopsies (770.1 ± 181.7 ng versus 42.5 ± 7.44, p = 0.0053), than FFPE comparators. RNA RT PCR mean C T values (n = 8) for β‐actin were lower (D) in PFPE lung parenchyma (31.48 ± 0.8484 versus 38.86 ± 0.6143, p = 0.002), block‐sized tumour samples (26.69 ± 0.2638 versus 38.01 ± 0.8508, p < 0.0001) and Temno biopsies (31.61 ± 0.8326 versus 39.24 ± 0.6544, p = 0.0002) compared to respective FFPE samples. EGFR mutation profiles were explored using the EGFR Therascreen® PCR assay (E). Paired samples from a single subject (Patient #8) were a technical failure and not included. Of the remaining n = 7 subjects, we identified a patient with a p. G719X EGFR mutation present in both PFPE and FFPE tumour blocks and Temno biopsies (and absent in both parenchyma blocks) and another subject harbouring two separate EGFR mutations (p.L858R and p.S768I) present in the block‐sized tumour samples and Temno biopsies but absent in both lung parenchyma indicating similar performance for PFPE and FFPE.
therascreen® EGFR PCR assay
Samples from a single subject (Patient #8) were a technical failure and not included. Of the remaining n = 7 subjects, we identified a patient with a p.G719X EGFR mutation present in both PFPE and FFPE tumour blocks and Temno tumour biopsies (and absent in the parenchyma block) and another subject harbouring two separate EGFR mutations (p.L858R and p.S768I) present in the block‐sized tumour samples and Temno biopsies and absent in the background lung parenchyma (Figure 3E).
QIAGEN GeneReader™ NGS workflow and UMI
We examined the suitability of paired samples for NGS by comparing common quality metrics (q scores, depth of coverage and read mapping efficiency) and used UMI technology to compare NGS performance of paired samples. Mean quality scores for PFPE and FFPE samples were generally good with over 90% of all reads having a q score > 25 (Figure 4A) and all samples having a mean % of reads within target regions >80 (Figure 4B). Percentage coverage >100x (Figure 4C) and >60x (Figure 4D) was superior in PFPE samples, reaching significance for PFPE Temno biopsies compared to FFPE counterparts (p = 0.0102 at >100x and p = 0.002 at >60x respectively). We compared the frequency of different UMI reads per million mapped UMI reads and found greater UMI read numbers in PFPE than FFPE comparators, reaching significance in PFPE tumour blocks and Temno biopsies (Figure 4E). In addition, the mean UMI read quality scores were higher in PFPE Temno biopsies compared to FFPE (Figure 4F).
Figure 4.
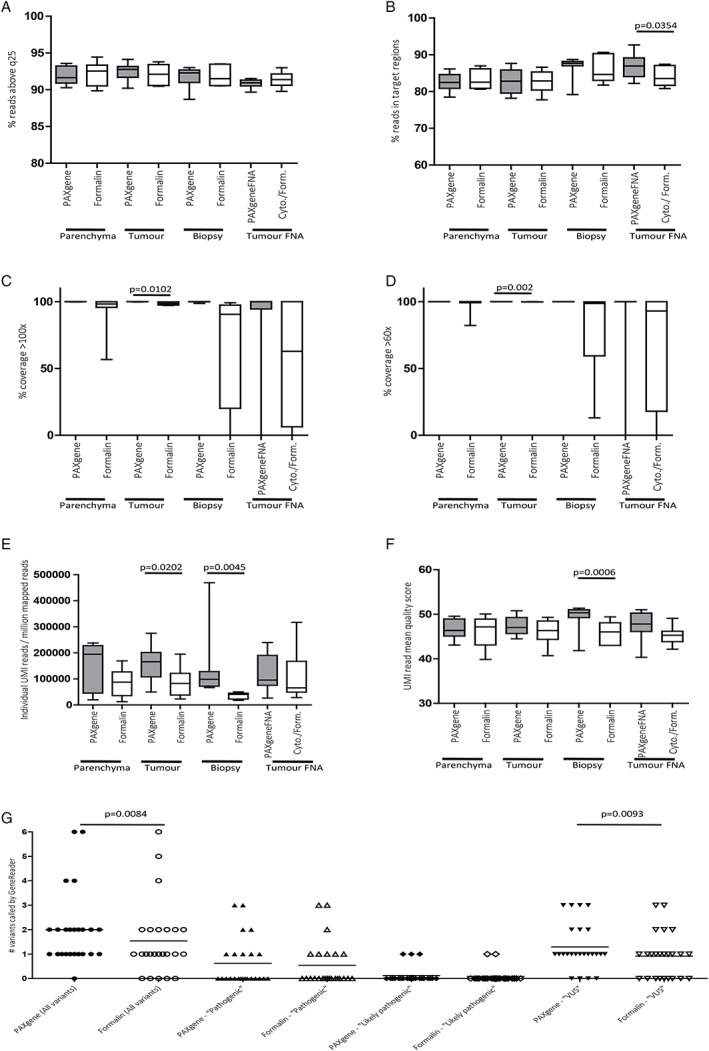
GeneReader™ NGS. Comparison of quality metrics for NGS indicated that mean quality scores for PFPE and FFPE samples were both good with over 90% of all samples having a q score > 20 (A) and all samples having a mean % of reads within target regions >80 and a significantly greater percentage of within‐target reads present in PFPE versus FFPE tumour FNA preparations (p = 0.0354) (B). Percentage coverage >100x (C) and >60x (D) was superior in PFPE samples and significantly so in Temno biopsies compared to FFPE counterparts (p = 0.0102 at >100x and p = 0.002 at >60x respectively). Greater numbers of individual UMI reads per million mapped reads were present in PFPE samples and reaching significance in PFPE tumour blocks (161 925 ± 24 464 versus 87 345 ± 19 776 individual reads/106 mapped reads, p = 0.0105) and Temno biopsies (141 552 ± 47 553 versus 36 117 ± 4679 individual reads/106 mapped reads, p = 0.0045 (E). Mean UMI read quality scores were higher in PFPE Temno biopsies compared to FFPE comparators (47.36 ± 0.7557 versus 46.03 ± 1.004, p = 0.0006) (F). The QIAGEN GeneReader™ analysis pipeline called a greater number of overall variants in PFPE samples than FFPE comparators (48 versus 37, p = 0.0084). Greater but non‐significant numbers of ‘pathogenic’ and ‘likely pathogenic’ variants were called in PFPE samples compared to FFPE samples (15 versus 13 and 3 versus 2 respectively). A significantly greater number of VUS were called in the PFPE samples compare to the FFPE equivalent (31 versus 22, p = 0.0093) (G).
GeneReader™ NGS variant calling
We used the QIAGEN QCI analysis pipeline to call variants in PFPE and FFPE samples. A greater number of overall variants were detected in PFPE samples (n = 48) compared to the equivalent FFPE samples (n = 37, p = 0.0084). Of these total variants, there were n = 15 ‘pathogenic’ variants called in PFPE samples compared to n = 13 in the FFPE samples, n = 3 ‘likely pathogenic' variants in PFPE samples compared to n = 2 in FFPE samples, and n = 31 ‘variants of unknown significance’ (VUS) in PFPE samples compared to n = 22 VUS in the FFPE samples (p = 0.0093; Figure 4G).
DNA extraction, quality metrics and targeted human lung cancer panel NGS at QUB
We prepared samples for DNA sequencing using the QIAseq targeted human lung cancer panel for NGS on Illumina platforms. Yields of extracted DNA were greater in PFPE lung parenchyma blocks (211.4 ± 40.72 ng/μl versus 123.5 ± 25.26 ng/μl, p = 0.042), tumour blocks (252.1 ± 44.0472 ng/μl versus 106.4 ± 16.1172 ng/μl, p = 0.0015), Temno biopsies (38.93 ± 4.648 ng/μl versus 13.68 ± 2.301 ng/μl, p = 0.0002) and PFPE tumour FNA preparations (20.8 ± 7.897 ng/μl versus 7.776 ± 2.972 ng/μl, p = 0.0431) measured by NanoDrop. Overall, values for DNA concentrations from QUBIT assessments were lower than those reported by NanoDrop but greater DNA concentrations were identified in PFPE Temno biopsies (17.08 ± 2.353 ng/μl versus 5.994 ± 1.438 ng/μl, p = 0.002) and PAXgene® FNA fixed tumour FNA samples (8.944 ± 2.358 ng/μl versus 2.621 ± 1.419 ng/μl, p = 0.005) than formalin or Cytolyt/formalin comparators. These findings are summarised in supplementary material, Figure S2.
We found mean fragment size (Figure 5A) was greater in PFPE lung parenchyma blocks (7069 ± 545.9 bp versus 4677 ± 1110 bp, p = 0.00357) and in PFPE tumour FNA preparations (5518 ± 626.2 bp versus 398.3 ± 320.0 bp, p = 0.0001). We quantified mean depth of coverage (Figure 5B) and found that coverage (x) was significantly deeper in PFPE parenchyma blocks (1165 ± 182.2x versus 951 ± 171.6x, p = 0.0365), Temno biopsies (917.9 ± 196.7x versus 551.1 ± 182.8x, p = 0.0017) and tumour FNA preparations (821.3 ± 166.7x versus 218.9 ± 114.0x, p = 0.0102). All PFPE samples were suitable for NGS whilst n = 1 FFPE tumour FNA was deemed a technical failure. Concordance between PFPE and FFPE samples was generally good but genomic findings were not identical with genetic variants present in resections that were absent in the smaller biopsy and tumour FNA samples and vice versa. We have summarised these findings in Figure 5C and in supplementary material, Figure S3 and Table S4. A total of n = 41 genetic variants were identified in PFPE tumour blocks compared to n = 48 variants in FFPE tumour blocks. Of these, n = 39 genetic variants were common to both PFPE and FFPE tumour blocks, with n = 2 variants unique to the PFPE tumour block and n = 9 unique variants in the FFPE tumour block. For summary of variants detected by sample type (see Figure 5D). In regards to the detection of actionable EGFR mutations, both PFPE and FFPE tumour blocks performed equally with a total of n = 5 EGFR variants identified in both fixation types, present in 3 of 8 patients (37.5% of tumours sampled).
Figure 5.
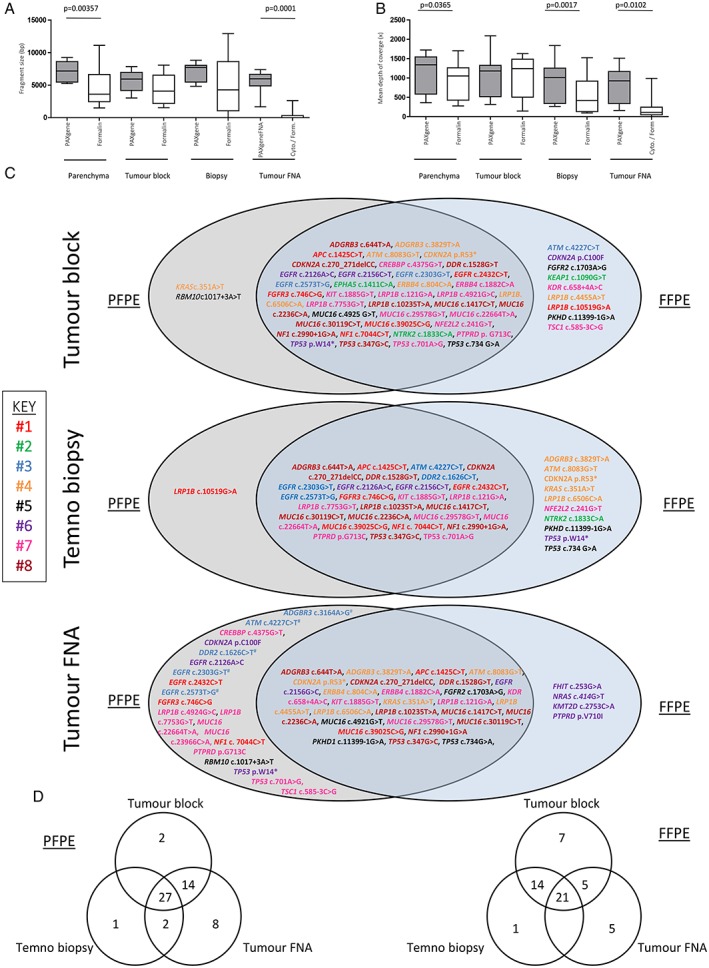
Illumina NGS. Mean fragment size was greater in PFPE lung parenchyma blocks (7069 ± 545.9 bp versus 4677 ± 1110 bp, p = 0.00357) and in tumour FNA preparations (5518 ± 626.2 bp versus 398.3 ± 320.0 bp, p = 0.0001) (A). Mean depth of coverage (x) was significantly deeper in PFPE parenchyma blocks (1165 ± 182.2x versus 951 ± 171.6x, p = 0.0365), Temno biopsies (1041 ± 194.4x versus 917.9 ± 196.7x, p = 0.0017) and in tumour FNA preparations (821.3 ± 166.7x versus 218.9 ± 114.0x, p = 0.0102) (B). All PFPE samples were suitable for NGS but n = 1 FFPE tumour FNA was deemed a technical failure. Venn diagrams summarising variant details and frequency in PFPE and FFPE tumour blocks, Temno biopsies and tumour FNA samples (C) and by sample type (D). We identified n = 4 EGFR mutations (EGFR c.2126A>C, EGFR c.2303G>T, EGFR c.2432C>T and EGFR c.2573T>G) in three individuals that were present in PFPE tumour FNA preparations but not identified in the FFPE comparator FNA. We should highlight that the FFPE tumour FNA from the subject harbouring n = 2 separate EGFR mutations was a technical failure and although we cannot exclude unintentional sampling variations, the potential underrepresentation of EGFR mutations in FFPE material is of note.
A total of n = 28 genetic variants were identified in PFPE Temno biopsies compared to n = 37 in the FFPE comparator Temno biopsies. In the PFPE Temno biopsies we detected an additional n = 3 variants absent in the corresponding PFPE tumour block, and n = 2 additional variants in the FFPE Temno biopsies that were absent in the corresponding FFPE tumour blocks. Only 60.96% of the variants present in the PFPE tumour block were also present in the corresponding PFPE Temno tumour biopsy and 72.92% of the variants identified in the FFPE tumour block were also present in the FFPE Temno biopsy. A single LRP1B variant was identified in the PFPE Temno biopsy that was absent in the FFPE Temno biopsy, compared to n = 10 variants present in the FFPE Temno biopsy but absent in the corresponding PFPE biopsy.
The PFPE tumour FNA samples contained n = 10 additional variants that were absent in the PFPE tumour blocks. Correspondingly, the FFPE tumour FNA samples contained n = 5 additional variants that were absent in the FFPE tumour blocks. In the PFPE tumour FNA preparations, a total of n = 48 genetic variants were identified compared to n = 32 in the FFPE tumour FNA samples. Critically, we identified n = 4 EGFR mutations (EGFR c.2126A>C, EGFR c.2303G>T, EGFR c.2432C>T and EGFR c.2573T>G) in three individuals that were present in PFPE tumour FNA preparations but not identified in the FFPE comparator FNA samples. We should highlight that the FFPE tumour FNA sample from the subject harbouring n = 2 separate EGFR mutations was a technical failure and although we cannot exclude unintentional sampling variations, the underrepresentation of EGFR mutations in FFPE material compared to those identified in PFPE material is noteworthy.
Discussion
Molecular testing for driver genetic mutations to guide clinical decision making is a key strategy for improving survival for NSCLC patients 34 and demand for genomic studies of diagnostic samples is likely to increase. Despite these advances, reliance on formalin remains widespread and is highly problematic for genomic studies, as exemplified by the preference of fresh‐frozen tissue for the 100 000 genomes project. The reasons for the continued use of formaldehyde are to a degree historical and understandably cost‐related but, whilst formaldehyde‐based fixatives adequately preserve morphology, closer attention should be paid to over/under‐fixation of samples 21, 22, 23, 24, 25, and overcoming formalin or processing related artefacts for key molecular or genomic applications is increasingly challenging 35, 36, 37. At the time of publication, the cost of the PAXgene® fixative and stabiliser is <£14 per block and requires a separate formalin‐free tissue processor (or full change of reagents prior to each batched PAXgene® run). This may seem expensive compared to the cost of formalin but represents only a fraction of the cost of the investigation, diagnosis and treatment of a patient on a lung cancer care pathway. For context, a PET scan is in the order of £850 per scan 38 and Gefitinib >£2000/month (https://bnf.nice.org.uk/medicinal-forms/gefitinib.html). Moreover, DNA sequencing remains an expensive modality and improving the failure rate of poorly fixed or degraded samples brings significant benefits, including financial; and avoiding/reducing the need for repeat sampling and additional costs/burden of additional patient intervention is crucial.
In the study of lung cancer, techniques to maximise high quality DNA yield from small samples are particularly relevant; hence our inclusion of biopsy and tumour‐FNA cell blocks. We demonstrate that PAXgene® use is suitable for histopathological assessment and broadly comparable to formalin fixation for most common histology and immunohistochemical tests used for the study of lung cancer. Although PFPE H&E‐stained sections were more eosinophilic, we found no interpretive challenges with PFPE prepared samples, in line with previous studies of colon cancer 39, prostate histomorphology 40, and within the field of veterinary pathology 33. Immunostaining of PFPE sections was similar to FFPE stained sections (without tailoring optimisation or protocols), highlighting how immunostaining of PFPE samples could potentially be performed alongside FFPE samples with little laboratory impact. In addition to lung, immunohistochemistry of PFPE and FFPE samples has already been explored in a number of tissues including the prostate 40, and by tissue microarrays of lung and colon adenocarcinomas 41. Previously, a comprehensive immunohistochemical evaluation compared 28 different antibody clones raised against 14 different antigens (2 antibody clones per antigen). Somewhat in contrast to our findings, these studies found comparable staining in only 7 of 28 antibodies. A further 10 of 28 antibodies were found to be ‘interpretable but sub‐optimally immunostained’, with the remaining 11 of 28 antibodies (including TTF‐1) showing insufficient staining quality 41. We have made no efforts to optimise our PAXgene® immunohistochemistry and are confident that antibody sensitivity and specificity could be improved by antibody titration for PAXgene® samples or the omission of antigen retrieval (as this is not theoretically required as PAXgene® is non‐cross‐linking). In addition, previous studies to evaluate PAXgene® suitability for Immunohistochemistry and to optimise staining specifically for PAXgene®‐Tissue‐fixed samples have proved successful 41, 42, 43.
Our preliminary data demonstrates the feasibility of successful PD‐L1 immunostaining in paired PFPE and FFPE samples (see supplementary material, Figure S4). Whilst outside of the scope of this study, we acknowledge that suitability and equivalence of alternative fixatives for biomarker expression, including PD‐L1, is essential for lung cancer samples 44, 45, 46, 47. Although validation of PD‐L1 staining to date has been performed on FFPE material 45, further work exploring variations in PD‐L1 (and other emerging biomarkers, e.g. ROS1) expression and careful validation in samples fixed in reagents other than formalin is warranted.
In line with existing reports 33, 41, our quantification of DNA/RNA yield and quality indicates PFPE to be superior to FFPE samples. We also found that both DNA integrity and fragment size were superior in PFPE specimens, again in keeping with previous studies 41, 48. Our β‐actin RT‐PCR also supports these findings as we found that, on average, fewer PCR amplification cycles were required to exceed the background cycle threshold for PAXgene® Tissue fixed samples; supporting superior biomolecule preservation.
We panel‐sequenced extracted DNA using a combination of approaches including the QIAGEN GeneReader™ NGS workflow and Illumina sequencing. We utilised the use of UMI barcoding to evaluate the precise effects of formalin and PAXgene® fixation upon sequencing fidelity, a technology particularly useful for distinguishing true, low frequency emerging clones of tumour cells from sequencing or PCR artefacts. We identified greater numbers of high quality UMI‐reads in PFPE tumour blocks and in biopsies compared to FFPE samples. Whilst not a key objective of this study, we identified a large degree of heterogeneity between variants found in different sample types from individual patients (as well as between differently fixed paired samples). It is plausible that some differences could be attributed to intra‐tumour heterogeneity, and quicker or improved DNA fixation in smaller samples (relative to blocks) resulting in superior NGS performance.
Depth of coverage is a useful parameter to help understand how well DNA sequencing has performed and is particularly relevant to investigations of lung cancer as deeper sequencing increases confidence in calling low frequency variants. Mean depth of coverage was greater in PFPE samples compared with FFPE comparators; an effect more evident in the smaller sized PAXgene®‐fixed tumour‐FNA samples and likely facilitated by the presence of greater yields of high‐quality, non‐fragmented DNA in PFPE samples. Distinction between low‐frequency tumour clones and PCR or sequencing artefacts in lung cancer is crucial as the acquisition and detection of resistance mutations can dictate a switch to a second or third line agent 49. Critically, we identified a greater number of EGFR mutations in our PFPE tumour FNA samples (n = 4 mutations present in n = 3 patients) that were not detected in the FFPE comparator (although present in the FFPE tumour block and biopsy). Despite one of the FFPE tumour FNA specimens being a technical failure, this observation again supports increased variant calling sensitivity using PAXgene® fixation and we should consider that many clinical FNA samples are physically small, relatively poorly cellular and often need to be extensively tested. Whilst we accept that this is a relatively small study using surgical resection material to replicate sample types used for the diagnosis and staging of lung cancer, the possibility of missing EGFR mutations through reliance upon formalin fixation may have profound consequences for the clinical management of patients harbouring actionable findings. We also confirmed that actionable findings from NGS were more comprehensive than the RT‐PCR‐based therascreen® platform as the EGFR c.2432C>T (p.S811F) mutation in patient#1 and the EGFR c.2126G>C (p.E709A) mutation in patient #7 were not detected by therascreen® due to these primers not being included in the assay. In addition, the study of circulating cell free DNA (ccfDNA) for resistance mutations will also become critical in guiding patient management,49,50 and a number of approaches, including the use of PAXgene® blood collection tubes for ccfDNA stabilisation, have been evaluated 50, 51, 52, 53. Given its comprehensiveness, it is most likely that NGS will emerge as the preferential modality for studying genomic changes, regardless of original sample type or location and that appropriate fixation will be a critical factor for molecular analysis. Here we provide compelling evidence for improved variant calling (potentially including detection of actionable EGFR mutations in PFPE Tumour FNA preparations) by the use of PAXgene® fixation that were absent in FFPE comparators.
In summary, biomarker‐driven molecular stratification of patients with the detection of driver mutations and epigenetic alterations, longitudinal monitoring for the acquisition of resistance mutations and potential molecular screening for malignancy in at‐risk individuals (e.g. current smokers) are key approaches in the current and future management of NSCLC. NSCLC is a highly dynamic condition and a combination of treatment approaches, including chemotherapy, along with a biomarker‐ (e.g. PD‐L1 expression) or molecular‐guided first‐ or second‐line therapy followed by prospective testing of the tumour or ccfDNA to monitor acquisition of resistance mutations may be of benefit. Key to the clinical implementation of these strategies is optimal DNA stabilisation. Here we demonstrate that PAXgene® fixation is suitable for the majority of existing workflows (with little or no optimisation) and brings significant advantages for the molecular study of samples typical of those collected from lung cancer patients. Although we feel that PAXgene® specific workflows are not justified for the majority of samples, a duelled‐laboratory approach, whereby small samples including biopsies and FNA samples (i.e. those most likely to require molecular testing) are processed using formalin‐free protocols. We feel this approach will optimise DNA suitability for molecular analysis and will prove beneficial for many conditions including lung cancer, where patients often present with advanced disease and identification of actionable mutations is key to guide clinical management and improve patient survival.
Author contributions statement
MS, TK, NC, PM, MS‐T, DG and DMR were involved in study design and conceived experiments. MS, TK, NC, MM, JG, PM, CM, EM, PS and SH carried out experiments and collected, analysed and interpreted data. MS and DR generated figures, searched literature and wrote the manuscript. All authors had final approval of the submitted and published versions.
Disclaimers
GeneReader NGS System is intended for research use only. Not for use in diagnostic procedures. PAXgene® Tissue and FNA fixative are intended for research use only.
Supporting information
Supplementary materials and methods
Figure S1. Summary of sample preparation, DNA extraction and sequencing and bioinformatic pipeline
Figure S2. DNA extraction and sample preparation for DNA sequencing using a QIAseq targeted human lung cancer panel
Figure S3. Summary of genetic findings from paired PFPE and FFPE tumour blocks, Temno tumour biopsies and tumour FNA samples
Figure S4. Representative PD‐L1 immunohistochemistry
Table S1. PAXgene® tissue processor schedule
Table S2. Formalin processing schedule
Table S3. Genes included on the human lung cancer QIAseq DNA panel
Table S4. Variant detection
Acknowledgements
This study was performed as part of the STRATfix study; ‘Enabling stratified medicine with novel fixatives for improved pre‐analytical pathology workflows’ and was funded by Innovate UK (Grant Ref. 38618‐281179). We also acknowledge the staff of the Royal Papworth Hospital Research Tissuebank for assistance with sample collection and patients for providing consent.
Conflict of interest statement: This study was funded by Innovate UK (Grant Ref 38 618‐281 179). As part of the Innovate UK programme (the aim of which is to accelerate use of commercial products with potential benefits for healthcare) an Industrial Partner (in this case QIAGEN) works with other consortium members. TK, NC and DG are employees of QIAGEN. All other authors have no other conflicts of interest.
References
- 1. Hiley C, LeQuesne J, Santis G, et al Challenges in molecular testing in non‐small‐cell lung cancer patients with advanced disease. Lancet 2016; 388: 1002–1010. [DOI] [PubMed] [Google Scholar]
- 2. Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 3. Bulzebruck H, Bopp R, Drings P, et al New aspects in the staging of lung cancer. Prospective validation of the International Union Against Cancer TNM classification. Cancer 1992; 70: 1102–1110. [DOI] [PubMed] [Google Scholar]
- 4. Kapo JM, Akqun KM. Integrating palliative care into the care of patients with advanced lung cancer. Cancer J 2015; 21: 434–439. [DOI] [PubMed] [Google Scholar]
- 5. Rossi A, Torri V, Garassino MC, et al The impact of personalised medicine on survival: comparisons of results in metastatic breast, colorectal and non‐small cell lung cancers. Cancer Treat Rev 2014; 40: 485–494. [DOI] [PubMed] [Google Scholar]
- 6. Hirsch FR, Suda K, Wiens J, et al New and emerging targeted treatments in advanced non‐small‐cell lung cancer. Lancet 2016; 388: 1012–1024. [DOI] [PubMed] [Google Scholar]
- 7. Zhang H. Osimertinib making a breakthrough in lung cancer targeted therapy. Onco Targets Ther 2016; 9: 5489–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan WL, Jain A, Takano A, et al Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol 2016; 17: e347–e362. [DOI] [PubMed] [Google Scholar]
- 9. Mok TS, Wu YL, Thongprasert S, et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957. [DOI] [PubMed] [Google Scholar]
- 10. Maemondo M, Inoue A, Kobayashi K, et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR . N Engl J Med 2010; 362: 2380–2388. [DOI] [PubMed] [Google Scholar]
- 11. Mitsudomi T, Morita S, Yatabe Y, et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–128. [DOI] [PubMed] [Google Scholar]
- 12. Sequist LV, Martins RG, Spigel D, et al First‐line Gefitinib in patients with advanced non‐small‐cell cancer harbouring somatic EGFR mutations. J Clin Oncol 2008; 26: 2442–2449. [DOI] [PubMed] [Google Scholar]
- 13. Zhou C, Wu YL, Chen G, et al Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol 2010; 12: 735–742. [DOI] [PubMed] [Google Scholar]
- 14. Rosell R, Carcereny E, Gervais R, et al Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–246. [DOI] [PubMed] [Google Scholar]
- 15. Soda M, Choi YL, Enomoto M, et al Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature 2007; 448: 561–566. [DOI] [PubMed] [Google Scholar]
- 16. Weickhardt AJ, Aisner DL, Franklin WA, et al Diagnostic assays for identification of anaplastic lymphoma kinase‐positive non‐small cell carcinoma. Cancer 2013; 119: 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salido M, Pijuan L, Martinez‐Avilies L, et al Increased ALK gene copy number and amplification are frequent in non‐small‐cell lung cancer. J Thorac Oncol 2011; 6: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zorzetto M, Ferrari S, Saracino L, et al MET genetic lesions in non‐small‐cell lung cancer; pharmacological and clinical implications. Transl Lung Cancer Res 2012; 1: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Endris V, Penzel R, Warth A, et al Molecular diagnostic profiling of lung cancer specimens with a semiconductor‐based massive parallel sequencing approach feasibility, costs, and performance compared with conventional sequencing. J Mol Diagn 2013; 15: 765–775. [DOI] [PubMed] [Google Scholar]
- 20. Idris SF, Ahmad SS, Scott MA, et al The role of high‐throughput technologies in clinical cancer genomics. Expert Rev Mol Diagn 2013; 13: 167–181. [DOI] [PubMed] [Google Scholar]
- 21. Hayat M. Microscopy, Immunohistochemistry and Antigen Retrieval Methods for Light and Electron Microscopy. Kluwer Academic: New York, NY, 2002. [Google Scholar]
- 22. Arnold MM, Srivastava S, Fredenburgh J, et al Effects of fixation and tissue processing on immunohistochemical demonstration of specific antigens. Biotech Histochem 1996; 71: 224–230. [DOI] [PubMed] [Google Scholar]
- 23. Ramos‐Vara JA. Technical aspects of immunohistochemistry. Vet Pathol 2005; 42: 405–426. [DOI] [PubMed] [Google Scholar]
- 24. Otali D, Stockard CR, Oelschlager DK, et al Combined effects of formalin fixation and tissue processing on immunorecognition. Biotech Histochem 2009; 84: 223–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Preusser M, Elezi L, Hainfellner JA. Reliability and reproducibility of PCR‐based testing of O6‐methylguanine‐DNA methyltransferase gene (MGMT) promoter methylation status in formalin‐fixed and paraffin‐embedded neurosurgical biopsy specimens. Clin Neuropathol 2008; 27: 388–390. [DOI] [PubMed] [Google Scholar]
- 26. Vollmer E, Galle J, Lang DS, et al The HOPE technique opens up a multitude of new possibilities in pathology. Rom J Morphol Embryol 2006; 47: 15–19. [PubMed] [Google Scholar]
- 27. Bolt HM, Degen GH, Hengstler JG. The carcinogenicity debate on formaldehyde: how to derive safe exposure limits? Arch Toxicol 2010; 84: 421–422. [DOI] [PubMed] [Google Scholar]
- 28. Bosetti C, McLaughlin JK, Tarone RE, et al Formaldehyde and cancer risk: a quantitative review of cohort studies through 2006. Ann Oncol 2008; 19: 29–43. [DOI] [PubMed] [Google Scholar]
- 29. Navani N, Brown JM, Nankivell M, et al Suitability of endobronchial ultrasound‐guided transbronchial needle aspiration specimens for subtyping and genotyping of non‐small cell lung cancer: a multicenter study of 774 patients. Am J Respir Crit Care Med 2012; 185: 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fielding DI, Kurimoto N. EBUS‐TBNA/staging of lung cancer. Clin Chest Med 2013; 34: 385–394. [DOI] [PubMed] [Google Scholar]
- 31. Zhang R, Ying K, Shi L, et al Combined endobronchial and endoscopic ultrasound‐guided fine needle aspiration for mediastinal lymph node staging of lung cancer: a meta‐analysis. Eur J Cancer 2013; 49: 1860–1867. [DOI] [PubMed] [Google Scholar]
- 32. Serra Fortuny M, Gallego M, Berna L, et al FDG‐PET parameters predicting mediastinal malignancy in lung cancer. BMC Pulm Med 2016; 16: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Craft WF, Conway JA, Dark MJ. Comparison of histomorphology and DNA preservation produced by fixatives in the veterinary diagnostic laboratory setting. Peer J 2014; 2: e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singh RR, Patel KP, Routbort MJ, et al Clinical validation of a next generation sequencing screen for mutational hotspots in 46 cancer‐related genes. J Mol Diagn 2013; 15: 607–622. [DOI] [PubMed] [Google Scholar]
- 35. Do H, Dobrovic A. Sequence artifacts in DNA from formalin‐fixed tissues: causes and strategies for minimization. Clin Chem 2015; 61: 64–71. [DOI] [PubMed] [Google Scholar]
- 36. Wong SQ, Li J, Tan AY, et al Sequence artefacts in a prospective series of formalin‐fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics 2014; 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ofner R, Ritter C, Ugurel S, et al Non‐reproducible sequence artifacts in FFPE tissue: an experience report. J Cancer Res Clin Oncol 2017; 143: 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wotton C, Allen K, Masani V. The cost effectiveness of PET scans in the radical treatment of lung cancer patients in a district general hospital. Thorax 2011; 66: A147. [Google Scholar]
- 39. Gündisch S, Slotta‐Huspenina J, Verderio P, et al Evaluation of colon cancer histomorphology: a comparison between formalin and PAXgene tissue fixation by an international ring trial. Virchows Arch 2014; 465: 509–519. [DOI] [PubMed] [Google Scholar]
- 40. Gillard M, Tom WR, Antic T, et al Next‐gen tissue: preservation of molecular and morphological fidelity in prostate tissue. Am J Transl Res 2015; 7: 1227–1235. [PMC free article] [PubMed] [Google Scholar]
- 41. Mathieson W, Marcon N, Antunes L, et al A critical evaluation of the PAXgene tissue fixation system: morphology, immunohistochemistry, molecular biology, and proteomics. Am J Clin Pathol 2016; 146: 25–40. [DOI] [PubMed] [Google Scholar]
- 42. Belloni B, Lambertini C, Nuciforo P, et al Will PAXgene substitute formalin? A morphological and molecular comparative study using a new fixative system. J Clin Pathol 2013; 66: 124–135. [DOI] [PubMed] [Google Scholar]
- 43. Stumptner C, Pabst D, Loibner M, et al The impact of crosslinking and non‐crosslinking fixatives on antigen retrieval and immunohistochemistry. N Biotechnol 2019; 52: 69–83. [DOI] [PubMed] [Google Scholar]
- 44. Büttner R, Gosney JR, Skov BG, et al Programmed death‐ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in non‐small‐cell lung cancer. J Clin Oncol 2017; 35: 3867–3876. [DOI] [PubMed] [Google Scholar]
- 45. Rebelatto MC, Midha A, Mistry A, et al Development of a programmed cell death ligand‐1 immunohistochemical assay validated for analysis of non‐small cell lung cancer and head and neck squamous cell carcinoma. Diagn Pathol 2016; 11: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cree IA, Booton R, Cane P, et al PD‐L1 testing for lung cancer in the UK: recognizing the challenges for implementation. Histopathology 2016; 69: 177–186. [DOI] [PubMed] [Google Scholar]
- 47. Herbst RS, Baas P, Kim DW, et al Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet 2016; 387: 1540–1550. [DOI] [PubMed] [Google Scholar]
- 48. Andersen GB, Hager H, Hansen LL, et al Improved reproducibility in genome‐wide DNA methylation analysis for PAXgene‐fixed samples compared with restored formalin‐fixed and paraffin‐embedded DNA. Anal Biochem 2015; 468: 50–58. [DOI] [PubMed] [Google Scholar]
- 49. Del Re M, Tiseo M, Bordi P, et al Contribution of KRAS mutations and c.2369C>T (p.T790M) EGFR to acquired resistance to EGFR‐TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget 2017; 8: 13611–13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pasquale R, Fenizia F, Esposito Abate R, et al Assessment of high‐sensitive methods for the detection of EGFR mutations in circulating free tumor DNA from NSCLC patients. Pharmacogenomics 2015; 16: 1135–1148. [DOI] [PubMed] [Google Scholar]
- 51. Bordi P, Del Re M, Danesi R, et al Circulating DNA in diagnosis and monitoring EGFR gene mutations in advanced non‐small cell lung cancer. Transl Lung Cancer Res 2015; 4: 584–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warton K, Yuwono NL, Cowley MJ, et al Evaluation of Streck BCT and PAXgene stabilised blood collection tubes for cell‐free circulating DNA studies in plasma. Mol Diagn Ther 2017; 21: 563–570. [DOI] [PubMed] [Google Scholar]
- 53. Schmidt B, Reinicke D, Reindl I, et al Liquid biopsy – performance of the PAXgene® blood ccfDNA tubes for the isolation and characterization of cell‐free plasma DNA from tumor patients. Clin Chim Acta 2017; 469: 94–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Summary of sample preparation, DNA extraction and sequencing and bioinformatic pipeline
Figure S2. DNA extraction and sample preparation for DNA sequencing using a QIAseq targeted human lung cancer panel
Figure S3. Summary of genetic findings from paired PFPE and FFPE tumour blocks, Temno tumour biopsies and tumour FNA samples
Figure S4. Representative PD‐L1 immunohistochemistry
Table S1. PAXgene® tissue processor schedule
Table S2. Formalin processing schedule
Table S3. Genes included on the human lung cancer QIAseq DNA panel
Table S4. Variant detection