

Abstract
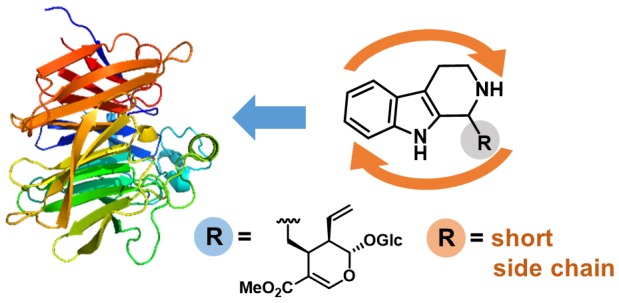
The Pictet–Spengler reaction is a valuable route to 1,2,3,4-tetrahydro-β-carboline (THBC) and isoquinoline scaffolds found in many important pharmaceuticals. Strictosidine synthase (STR) catalyzes the Pictet–Spengler condensation of tryptamine and the aldehyde secologanin to give (S)-strictosidine as a key intermediate in indole alkaloid biosynthesis. STRs also accept short-chain aliphatic aldehydes to give enantioenriched alkaloid products with up to 99% ee STRs are thus valuable asymmetric organocatalysts for applications in organic synthesis. The STR catalysis of reactions of small aldehydes gives an unexpected switch in stereopreference, leading to formation of the (R)-products. Here we report a rationale for the formation of the (R)-configured products by the STR enzyme from Ophiorrhiza pumila (OpSTR) using a combination of X-ray crystallography, mutational, and molecular dynamics (MD) studies. We discovered that short-chain aldehydes bind in an inverted fashion compared to secologanin leading to the inverted stereopreference for the observed (R)-product in those cases. The study demonstrates that the same catalyst can have two different productive binding modes for one substrate but give different absolute configuration of the products by binding the aldehyde substrate differently. These results will guide future engineering of STRs and related enzymes for biocatalytic applications.
Introduction
Isoquinoline and β-carboline derivatives are both widespread structural motifs found in alkaloids isolated from various plants and animals. The isoquinoline scaffold is a building block of many pharmaceutically important drugs such as the antispasmodic papaverine and the analgesic codeine.1 β-Carboline alkaloids exhibit a wide range of psychopharmacological effects through inhibition of monoamine oxidases or by binding to benzodiazepine receptors.2
The Pictet–Spengler reaction, by which 1,2,3,4-tetrahydro-β-carbolines (THBCs) and isoquinolines are synthesized from amines and aldehydes, represents an important synthetic procedure for the preparation of alkaloid derivatives and pharmaceuticals since its discovery more than a century ago.3 The development of asymmetric synthetic versions of the reaction has sparked great interest. In addition to diastereoselective approaches, where stereocontrol can be exerted by either the amine4 or the carbonyl substrate,5 using a chiral auxiliary,6 several procedures employing chiral catalysts, such as Bronsted acids,7 Lewis acids,8 or thiourea compounds,9 have also been described in literature.
In nature, the equivalent reaction is catalyzed by enzymes known as “Pictet–Spenglerases” (PSases). Since the identification of the first PSase in 197610 this enzyme family has been constantly growing. In addition to the well-investigated representatives norcoclaurine synthase (NCS)11 and strictosidine synthase (STR), many PSases, such as deacetylipecoside synthase (DIS) and deacetylisoipecoside synthase (DIIS) from Alangium lamarckii,12 SfmC-C from Streptomyces lavendulae,13 McbB from Marinactinospora thermotolerans,14 and NscbB (from Nocardiopsis synnemataformans DSM 44143),15 have been identified; however, structural, mechanistic, or biosynthetic applications of the latter have not been intensively studied.
The first identified representative of “Pictet–Spenglerases”, STR from Rauvolfia serpentina (RsSTR, EC 4.3.3.2), catalyzes the stereoselective condensation of tryptamine (1) and secologanin, leading to the formation of (S)-strictosidine, a central intermediate in the biosynthetic pathway of indole alkaloids (Scheme 1A).16 Crystal structures of RsSTR and mechanistic studies revealed that the reaction is catalyzed by a glutamate residue, Glu309, in the active site pocket, likely assisted by neighboring tyrosine and histidine.17 Evaluation of the synthetic potential of this and other STRs revealed a relatively broad applicability of various tryptamine analogues as substrates.17c,18 In addition to secologanin analogues,18c,19 selected aldehydes bearing shorter side chains were accepted by the enzyme, although with lower activities.20 However, interestingly, we discovered that for small aliphatic aldehydes (e.g., 2a–d) the corresponding (R)-configured THBC products were obtained employing, for example, the STR from Ophiorrhiza pumila (OpSTR) or RsSTR with an ee of >98% for 3a (Scheme 1B), in contrast to the absolute configuration of the natural (S)-configured product (S)-strictosidine.20b
Scheme 1. Reactions Catalyzed by Strictosidine Synthase (STR).

Results published previously.20b
A: natural reaction of tryptamine with secologanin yielding (S)-strictosidine; B: reactions between tryptamine and small aliphatic aldehydes 2a–d resulted in the formation of the corresponding (R)-configured tetrahydro-β-carbolines 3.
We have now investigated the origin of this switch in stereopreference from (S) for the natural reaction (tryptamine with secologanin) to (R) when using non-native aldehydes 2a–d as substrates with tryptamine employing the STR from Ophiorrhiza pumila (OpSTR). We use X-ray crystallography, mutational analysis, and MD simulations to determine the origin of this unexpected phenomenon.
Results and Discussion
In a first attempt to elucidate the origin of the (R)-stereoselectivity of OpSTR with aldehydes 2b–d (Scheme 1B), X-ray crystallographic studies were performed with OpSTR cocrystallized with racemic THBC products 3a–c as ligands. The overall architecture of OpSTR is a six-bladed β-propeller fold (Figure 1), as previously observed in crystal structures of related enzymes such as RsSTR. Here each blade consists of a four-stranded antiparallel β-sheet arranged around a central axis. The individual strands are twisted, resulting in an almost perpendicular orientation of the fourth strand to the first inner strand. One of these β-sheets (β-sheet 1, Figure 1) is formed from one strand from the N-terminus and three strands from the C-terminus, forming the so-called “velcro closure” present in most β-propeller proteins. Additionally, four short α-helices located at the outer surface of the propeller are present in the structure of OpSTR. One of these helices situated on top of the central axis of the enzyme forms the entrance to the active site pocket. Two of these helices are connected by a conserved disulfide bridge formed between residues Cys60 and Cys72 in OpSTR (numbering of residues is adapted to the amino acid sequence devoid of a N-terminal signal peptide that is removed for improved expression20b).
Figure 1.
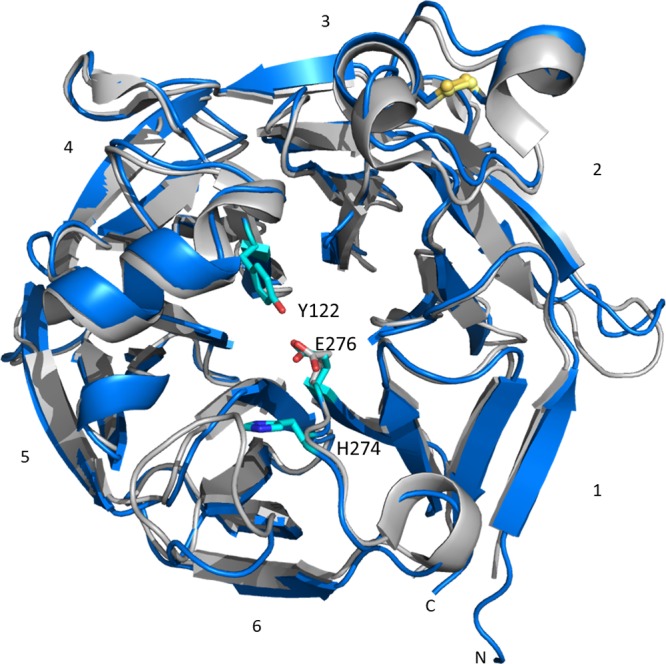
Overlay of structures of OpSTR (blue) and RsSTR (gray, PDB 2v91). Residues mentioned in this paper are highlighted in cyan. Ligands are not displayed.
Soaking experiments with apo-crystals of OpSTR with THBCs were unsuccessful. However, OpSTR readily cocrystallized in the presence of racemic THBCs rac-3a–c and yielded diffracting crystals with the corresponding ligand coordinated in the active site pocket. In the observed orientation of ligand 3 in the active site, the aliphatic substituent of the THBCs (isobutyl for 3a) points into the active site pocket; thus, the β-carboline is pointing outside. This is in contrast to the structures of STR from Rauvolfia serpentina with the natural product (S)-strictosidine (PDB code: 2v91), where the β-carboline points into the active site pocket and the aldehyde side chain protrudes from the active site. Despite the switched orientation of the molecule, the position of the nitrogen atom of the indole moiety of the ligands (S)-strictosidine and 3a is similar and in close proximity to the active site glutamate residue (Glu309 in RsSTR, Glu276 in OpSTR; Figure 2A). All ligands found in crystal structures determined from cocrystallization experiments with racemic THBCs (3a–c) adopted a similar orientation in the active site (Figure 2B). The distances from the N atom to Glu276 found in the crystal structures range between 2.8 Å (structure with 3b) and 3.6 Å (structure with 3c). The most surprising finding however was that in all cases exclusively the (S)-enantiomers of 3a–c were identified in the active site of OpSTR, which represents the opposite enantiomer of the preferred (R)-product formed in the corresponding enzymatic reactions. However, no crystals were formed when using optically pure (R)-3a, which is produced by the enzyme with >98% ee, as ligand for cocrystallization,
Figure 2.

OpSTR and RsSTR with THBCs as ligands. (A) Comparison of OpSTR (blue) containing 3a (orange) with RsSTR (gray mesh) containing (S)-strictosidine (gray, PDB: 2V91). (B) Alignment of different THBC ligands coordinated in OpSTR. The active site glutamate Glu276 coordinating the N atom is displayed as stick representation. The ligands are depicted in the following colors: 3a, orange; 3b, yellow; 3c, cyan.
Because the obtained crystal structures contained the (S)- and not the expected (R)-enantiomers, it was suspected that the (S)-THBC may act as inhibitor, due to the higher affinity of this enantiomer to the enzyme. Indeed, when testing the OpSTR in a reaction between 1 and 2a adding (S)-, (R)-, or rac-3d, the IC50 values calculated from experimentally determined reaction rates indicate that 3d has inhibitory effects on OpSTR (Table 1).
Table 1. Summary of IC50 Values Determined from Curve Fits of Experimentally Determined Values (determined from Figures S4–S6, Supporting Information)a.
| compound | IC50 [mM] |
|---|---|
| rac-3d | 3.4 ± 0.3 |
| (S)-3d | 1.6 ± 0.1 |
| (R)-3d | n.a. |
Reactions were run with 10 mM tryptamine 1, 50 mM isovaleraldehyde 3a in PIPES (500 μL, 50 mM, pH 7.0, 10% v/v DMSO) in the presence of 3d (0.01–20 mM) catalyzed by 100 μg of purified OpSTR (∼5.5 μM) at 35 °C, 650 rpm. Velocities were determined by following the formation of product for 3 h. Normalized reaction rates were plotted in a semilogarithmic plot using the SigmaPlot Standard Curves Analysis macro to determine the IC50 values. n.a.: not applicable.
While the (S)-enantiomer (S)-3d led to inhibition of the reaction already at low concentration, the mirror image (R)-3d led to inhibition only at higher concentrations. Thus, product inhibition is mainly caused by the (S)-enantiomer. On the basis of this observation, we concluded that the crystal structures obtained most likely did not show the product ligand in a catalytically productive conformation but rather revealed the inhibition of the enzyme by (S)-THBCs.
Despite the acquisition of the OpSTR structures, the ligand complexes did not provide information on the binding mode of (R)-THBCs and thus did not explain the observed stereopreference. To gain more insight into the basis for selective transformation by OpSTR, a stable amine mimic 4 of the imine intermediate int-I formed from tryptamine and isobutyraldehyde 2a was designed (Figure 3). In the structure of OpSTR obtained by cocrystallization with 4, two ligand molecules were found in each protein monomer. The active site was occupied by one ligand, while another ligand was bound to the periphery of the enzyme (Figure 3). In the peripheral site, the amine nitrogen was approximately 3.0 Å from the side chain of Glu6, but mutation of this residue to alanine gave a mutant with activity comparable to the wild type, suggesting that this is not a secondary active site. In contrast, the Glu276Ala variant was not active, showing that this is the residue responsible for activity. In the active site, the aliphatic side chain of 4 was pointing into the active site pocket, while the indole moiety was pointing toward the enzyme surface, in different orientations in each monomer dictated by rotation around the side chain, suggestive of the mobility required to achieve proximity to the imine bond for ring closure in a reactive complex. The alicyclic nitrogen atom is located at a distance of 4.2–4.7 Å from the side chain of Glu279, in approximately the same location as the endocyclic nitrogen of (S)-3a, with the isobutyl group again held in a hydrophobic pocket formed by Phe197, Trp120, and Tyr122, in a similar mode to that seen in (S)-3a.
Figure 3.

OpSTR cocrystallized with amine 4 as ligand. The figure shows a comparison between OpSTR with amine 4 (green) bound (displayed in blue) and OpSTR with THBC 3a (protein depicted in gray, ligand in orange). Furthermore, the orientations of 4 found in the active site of OpSTR and in the outer coordination site at Glu6 are displayed.
To investigate the influence of these active site residues including the essential Glu276 on the reaction, variants were generated (Table S5). The exchange of Glu276 to aspartate yielded an active variant; however, the stereoselectivity in reactions between 1 and 2a or 2d dropped significantly from >98% ee and 76% ee to 92% and 8%, respectively. The same effect was observed for variants of Trp120 with sterically less demanding residues such as alanine, phenylalanine, and tyrosine and for Tyr122Phe. Variant Tyr122Leu was not active. Actually, also products with lower ee were obtained for the variants Phe197Tyr/Trp/Leu. Thus, the variants generated indicate that all these active side residues contribute to the high stereoselectivity observed by the wild type enzyme for the transformation of isovaleraldehyde 2a with 1, but they do not give a hint for the switch of stereopreference compared to the reaction of secologanin with 1.
To better understand the switch of stereopreference, a combined density functional theory (DFT) and molecular dynamics (MD) study was undertaken. The Pictet–Spengler reaction follows a well-studied mechanism,3,17c,17d,20a,21 and we computed the free energy barrier for the analogue to confirm the rate-determining step (Figure 4) at the M06-2X/6-311++G(2d,2p)-SMD // M06-2X/6-31+G(d,p)-SMD level of theory with the water solvation model. All intermediates and transition structures were computed from exhaustive conformational searches, and the lowest-energy conformations are shown in Figure 4. In agreement with previous computational studies,9b we found the cyclization barrier from iminium int-I to be low and reversible (ΔG⧧ = 13.9 kcal/mol), leading to the carbocation intermediate int-II. This is consistent with the structural data that showed the mobility of the indole in the binding site. We modeled the deprotonation and rearomatization step with a free acetate molecule to simulate the catalytic glutamate residue. The deprotonation transition structure TS-II has a barrier of 23.8 kcal/mol, leading directly to the product 3a. It is well-known that the deprotonation step is rate-determining, while the cyclization establishes the stereochemistry of the new stereogenic center. Based on these results, and previous studies, the cyclization step is reversible, and the enzyme deprotonates only one of the stereoisomeric intermediates to form the product. We chose to model the carbocation intermediate int-II for both the analogue and strictosidine in molecular dynamics simulations to determine which stereoisomeric intermediate is able to acquire the best near-attack-conformation for deprotonation.22
Figure 4.
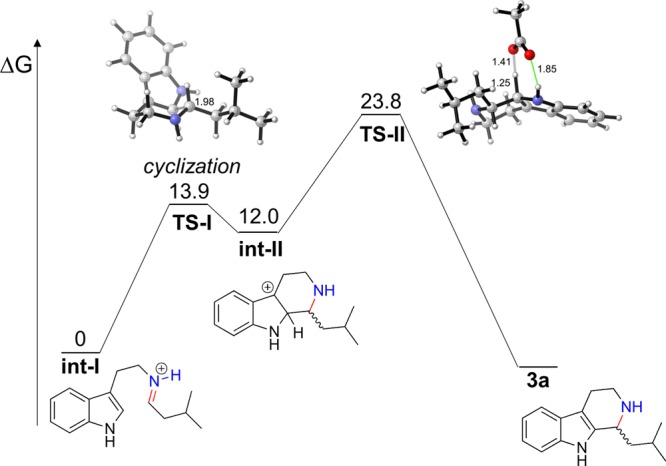
Free energy profile of the Pictet–Spengler reaction between tryptamine (1) and 2a calculated at the M06-2X/6-311++G(2d,2p)-SMD // M06-2X/6-31+G(d,p)-SMD level of theory. Distances are in angstroms (Å). Energies are shown in kcal/mol.
To understand first the stereopreference of the STR for the natural strictosidine substrate, we performed molecular dynamics simulations on the carbocation intermediate int-II for (S)- and (R)-strictosidine. The intermediates were docked into the binding pocket with Autodock Vina,23 and the best-scoring pose was selected as the input for 500 ns of MD simulations using Amber.24 The purpose of the MD simulations was to determine which stereoisomeric intermediate is stable in the binding pocket and best resembles the near-attack-conformation for deprotonation. In principle, the intermediate with the optimal near-attack-conformation between the hydrogen (of the carbocation) and the catalytic glutamate residue would be predicted to have a lower energy barrier for rearomatization.
Snapshots for (S)- and (R)-strictosidine-int, the analogous carbocation intermediates of int-II for strictosidine, are shown in Figure 5. Figure 5A illustrates a snapshot of the carbocation (S)-strictosidine-int in the protein pocket. The interaction between the glutamate oxygen and hydrogen for deprotonation is shown in blue dashed lines. The catalytic triad (tyrosine, glutamate, and histidine) as well as residues that interact strongly with the glucose side chain of strictosidine are shown. Figure 5B shows a snapshot of the carbocation (R)-strictosidine-int in the protein pocket. The interaction between the glutamate oxygen and hydrogen for deprotonation is shown in red dashed lines. Similarly, only the catalytic triad and residues interacting with strictosidine are shown for clarity.
Figure 5.

Results from MD simulation with (S)- and (R)-strictosidine-int. (A) Snapshot of MD trajectory of (S)-strictosidine-int. (B) Snapshot of MD trajectory of (R)-strictosidine-int. (C) Plot of hydrogen (of (R/S)-strictosidine-int) to oxygen (of glutamate) distances vs time in nanoseconds throughout the MD trajectory. The blue line represents the distance for (S)-strictosidine-int and the red line represents the distance for (R)-strictosidine-int.
The distance between the hydrogen HA and the oxygens on the catalytic glutamate were monitored throughout the MD simulation, which is plotted in Figure 5C. For the (S)-strictosidine-int carbocation, the trajectory showed that conformations are sampled for roughly 150 ns until a highly stable conformation is located at 175 ns, which remains for the 500 ns simulation. This is shown for one trajectory in Figure 5C by the blue line. These results are found in triplicate MD simulations. The (R)-strictosidine-int carbocation, by contrast, is unable to find a stable conformation in the binding pocket and adopts a binding pose to the surface of the protein. The red line in Figure 5C shows the H···O distance and illustrates how the intermediate changes conformations roughly every 50 ns until the 150 ns region where the intermediate exits from the binding pocket. These results are found in triplicate, unless a different starting geometry is used for (R)-strictosidine-int where the hydrogen for deprotonation is on the opposite face of the indole ring.
In these MD simulations, the polar interactions between the residues in Figure 5A and 5B with the strictosidine glucose unit were observed. For the (S)-intermediate, the interactions are maintained throughout the 500 ns of the MD simulation. In contrast, the (R) intermediate cannot accommodate those interactions while being in a near-attack-conformation for HA and the glutamate oxygen. When the carbocation is flipped, a stable conformation is found with proximal polar interactions in the MD simulation, but deprotonation cannot occur from this geometry. Thus, we can extrapolate that the strictosidine synthase shows the known stereopreference because the polar glucose unit anchors the substrate into the binding pocket by binding to a specific region of the protein surface. This conformation places the tryptamine unit of the carbocation tightly in the hydrophobic region of the binding pocket. The observed stereoisomer, (S)-strictosidine, forms because a near-attack-conformation is highly preferred. The alternative stereoisomer is not formed because there are insufficient stabilizing interactions for the (R)-strictosidine intermediate around the binding pocket to anchor the substrate to the catalytic residue, and consequently the deprotonation is very slow.
While the stereospecificity of strictosidine was established with MD simulations, we continued to investigate why the stereoselectivity switches with small aliphatic aldehydes such as isovaleraldehyde 2a. We performed MD simulations on the stereoisomers of the analogue substrate carbocations int-II. Snapshots for each 500 ns MD simulation [(R)- and (S)-int-II] are shown in Figure 6A and 6B. In both MD simulations, the isobutyl substituent on the chiral center prefers to bind on the inside of the binding pocket, in contrast to strictosidine which prefers tryptamine in the binding pocket. In each snapshot, only the catalytic triad is shown. During the 500 ns trajectories, stable conformations are found for both stereoisomers within close distances to the catalytic glutamate.
Figure 6.

Results from MD simulation with (R)- and (S)-int-II. (A) Snapshot of MD trajectory of (R)-int-II. (B) Snapshot of MD trajectory of (S)-int-II. (C) Plot of hydrogen (of int-II) to oxygen (of glutamate) distances vs C–H–O angles throughout the MD trajectory, with the transition state geometry shown in green. The blue data points are results from the MD trajectory with (R)-int-II, and the red data points are results from the MD trajectory with (S)-int-II.
However, the trajectories differ for the stereoisomeric intermediates at the near-attack-conformation geometry. In snapshot A, the (R)-int-II H–O distance is proximal, and the catalytic triad is conserved with close interactions. However, (S)-int-II, shown in snapshot B, the catalytic triad fails to form a strong hydrogen bond consistently for a majority of the 500 ns trajectory. This occurs for a majority of the 500 ns trajectory. The glutamate, while proximal to the substrate, develops a C–H–O angle between 60° and 120° throughout the MD simulation. Few snapshots reach the near-attack-conformation and a geometry near the quantum chemical transition structure for the (S)-analogue. The distances between the hydrogen of the carbocations and the oxygen of the glutamates were plotted against the C–H–O angle, which are both required parameters for the deprotonation, shown in Figure 6C. The green data point represents the ideal transition structure optimized with density functional theory in Figure 4. The data in Figure 6C show a clear trend in which (R)-int-II often achieves a conformation close to the transition structure, or an optimal near-attack-conformation, whereas (S)-int-II has a poor geometry for deprotonation despite being proximal to the catalytic residue. The difference in geometry between (R)-int-II and (S)-int-II is the location of the isobutyl group. In (R)-int-II, the isobutyl group moves into the area that is occupied by the tryptamine core in strictosidine, whereas in (S)-int-II the isobutyl group adopts a position close to the backbone of the protein and interferes sterically with the glutamate residue, similar to that observed for the inhibiting enantiomer in the X-ray complex with (S)-3a as well as for amine 4. This illustrates the switch in stereopreference, as well as the lack of stereospecificity with the smaller aldehydes. Because both stereoisomers can reach a near-attack-conformation with a stable geometry, both stereoisomers are formed but (R)-int-II is preferred.
Due to the suggested changed binding mode of tryptamine 1 in the reaction with 2a compared to the natural reaction with secologanin, the KM for tryptamine 1 was determined in the reaction between 1 and 2a and found to be 1.24 ± 0.13 mM. This value is just about twice the value reported for the KM of tryptamine 1 with secologanin (0.5 mM) for OpSTR.25 Literature values for the KM for the natural substrate secologanin vary between 3.9 × 10–2 mM17c and 4 mM18e for STR wild types, while the here determined KM for the non-natural substrate isovaleraldehyde 2a is slightly higher at 5.36 ± 0.83 mM.
It is also worth to mention that Glu276 is the active residue in the active site in the natural reaction (tryptamine with secologanin) as well as in the here-described inverted binding mode. This was demonstrated by the exchange of Glu276 to alanine, which resulted in an inactive protein.
Conclusions
In the (organo)catalytic Pictet–Spengler reaction investigated, the stereochemistry-determining reaction involves the deprotonation of the (S)- and (R)-intermediates. The MD simulations indicate clearly a binding mode of the natural intermediate and the substrate in which the indole part is pointing to the periphery of the enzyme, a conformation that was also observed for the inhibiting enantiomer in the X-ray complex with (S)-3a as well as for amine 4 mimicking the iminium int-II. By contrast, the alkyl group of the aldehyde, not bound by hydrogen binding present for secologanin, dominates binding with unnatural aldehydes. Thus, it can be concluded that the observed switch in stereopreference is due to a switch of positioning of the reacting substrate parts compared to the natural reaction of tryptamine with secologanin. Remarkably, the same catalyst can have two different productive binding modes for one substrate, here tryptamine, which lead finally to opposite absolute configurations of the product. Furthermore, as tryptamine serves as one reaction partner for both reactions, the one with the natural substrate secologanin as well as the reaction with isovaleraldehyde, the binding mode seems to be controlled by the aldehyde and not the tryptamine part. Furthermore, the investigations revealed that the minor enantiomer formed in the reaction may act as an inhibitor. These insights have profound significance for the future engineering of STRs to ensure on the one hand that the amino acids in the appropriate region are addressed for tuning the catalyst and on the other hand to consider that the minor enantiomer is either not formed or cannot bind. STRs thus present an unusual set of considerations for the engineering of enzyme activity that may have relevance to other systems where enzymes are required to transform substrates very different from those accepted in the native reaction.
Acknowledgments
The research leading to these results was funded partly by the Austrian Science Fund (FWF, project W9-01, DK Molecular Enzymology). M.S. was funded by grant BB/M006832/1 from the UK Biotechnology and Biological Sciences Research Council. We thank Dr. Johan P. Turkenburg and Mr. Sam Hart for assistance with X-ray data collection, and the Diamond Light Source for access to beamline I04-1 under proposal number mx-9948. We are grateful to the National Institutes of General Medical Sciences, National Institutes of Health, for research grant GM-124480 to K.N.H., and Chemistry-Biology Interface Research Training Grant (T32GM008496) for a traineeshipt to A.S. Computational resources from the UCLA Institute of Digital Research and Education (IDRE) are gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b08704.
Detailed experimental procedures and information on crystallographic data collection, structure refinement, and MD simulations (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Khan A. Y.; Suresh Kumar G. Natural isoquinoline alkaloids: binding aspects to functional proteins, serum albumins, hemoglobin, and lysozyme. Biophys. Rev. 2015, 7, 407–420. 10.1007/s12551-015-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Peng W.; Wang Z.; Xu A. β-Carboline Alkaloids: Biochemical and Pharmacological Functions. Curr. Med. Chem. 2007, 14, 479–500. 10.2174/092986707779940998. [DOI] [PubMed] [Google Scholar]
- Pictet A.; Spengler T. Über die Bildung von Isochinolin-derivaten durch Einwirkung von Methylal auf Phenyl-äthylamin, Phenyl-alanin und Tyrosin. Ber. Dtsch. Chem. Ges. 1911, 44, 2030–2036. 10.1002/cber.19110440309. [DOI] [Google Scholar]
- a Cox E. D.; Hamaker L. K.; Li J.; Yu P.; Czerwinski K. M.; Deng L.; Bennett D. W.; Cook J. M.; Watson W. H.; Krawiec M. Enantiospecific formation of trans 1,3-Disubstituted Tetrahydro-β-carbolines by the Pictet–Spengler reaction and Conversion of Cis Diastereomers into their trans counterparts by Scission of the C-1/N-2 Bond. J. Org. Chem. 1997, 62, 44–61. 10.1021/jo951170a. [DOI] [PubMed] [Google Scholar]; b Bailey P. D.; Moore M. H.; Morgan K. M.; Smith D. I.; Vernon J. M. Enhancing the yield and diastereoselectivity of the Pictet–Spengler reaction: A highly efficient route to Cis-1,3-disubstituted tetrahydro-β-carbolines. Tetrahedron Lett. 1994, 35, 3587–3588. 10.1016/S0040-4039(00)73247-3. [DOI] [Google Scholar]; c Alberch L.; et al. The cis-Specific Pictet–Spengler Reaction. Eur. J. Org. Chem. 2004, 2004, 1887–1890. 10.1002/ejoc.200400017. [DOI] [Google Scholar]; d Bailey P. D.; Beard M. A.; Phillips T. R. Unexpected cis selectivity in the Pictet–Spengler reaction. Tetrahedron Lett. 2009, 50, 3645–3647. 10.1016/j.tetlet.2009.03.121. [DOI] [Google Scholar]
- Ducrot P.; Rabhi C.; Thal C. Synthesis of tetrahydro-β-carbolines and studies of the pictet–spengler reaction. Tetrahedron 2000, 56, 2683–2692. 10.1016/S0040-4020(00)00165-4. [DOI] [Google Scholar]
- a Kawate T.; Yamanaka M.; Nakagawa M. Chiral auxiliary approach to the asymmetric Pictet–Spengler reaction of tryptamines. Heterocycles 1999, 50, 1033–1039. 10.3987/COM-98-S(H)106. [DOI] [Google Scholar]; b Soe T.; Kawate T.; Fukui N.; Nakagawa M. Asymmetric Pictet–Spengler reaction with chiral-N-(β-3-indoly) ethyl-1-methylbenzylamine. Tetrahedron Lett. 1995, 36, 1857–1860. 10.1016/0040-4039(95)92843-3. [DOI] [Google Scholar]; c Gremmen C.; Willemse B.; Wanner M. J.; Koomen G.-J. Enantiopure Tetrahydro-β-carbolines via Pictet–Spengler Reactions with N-Sulfinyl Tryptamines. Org. Lett. 2000, 2, 1955–1958. 10.1021/ol006034t. [DOI] [PubMed] [Google Scholar]; d Waldmann H.; Schmidt G.; Henke H.; Burkard M. Asymmetric Pictet–Spengler reactions employing N,N-phthaloyl amino acids as chiral auxiliary groups. Angew. Chem., Int. Ed. Engl. 1995, 34, 2402–2403. 10.1002/anie.199524021. [DOI] [Google Scholar]
- a Seayad J.; Seayad A. M.; List B. Catalytic Asymmetric Pictet–Spengler Reaction. J. Am. Chem. Soc. 2006, 128, 1086–1087. 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]; b Wanner M. J.; van der Haas R. N.; de Cuba K. R.; van Maarseveen J. H.; Hiemstra H. Catalytic asymmetric Pictet–Spengler reactions via sulfenyliminium ions. Angew. Chem., Int. Ed. 2007, 46, 7485–7487. 10.1002/anie.200701808. [DOI] [PubMed] [Google Scholar]; c Sewgobind N. V.; Wanner M. J.; Ingemann S.; de Gelder R.; van Maarseveen J. H.; Hiemstra H. Enantioselective Binol-phosphoric acid catalyzed Pictet–Spengler reactions of N-benzyltryptamine. J. Org. Chem. 2008, 73, 6405–6408. 10.1021/jo8010478. [DOI] [PubMed] [Google Scholar]; d Muratore M. E.; Holloway C. A.; Pilling A. W.; Storer R. I.; Trevitt G.; Dixon D. J. Enantioselective Brønsted acid-catalyzed N-acyliminium cyclization cascades. J. Am. Chem. Soc. 2009, 131, 10796–10797. 10.1021/ja9024885. [DOI] [PubMed] [Google Scholar]; e Holloway C. A.; Muratore M. E.; Storer R. l.; Dixon D. J. Direct Enantioselective Brønsted Acid Catalyzed N-Acyliminium Cyclization Cascades of Tryptamines and Ketoacids. Org. Lett. 2010, 12, 4720–4723. 10.1021/ol101651t. [DOI] [PubMed] [Google Scholar]; f Wang S. G.; Xia Z. L.; Xu R. Q.; Liu X. J.; Zheng C.; You S. L. Construction of Chiral Tetrahydro-β-Carbolines: Asymmetric Pictet–Spengler Reaction of Indolyl Dihydropyridines. Angew. Chem., Int. Ed. 2017, 56, 7440–7443. 10.1002/anie.201703178. [DOI] [PubMed] [Google Scholar]
- a Ishihara K.; Miyata M.; Hattori K.; Tada T.; Yamamoto H. A new chiral BLA promoter for asymmetric aza Diels-Alder and Aldol-type reactions of imines. J. Am. Chem. Soc. 1994, 116, 10520–10524. 10.1021/ja00102a019. [DOI] [Google Scholar]; b Yamada H.; Kawate T.; Matsumizu M.; Nishida A.; Yamaguchi K.; Nakagawa M. Chiral Lewis Acid-Mediated Enantioselective Pictet–Spengler Reaction of N b-Hydroxytryptamine with Aldehydes. J. Org. Chem. 1998, 63, 6348–6354. 10.1021/jo980810h. [DOI] [PubMed] [Google Scholar]; c Bou-Hamdan F. R.; Leighton J. L. Highly Enantioselective Pictet–Spengler Reactions with α-Ketoamide-Derived Ketimines: Access to an Unusual Class of Quaternary α-Amino Amides. Angew. Chem., Int. Ed. 2009, 48, 2403–2406. 10.1002/anie.200806110. [DOI] [PubMed] [Google Scholar]
- a Taylor M. S.; Jacobsen E. N. Highly enantioselective catalytic acyl-Pictet–Spengler reactions. J. Am. Chem. Soc. 2004, 126, 10558–10559. 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]; b Klausen R. S.; Kennedy C. R.; Hyde A. M.; Jacobsen E. N. Chiral Thioureas Promote Enantioselective Pictet–Spengler Cyclization by Stabilizing Every Intermediate and Transition State in the Carboxylic Acid-Catalyzed Reaction. J. Am. Chem. Soc. 2017, 139, 12299–12309. 10.1021/jacs.7b06811. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Raheem I. T.; Thiara P. S.; Peterson E. A.; Jacobsen E. N. Enantioselective Pictet–Spengler-type cyclizations of hydroxylactams: H-bond donor catalysis by anion binding. J. Am. Chem. Soc. 2007, 129, 13404–13405. 10.1021/ja076179w. [DOI] [PubMed] [Google Scholar]; d Mergott D. J.; Zuend S. J.; Jacobsen E. N. Catalytic asymmetric total synthesis of (+)-yohimbine. Org. Lett. 2008, 10, 745–748. 10.1021/ol702781q. [DOI] [PubMed] [Google Scholar]
- Stockigt J.; Zenk M. H. Strictosidine (isovincoside): the key intermediate in the biosynthesis of monoterpenoid indole alkaloids. J. Chem. Soc., Chem. Commun. 1977, 646–648. 10.1039/C39770000646. [DOI] [Google Scholar]
- a Rueffer M.; El-Shagi H.; Nagakura N.; Zenk M. H. (S)-norlaudanosoline synthase: The first enzyme in the benzylisoquinoline biosynthetic pathway. FEBS Lett. 1981, 129, 5–9. 10.1016/0014-5793(81)80742-9. [DOI] [Google Scholar]; b Samanani N.; Facchini P. J. Purification and Characterization of Norcoclaurine Synthase: the First Committed Enzyme in Benzylisoquinoline Alkaloid Biosynthesis in Plants. J. Biol. Chem. 2002, 277, 33878–33883. 10.1074/jbc.M203051200. [DOI] [PubMed] [Google Scholar]; c Luk L. Y. P.; Bunn S.; Liscombe D. K.; Facchini P. J.; Tanner M. E. Mechanistic Studies on Norcoclaurine Synthase of Benzylisoquinoline Alkaloid Biosynthesis: An Enzymatic Pictet–Spengler Reaction. Biochemistry 2007, 46, 10153–10161. 10.1021/bi700752n. [DOI] [PubMed] [Google Scholar]; d Ilari A.; Franceschini S.; Bonamore A.; Arenghi F.; Botta B.; Macone A.; Pasquo A.; Bellucci L.; Boffi A. Structural Basis of Enzymatic (S)-Norcoclaurine Biosynthesis. J. Biol. Chem. 2009, 284, 897–904. 10.1074/jbc.M803738200. [DOI] [PubMed] [Google Scholar]; e Lichman B. R.; Gershater M. C.; Lamming E. D.; Pesnot T.; Sula A.; Keep N. H.; Hailes H. C.; Ward J. M. ‘Dopamine-first’ mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J. 2015, 282, 1137–1151. 10.1111/febs.13208. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pesnot T.; Gershater M. C.; Ward J. M.; Hailes H. C. The Catalytic Potential of Coptis japonica NCS2 Revealed – Development and Utilisation of a Fluorescamine-Based Assay. Adv. Synth. Catal. 2012, 354, 2997–3008. 10.1002/adsc.201200641. [DOI] [Google Scholar]; g Lechner H.; Soriano P.; Poschner R.; Hailes H. C.; Ward J. M.; Kroutil W. Library of Norcoclaurine Synthases and Their Immobilization for Biocatalytic Transformations. Biotechnol. J. 2018, 13, 1700542. 10.1002/biot.201700542. [DOI] [PubMed] [Google Scholar]
- a De-Eknamkul W.; Ounaroon A.; Tanahashi T.; Kutchan T. M.; Zenk M. H. Enzymatic condensation of dopamine and secologanin by cell-free extracts of Alangium lamarckii. Phytochemistry 1997, 45, 477–484. 10.1016/S0031-9422(96)00828-X. [DOI] [Google Scholar]; b De-Eknamkul W.; Suttipanta N.; Kutchan T. M. Purification and characterization of deacetylipecoside synthase from Alangium lamarckii Thw. Phytochemistry 2000, 55, 177–181. 10.1016/S0031-9422(00)00260-0. [DOI] [PubMed] [Google Scholar]
- a Koketsu K.; Minami A.; Watanabe K.; Oguri H.; Oikawa H. Pictet–Spenglerase involved in tetrahydroisoquinoline antibiotic biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 142–149. 10.1016/j.cbpa.2012.02.021. [DOI] [PubMed] [Google Scholar]; b Koketsu K.; Watanabe K.; Suda H.; Oguri H.; Oikawa H. Reconstruction of the saframycin core scaffold defines dual Pictet–Spengler mechanisms. Nat. Chem. Biol. 2010, 6, 408. 10.1038/nchembio.365. [DOI] [PubMed] [Google Scholar]
- a Mori T.; Hoshino S.; Sahashi S.; Wakimoto T.; Matsui T.; Morita H.; Abe I. Structural Basis for β-Carboline Alkaloid Production by the Microbial Homodimeric Enzyme McbB. Chem. Biol. 2015, 22, 898–906. 10.1016/j.chembiol.2015.06.006. [DOI] [PubMed] [Google Scholar]; b Chen Q.; Ji C.; Song Y.; Huang H.; Ma J.; Tian X.; Ju J. Discovery of McbB, an Enzyme Catalyzing the β-Carboline Skeleton Construction in the Marinacarboline Biosynthetic Pathway. Angew. Chem., Int. Ed. 2013, 52, 9980–9984. 10.1002/anie.201303449. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Zhang S.; Xie Y. Characterization of a new microbial Pictet–Spenglerase NscbB affording the β-carboline skeletons from Nocardiopsis synnemataformans DSM 44143. J. Biotechnol. 2018, 281, 137–143. 10.1016/j.jbiotec.2018.07.007. [DOI] [PubMed] [Google Scholar]
- Brown R. T.; Leonard J.; Sleigh S. K. The role of strictosidine in monoterpenoid indole alkaloid biosynthesis. Phytochemistry 1978, 17, 899–900. 10.1016/S0031-9422(00)88642-2. [DOI] [Google Scholar]
- a Ma X.; Koepke J.; Fritzsch G.; Diem R.; Kutchan T. M.; Michel H.; Stöckigt J. Crystallization and preliminary X-ray crystallographic analysis of strictosidine synthase from Rauvolfia: the first member of a novel enzyme family. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1702, 121–124. 10.1016/j.bbapap.2004.06.013. [DOI] [PubMed] [Google Scholar]; b Koepke J.; Ma X.; Fritzsch G.; Michel H.; Stockigt J. Crystallization and preliminary X-ray analysis of strictosidine synthase and its complex with the substrate tryptamine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 690–693. 10.1107/S0907444904029348. [DOI] [PubMed] [Google Scholar]; c Ma X.; Panjikar S.; Koepke J.; Loris E.; Stöckigt J. The Structure of Rauvolfia serpentina Strictosidine Synthase Is a Novel Six-Bladed β-Propeller Fold in Plant Proteins. Plant Cell 2006, 18, 907–920. 10.1105/tpc.105.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Maresh J. J.; Giddings L.-A.; Friedrich A.; Loris E. A.; Panjikar S.; Trout B. L.; Stöckigt J.; Peters B.; O’Connor S. E. Strictosidine synthase: Mechanism of a Pictet–spengler catalyzing enzyme. J. Am. Chem. Soc. 2008, 130, 710–723. 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Stöckigt J.; Barleben L.; Panjikar S.; Loris E. A. 3D-Structure and function of strictosidine synthase – the key enzyme of monoterpenoid indole alkaloid biosynthesis. Plant Physiol. Biochem. 2008, 46, 340–355. 10.1016/j.plaphy.2007.12.011. [DOI] [PubMed] [Google Scholar]
- a Bernhardt P.; McCoy E.; O’Connor S. E. Rapid Identification of Enzyme Variants for Reengineered Alkaloid Biosynthesis in Periwinkle. Chem. Biol. 2007, 14, 888–897. 10.1016/j.chembiol.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yamazaki M.; Asano T.; Yamazaki Y.; Sirikantaramas S.; Sudo H.; Saito K. Biosynthetic system of camptothecin: an anticancer plant product. Pure Appl. Chem. 2010, 82, 213–218. 10.1351/PAC-CON-09-02-08. [DOI] [Google Scholar]; c McCoy E.; Galan M. C.; O’Connor S. E. Substrate specificity of strictosidine synthase. Bioorg. Med. Chem. Lett. 2006, 16, 2475–2478. 10.1016/j.bmcl.2006.01.098. [DOI] [PubMed] [Google Scholar]; d Loris E. A.; Panjikar S.; Ruppert M.; Barleben L.; Unger M.; Schübel H.; Stöckigt J. Structure-Based Engineering of Strictosidine Synthase: Auxiliary for Alkaloid Libraries. Chem. Biol. 2007, 14, 979–985. 10.1016/j.chembiol.2007.08.009. [DOI] [PubMed] [Google Scholar]; e Hampp N.; Zenk M. H. Homogeneous strictosidine synthase from cell suspension cultures of Rauvolfia serpentina. Phytochemistry 1988, 27, 3811–3815. 10.1016/0031-9422(88)83023-1. [DOI] [Google Scholar]; f Wu F.; Zhu H.; Sun L.; Rajendran C.; Wang M.; Ren X.; Panjikar S.; Cherkasov A.; Zou H.; Stöckigt J. Scaffold tailoring by a newly detected Pictet–Spenglerase activity of strictosidine synthase: from the common tryptoline skeleton to the rare piperazino-indole framework. J. Am. Chem. Soc. 2012, 134, 1498–1500. 10.1021/ja211524d. [DOI] [PubMed] [Google Scholar]; g Yang L.; Zou H.; Zhu H.; Ruppert M.; Gong J.; Stöckigt J. Improved Expression of His6-Tagged Strictosidine Synthase cDNA for Chemo-Enzymatic Alkaloid Diversification. Chem. Biodiversity 2010, 7, 860–870. 10.1002/cbdv.201000052. [DOI] [PubMed] [Google Scholar]
- a Treimer J. F.; Zenk M. H. Purification and Properties of Strictosidine Synthase, the Key Enzyme in Indole Alkaloid Formation. Eur. J. Biochem. 1979, 101, 225–233. 10.1111/j.1432-1033.1979.tb04235.x. [DOI] [PubMed] [Google Scholar]; b Chen S.; Galan M. C.; Coltharp C.; O’Connor S. E. Redesign of a Central Enzyme in Alkaloid Biosynthesis. Chem. Biol. 2006, 13, 1137–1141. 10.1016/j.chembiol.2006.10.009. [DOI] [PubMed] [Google Scholar]
- a Bernhardt P.; Usera A. R.; O’Connor S. E. Biocatalytic asymmetric formation of tetrahydro-β-carbolines. Tetrahedron Lett. 2010, 51, 4400–4402. 10.1016/j.tetlet.2010.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pressnitz D.; Fischereder E.-M.; Pletz J.; Kofler C.; Hammerer L.; Hiebler K.; Lechner H.; Richter N.; Eger E.; Kroutil W. Asymmetric Synthesis of (R)-1-Alkyl-Substituted Tetrahydro-ß-carbolines Catalyzed by Strictosidine Synthases. Angew. Chem., Int. Ed. 2018, 57, 10683–10687. 10.1002/anie.201803372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ungemach F.; Cook J. M. The Spiroindolenine Intermediate. A Review. Heterocycles 1978, 9, 1089–1119. 10.3987/R-1978-08-1089. [DOI] [Google Scholar]; b Bailey P. Direct proof of the involvement of a spiro intermediate in the Pictet–Spengler reaction. J. Chem. Res., Synop 1987, 202–203. [Google Scholar]
- Hur S.; Bruice T. C. The near attack conformation approach to the study of the chorismate to prephenate reaction. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12015–12020. 10.1073/pnas.1534873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Izadi S; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin S.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2018, University of California: San Francisco, 2018.
- Yamazaki Y.; Urano A.; Sudo H.; Kitajima M.; Takayama H.; Yamazaki M.; Aimi N.; Saito K. Metabolite profiling of alkaloids and strictosidine synthase activity in camptothecin producing plants. Phytochemistry 2003, 62, 461–470. 10.1016/S0031-9422(02)00543-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.