

Abstract
Introduction
Carboxyalkyl flavonoids derivatives are considered as effective inhibitors in reducing post-prandial hyperglycaemia.
Methods
Combined with Density Functional Theory (DFT) and the theory of Atoms in Molecules (AIM), molecular docking and charge density analysis are carried out to understand the molecular flexibility, charge density distribution and the electrostatic properties of these carboxyalkyl derivatives.
Results
Results show that the electron density of the chemical bond C14-O17 on B ring of molecule II increases while O17-H18 decreases at the active site, suggesting the existence of weak non-covalent interactions, most prominent of which are H-bonding and electrostatic interaction. When hydroxyl groups are introduced, the highest positive electrostatic potentials are distributed near the B ring hydroxyl hydrogen atom and the carboxyl hydrogen atom on the A ring. It was reported that quercetin has a considerably inhibitory activity to S. cerevisiae α-glucosidase, from the binding affinities, it is suggested that the position and number of hydroxyl groups on the B and C rings are also pivotal to the hypoglycemic activity when the long carboxyalkyl group is introduced into the A ring.
Conclusion
It is concluded that the presence of three well-defined zones in the structure, both hydrophobicity alkyl, hydrophilicity carboxyl and hydroxyl groups are necessary.
Keywords: Molecular docking, quantum chemical calculations, α-glucosidase, charge density distribution, carboxyalkyl flavonoids, density functional theory (DFT)
1. INTRODUCTION
Diabetes mellitus is considered a paradigm of a 21st-century chronic disease. Actually, the treatment is significantly dependent on the patients’ lifestyle and self- management behaviors. Gestational diabetes becomes a major public health concern, because its rate is increasing quickly worldwide [1]. WHO proposed that diabetes will be the 7th major disease responsible for most of the deaths by 2030. Insulin is considered to be the American Diabetes Association-recommended first-line therapy and the only pharmacologic treatment approved by the US Food and Drug Administration. In addition, several categories of drugs, such as sulfonylureas, biguanides, thiazolidinediones, meglitinides and α-glucosidase inhibitors, are used for type 2 diabetes [2]. Although good efficacies were received, however, some deleterious side effects are followed, such as liver toxicity and harmful gastrointestinal symptoms [3, 4].
Due to the commonly used α-glucosidase inhibitors were reported to be associated with rare adverse hepatic events [5], these drugs have low efficacy and high IC50 values [6, 7]. Therefore, much effort has been undertaken to search for more effective and safer inhibitors. Recently, new effective and safer inhibitors from natural products and their derivatives have continued [8, 9]. Many flavonoids have shown some considerable inhibitory effects against α-glucosidase enzymes, such as morin [10], luteolin [11], baicalein [12], kaempferol [13] and apigenin [7]. From the structure-activity analysis of flavonoids, it can be concluded that the position and number of hydroxyl groups on B rings can affect the inhibitory activity towards the α-glucosidase [14]. However, due to the low solubility in both water and lipid, the clinical utilities of most flavonoids are limited [15, 16].
Various derivatives of flavonoids are thus produced in order to improve their biological activities or bioavailabilities [17-19]. As an amphipathic group, carboxyl is always used to introduce into the molecules to improve their solubility in both water and lipid. It was reported that apigenin can increase its α-glucosidase inhibitory activities by coupling the carboxyalkyl group to the 7-position of the A ring, especially the long octanoic acid -(CH2)7 COOH group [20]. In fact, reports showed that many flavonoids connected with different hydroxyl groups have some inhibitory activity of α-glucosidase [14-22]. In the present study, the inhibitory activity and molecular interactions of four 7-carboxyalkyl flavonoids derivatives (Fig. 1) with different hydroxyl groups on B rings are compared, In addition, the molecular flexibility, charge density distribution and the molecular Electrostatic Potential (ESP) are also analyzed. Combined with quantum chemical calculations [23], Bader’s theory of atoms [24] in molecules and molecular docking [25], the structure and property of inhibitors are revealed, which are important to design new carboxyalkyl flavonoids derivatives with enhanced efficiencies and lower toxicities.
Fig. (1).
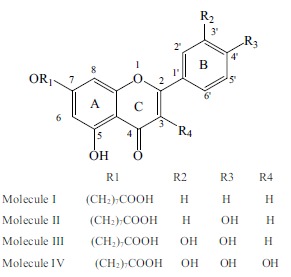
The planar structure of molecules I~IV.
2. MATERIALS AND METHODS
The geometry and energy of the molecules (I-IV) were optimized by using Gaussian 09 software [26]. Results indicated that DFT-B3LYP calculations have afforded excellent agreement with experimental analysis [27-29]. Based on the B3LYP (6-311G (d, p) basis) method, the optimized geometries were used for further analysis. In addition, tomasi's Polarized Continuum model (PCM) is employed in this text, which has emerged in the last two decades as the most effective tool to treat bulk solvent effects for both ground and excited states [30, 31]. By using the Multiwfn program, topology analysis of electron density and the molecular electrostatic potential surfaces in both forms were conducted [32]. The binding modes of molecules were detected by AutoDock 4.2 [33]. The docking grid box was set at 40 × 40 × 40 points with a spacing value of 0.375 Å. Genetic Algorithm with default settings was employed in the search parameter, the maximum number of evals was 2,500,000, and the number of runs was set at 100.
Because the crystal structure for S. cerevisiae α-glucosidase has not been available, the identity and similarity between S. cerevisiae isomaltase and target α-glucosidase enzyme are 72% and 85%, respectively. Homology modeling was proposed by using the crystal structure of S.cerevisiae isomaltase as the template [34, 35]. By using PROCHECK, the final structure of α-glucosidase generated from homology modeling was evaluated and suggested reliable results [36, 37]. It can be seen from the Ramachandran plot that 87.5% of residues of the final 3D structure lay in most favored regions (Supplementary Fig. 1). The protein of the S. cerevisiae isomaltase (PDBID: 3A4A) was download from the PDB website (http://www.rcsb.org). Protein sequence for Baker's yeast α-glucosidase (MAL12) was obtained from UniProt (http://www.uniprot.org). The developed structure was subjected to energy minimization up to 0.05 RMS gradients, and then used for the purpose of molecular docking.
Due to the lowest energy conformations from the docking study rarely represent bioactive conformations of ligands in the active site [38], 9 variables generated in the docking analysis were evaluated by chemometric techniques. Factors that can affect the bonding energies were: binding_energy, ligand_efficiency, intermol_energy, vdw_hb_desolv_energy, electrostatic_energy, unbounded_energy, cluster_RMS (the root mean square difference rms between this individual and the seed for the cluster) and reference_RMS (the rms between the specified reference structure) (marked as VAR00001-9, respectively). Chemometric techniques based on the principal component analysis (PCA) were used in this study [39]. The principal components (Yi) are given by the linear combination of the original variables (Xi) Eq. (1) [40]. Now, the system can be investigated by three new components (PC3, PC2 and PC1).
(1)
Usually, a well-known index of goodness of fit in multivariate data analysis is the percentage of explained variance by each PC (PC3, PC2 and PC1) for each molecule.
3. RESULTS AND DISCUSSION
3.1. Selection of the Molecular Conformations
Using molecular docking procedure, system configurations were generated. The lowest energy conformations cannot be used to investigate the reaction mechanism and further quantum chemistry analysis [41], because they rarely represent bioactive conformations in the active site. In addition, in the absence of the bioactive conformation from experimental data, it is desirable that the elected conformation can represent the average structure for interaction between ligand and receptor. Thus, the chemometric techniques were usually used to select the active conformation by using the principal component analysis (PCA) [42, 43].
In this text, these variables were evaluated in order to investigate the influence of the interaction energy values. As a result, the percentage of explained variance by each PC (PC1, PC2 and PC3) for molecule I, molecule II, molecule III and molecule IV are (55.6%, 13.9% and 11.8%), (59.6%, 12.6% and 11.4%), (54.2%, 12.3% and 11.8%) and (50.0%, 14.2% and 11.8%), respectively (Fig. 2). These values were regarded as well-known indexes of goodness of fit in multivariate data analysis. Based on the occupancy frequency, configurations that contribute less to the root-mean-square deviation of the interaction between ligand and receptor were proposed [43]. Fig. (3) displays the PCA results for the molecule I~IV with the presence of three principal components (81.3%, 83.6%, 78.3% and 76.0%, respectively). As a result, 42, 50, 60 and 36 were elected for molecules I~IV in the active site for further quantum chemistry calculations, respectively.
Fig. (2).
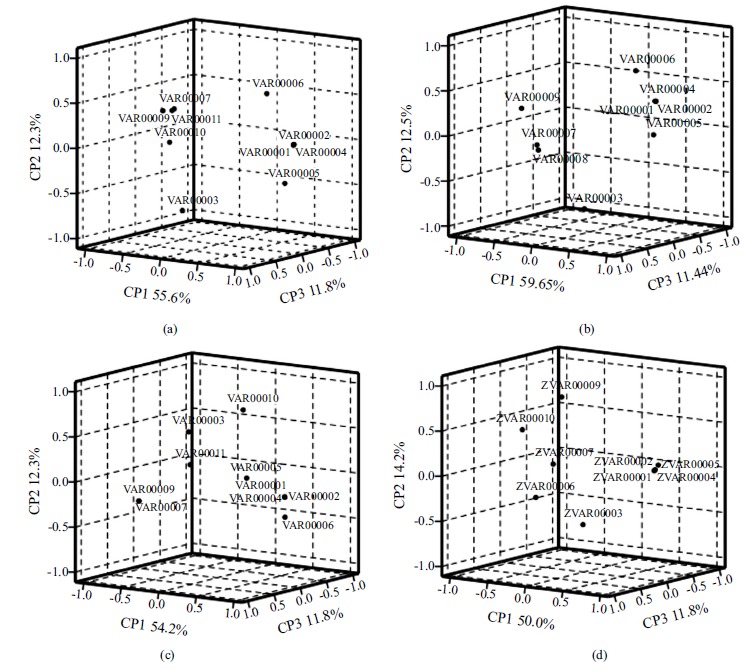
Loading graph of three molecules molecule I (a), molecule II (b), molecule III (c) and molecule IV (d).
Fig. (3).
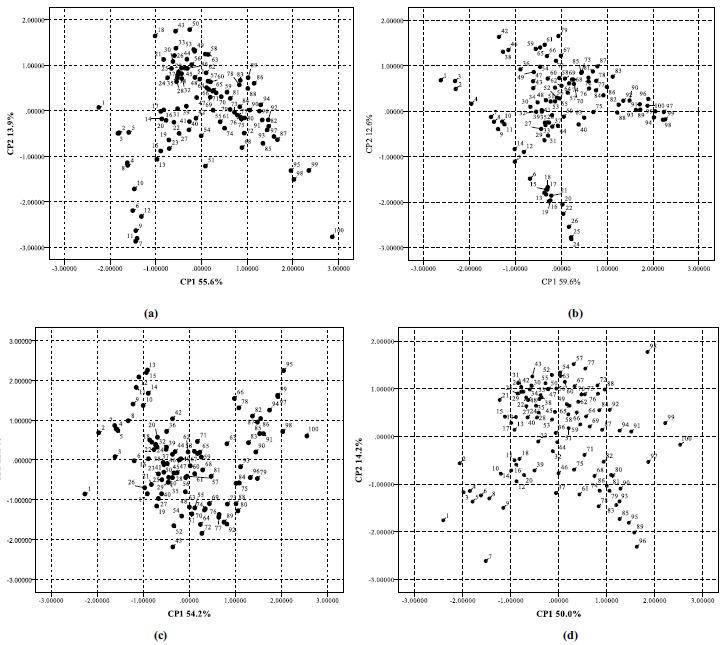
PC1 × PC2 projection to the yeast α-glucosidase and four molecules I~IV (a-d) systems.
3.2. Intermolecular Interactions
Results indicated that the selected binding affinities for the four complexes are -7.21, -7.52, -6.64 and -6.66 kcal/mol for molecule I~IV, respectively. Besides the number of hydroxyl groups, it is shown that their position also plays an important role in the inhibitory activity. According to the literature [44], the inhibition is reported to be related to the binding modes and binding affinities of the small molecules to the enzyme. Key residues are also considered to be a significant factor that can affect dynamical correlated motions, such as binding activities [45]. Nearest neighbors and short contact distances (Å) of the four flavones derivatives with the amino acid residues of S. cerevisiae α-glucosidase are shown in Table (1). It can be seen from Fig. (4), that the binding sites of the molecules are greatly consistent. However, the directions of the molecules are different, among which molecule II and IV are identical. Hydrogen bonds are depicted as green dotted lines, and the compounds are displayed as pink lines.
Table 1. Nearest neighbors and short contact distances (Å) of the four molecules and its derivatives with the amino acid residues of S. cerevisiae α-glucosidase.
| Name | Distance | Name | Distance | |
|---|---|---|---|---|
| Molecule I | Molecule II | |||
| H17...LEU176/CD | 3.010 | H18...ASP214/OD2 | 2.079 | |
| H17...SER244/HG | 2.256 | O42...HIS239/HE2 | 1.917 | |
| H31...SER244/HG | 3.136 | O38...ARG312/H | 1.675 | |
| H33...LYS/O | 2.947 | H21... GLN350/ OE1 | 2.194 | |
| H28...PHE311/H | 1.354 | H34... ASP349/OD1 | 2.528 | |
| H47...GLU304/CD | 2.277 | H30... ASP349/ OD1 | 2.373 | |
| H46...HIS279/CE1 | 2.685 | O19... GLN350/ OE1 | 2.500 | |
| H48...ARG312/HH12 | 1.946 | H29... ARG312/ HH11 | 2.983 | |
| Name | Distance | Name | Distance | |
| Molecule I | Molecule II | |||
| O41...VAL303/O | 3.447 | H47... ARG312 / CB | 2.399 | |
| H38...GLN350/OE1 | 2.034 | O38...ARG312/H | 1.675 | |
| - | - | H28... PHE157 / CE1 | 2.657 | |
| - | - | H32... PHE177 / CE1 | 3.498 | |
| - | - | O42...HIS239/HE1 | 1.917 | |
| Molecule III | - | Molecule IV | - | |
| O20...THR215/HG1 | 1.702 | H1...ASP214/OD1 | 2.037 | |
| H21...THE215/HG1 | 1.785 | H55...ASP214/OD1 | 1.954 | |
| O19...GLU276/OE1 | 3.079 | O32...GLU276/OE2 | 3.457 | |
| H55...GLU350/OE1 | 2.151 | O22...HIS279/HE2 | 2.159 | |
| H18...ASP349/O | 2.083 | O7...PHE157/CE2 | 3.076 | |
| H33...ARG439/HH11 | 1.519 | O30...ASP349/OD2 | 2.775 | |
| H31...PHE300/CE2 | 2.424 | O19...GLN350/OE1 | 2.731 | |
| O42...HIS239/HE2 | 1.920 | H21...GLN350/OE1 | 2.977 | |
| O42...LYS155/HZ1 | 2.716 | O20...ARG312/HH12 | 3.328 | |
| H59...PHE157/O | 2.945 | O38...HIS239/HE2 | 1.976 | |
| - | - | H39...PHE300/O | 2.587 | |
Fig. (4).
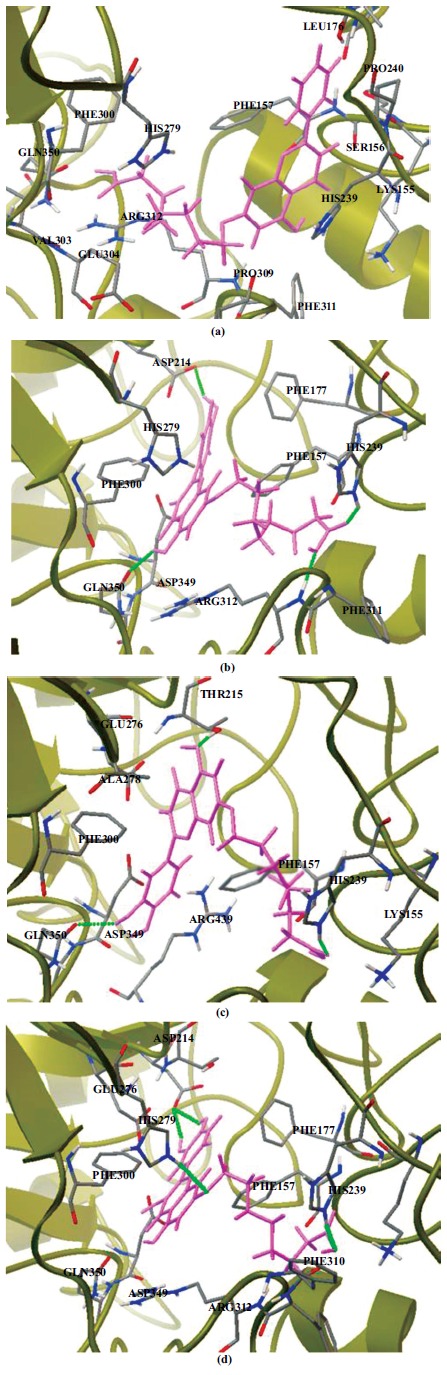
Intermolecular interactions between the selected compounds of molecule I~IV (a-d) and the α-glucosidase of S. cerevisiae, respectively. Hydrogen bonds are depicted as green dotted lines, and the compounds are displayed by pink lines.
The geometrical conformations of molecules in the optimal structure and the docking configurations are presented in Fig. (5). It can be seen from Fig. (5) that the conformations of molecules in solution phase are significantly different from the same found at the active site, indicating that these molecules are highly flexible. The most obvious change in these structures mainly occurs on the side chain of carboxyalkyl group. Detailed structural parameters are shown in Supplementary Table S1 (247.1KB, pdf) . For molecule I, there are obvious hydrophobic interactions between the hydrogen atoms of the B ring with the surrounding amino acids, such as Pro240, Leu176 and Phe157. The carboxyalkyl group of the side chain on the A-ring is embedded deep inside another binding cavity, which is composed ofHis279, Glu304, Phe300, Val303, Arg312 and Gln350. It is showed that almost all of the bond lengths and bond angles remain basically identical whether they interact with the protein or not. However, the dihedral angles change obviously. In the active site, although the carboxyalkyl group of the A ring is folded obviously, the A, B and C rings of molecule I are essentially planar. This leads to the intermolecular electrostatic interaction between the terminal carboxyl group and the amino acids Gln350, Arg312 and Val303.
Fig. (5).
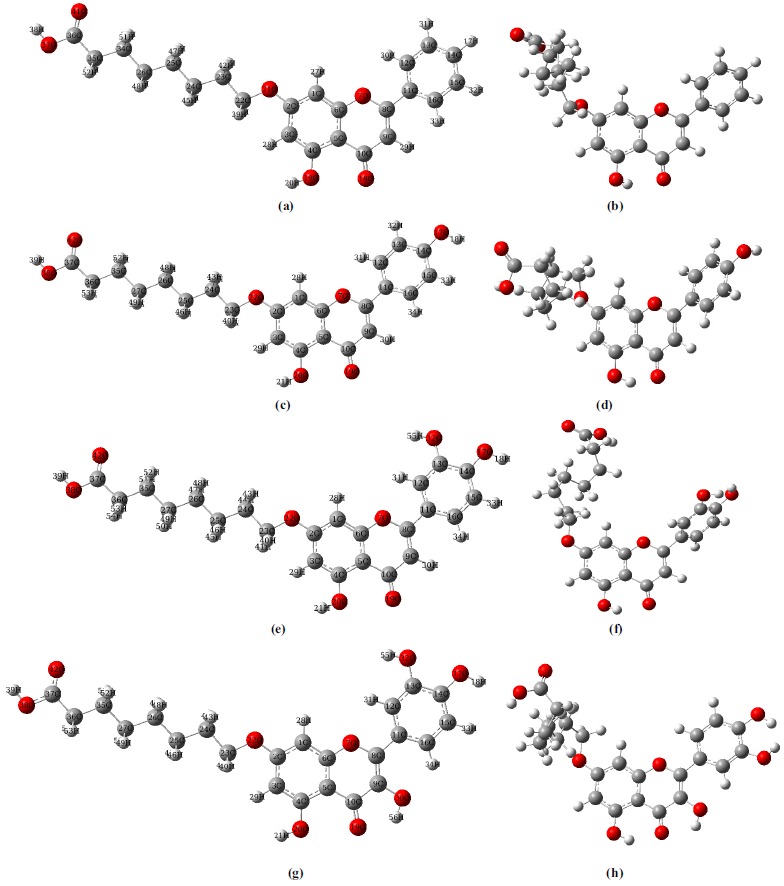
The geometrical conformations of molecules I~IV in the optimal structure (a, c, e, g) and the active phase (b, d, f, h). The atom numbering scheme of the molecules are correspondingly labeled in the optimal structure.
For molecule II, B ring is embedded deep inside a cavity consisted of Glu276, Thr215 and Phe177. The angle between B ring and A/C ring is significantly increased because of the torsion. Thus, the hydrogen atom H18 on B ring can interact with the oxygen atom OD2 of the amino acid Asp214 by strong hydrogen bonding interaction (2.079 Å). Similarly, the carboxyalkyl group of the A ring is folded obviously. There are two strong hydrogen-bonding interactions between the carbonyl oxygen atom O42 and the HE1 atom of His239 (1.917 Å), terminal oxygen atom O38 and the atom H of amino acid Arg312 (1.675 Å). The B ring of molecule III is embedded deep inside the binding cavity, which is surrounded by amino acids Asp349, Arg439 and Gln350. Compared with the solution phase, the rotational degree of the B ring is the largest and almost perpendicular to the plane of the A/C ring. Thus, the hydroxyl hydrogen atom on the B ring can form a strong hydrogen bonding interaction with the oxygen atom OE1 of the amino acid Gln350. In addition, there is an obvious π-π interaction between the B ring and His239.
The B ring of molecule IV is also embedded deep inside a binding cavity composed by the amino acids Asp349, Glu276, Asp214 and Phe177. Two hydrogen bonding interactions can be formed between the OD1 oxygen atom of Asp214 and the hydroxyl hydrogen atoms H55 (1.954 Å) and H18 (2.037 Å) of B ring, respectively. For molecules II~IV, generally, the hydroxyl groups of the B ring can form intermolecular hydrogen bonding forces with the surrounding amino acids, such as Asp214 and Gln350. This is consistent with the reported studies, which suggested that the position and number of hydroxyl groups may have the ability to make molecules bind into the binding pocket properly to maintain a high inhibitory activity [14-16]. The terminal hydroxyl oxygen O38 atom of the side chain on the molecule IV can also interact with the surrounding amino acid His239 by hydrogen bonding interaction (1.976 Å), which is similar to the molecule III. It was reported that quercetin has a considerable inhibitory activity to S. cerevisiae α-glucosidase (15±3μM) [14]. From these binding affinities, it is indicated that the position and number of hydroxyl groups on the B and C rings are also pivotal to the hypoglycemic activity when the long carboxyalkyl group is introduced into the A ring.
3.3. Topology Analysis of Electron Density
By using the Multiwfn program, the (3-1) type of bond critical point (bcp) for all bonds of the inhibitors (I~IV) were found, which suggested the existence of the covalent interactions [46]. Based on further topological analysis, the electron density (ρbcp), their Laplacian values (∇2ρbcp) at the bcp of all bonds were listed (Supplementary Table S2 (247.1KB, pdf) ). It can be seen from Figs. (6 a-d) that the Laplacian values of electron densities of the four molecules at active site are very close to the solution phase. The electron densities (ρbcp) of the C-C bonds on the aromatic ring for all molecules in solution forms ranged from 1.996 to 2.126eÅ-3, which are smaller than the reported values [47]. Most of the Laplacian values of electron densities decreased at the active site, whereas the changes were not significant. The highest electron density values of the four molecules (2.796~2.797 eÅ-3) can be found on the carbonyl C-O bonds of the parent ring in both situations. This is consistent with the reported results [48, 49].
Fig. (6).
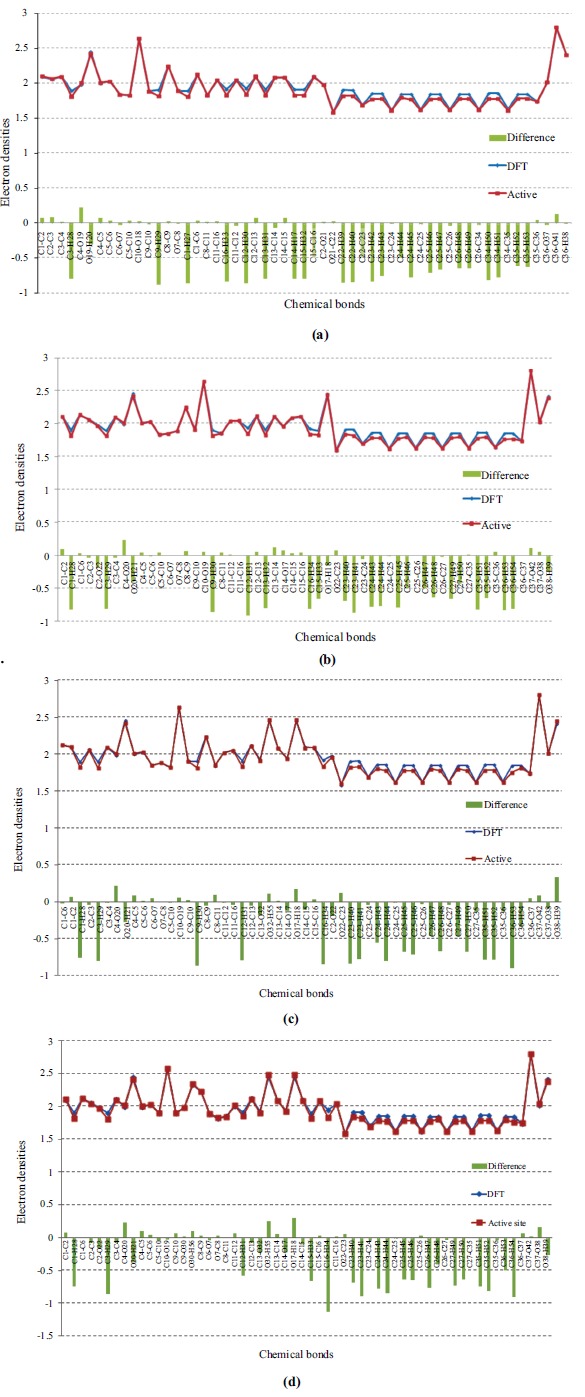
The electron densities of molecule I~IV (a-d) in the gas optimal phase (blue) and in the active site of S. cerevisiae α-glucosidase (red). Tenfold of the differences between the two states are also denoted as green columns correspondingly under the electron densities. The ordinate means the value of electron density, and the abscissa means the type of the bond sequentially.
It is suggested that the charge concentration/depletion at the bcp of the chemical bond can provide interesting information about the chemical bonds by using the Laplacian of electron density ∇2ρbcp [50, 51]. For molecule I, the Laplacian values of electron density for C-H bonds on the B ring increased about 1.863~2.039 eÅ-5. Acted as a hydrogen bond acceptor, the hydrogen atom H18 of the B ring on the molecule II can form intermolecular hydrogen bonding interaction (2.079 Å) with the OD2 atom of Asp214. Thus, the electron density of the chemical bond O17-H18 decreased slightly (~0.162 eÅ-3), whereas the corresponding Laplacian value increased about 2.251 eÅ-5. It can be concluded that the electron densities of chemical bonds became more depleted after interacting with the S. cerevisiae α-glucosidase, implying strong interactions with the surrounding amino acids [52].
For molecule III, the Laplacian value of the electron density of almost all chemical bonds on B ring increased, especially the C13-O32 (2.014 eÅ-5) and O32-H55 (2.861 eÅ-5), which may be related to the formation of intermolecular hydrogen bonds between H55 atom and the oxygen atom OE1 of amino acid Glu350. Due to the electrostatic interaction with the amino acids Asp349, Arg439 and Gln350, the electron density of the other hydroxyl group O17-H18 on B ring also increased about 2.076 eÅ-5, whereas the electron density of C14-O17 did not change significantly. The electron densities of B ring on molecule IV decreased, which indicated strong intermolecular interactions with the surrounding amino acids Phe177, Asp214, Glu276 and Asp349. The increased extents of the Laplacian values for the two hydroxyl groups O32-H55 and O17-H18 are higher than other molecules (about 3.396 and 2.766 eÅ-5), indicating that the electron densities become more depleted after docking with the S. cerevisiae α-glucosidase. This is due to the strong hydrogen bonding interactions between the OD1 atom of Asp214 and the two hydrogen atoms of the hydroxyl groups on B ring, which act as electron acceptors. Compared with others, molecule IV has an additional hydroxyl group on C ring; the electron density value of C9-O30 is higher than C9-H30 of other molecules, which may be related to the electrostatic interactions with amino acids Asp349, Gln350 and Phe300.
3.4. Molecular Electrostatic Potential
The molecular electrostatic potential is usually used to anticipate reactive sites for electrophilic and nucleophilic attacks [53]. In fact, the molecular electrostatic potential is related to the electronic density [54]. Molecular electrostatic potential can provide a three-dimensional visual method to understand the net electrostatic effect of a molecule. Two negative (red) and positive (blue) regions can be considered as electrophilic and nucleophilic reactivity regions, respectively (Fig. 7). The white regions are named as zero potential. It is important to note that the negative region for the electrophilic attack is at the vicinity of all oxygen atoms, which corresponds to an attraction of the proton by aggregating electron density in the molecules.
Fig. (7).
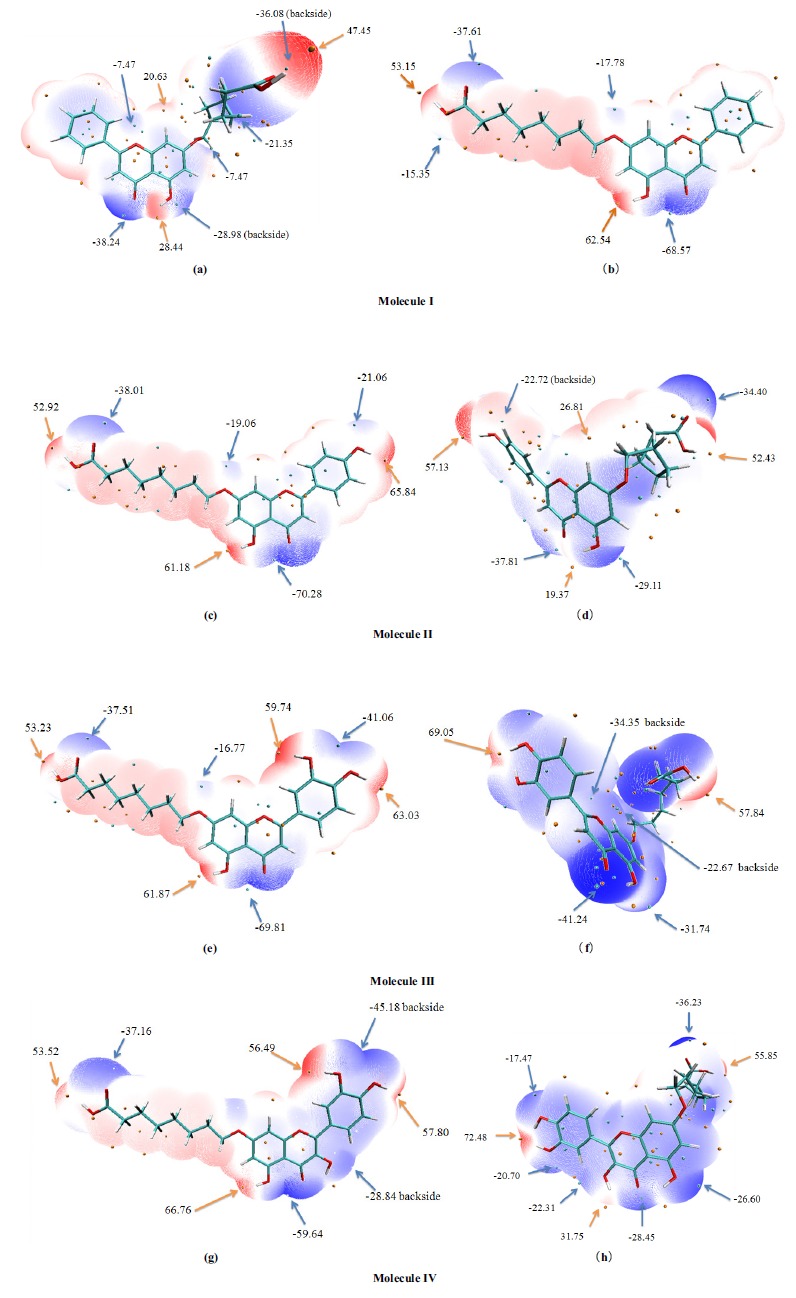
Molecular electrostatic potential on vdW surface of molecules I~IV (a-h). The unit is in Kcal/mol. Surface local minima and maxima of molecular electrostatic potential are represented as blue and orange spheres, respectively. The local surface maxima values and the global minimum and maximum are labeled by bold font. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
The distributions of molecular electrostatic potential on vdW surface for molecule I~IV in the solution phase are extremely similar. For molecule I, the minimum value corresponds to the carbonyl oxygen atom of the C ring (-68.57 kcal/mol). The maximum values for these four molecules are all at the vicinity of the hydroxyl hydrogen atom of the ring A (61.18~66.76 kcal/mol), among which, molecule IV remains the largest one. It was found that the largest molecular electrostatic potential surface minimum value of molecule IV is -59.64 kcal/mol, which is located near the carbonyl oxygen atom of the C ring. This may be related to the lone pair electrons of the oxygen atom. With the increase of the hydroxyl numbers on B ring, the molecular electrostatic potential on vdW surfaces of the molecule I~IV are significantly different. Due to none of the hydroxyl oxygen atom connected to the B ring of molecule I, there is no obvious distribution of the positive and negative molecular electrostatic potential. However, with the increase of the hydroxyl numbers, it can be seen from the figure, that the positive electrostatic potential is distributed near the hydroxyl hydrogen atom (57.80~65.84 kcal/mol), and the negative one is located at the vicinity of the hydroxyl oxygen atom. There are two hydroxyl groups connected to molecule III and IV; the negative electrostatic potential energy near the hydroxyl oxygen atom is significantly increased (-41.06 and -45.18 kcal/mol, respectively).
After interacting with the S. cerevisiae α-glucosidase, it can be found from the Fig. (4) that all geometric conformations of these molecules changed obviously. The side chain of the A ring has been folded at different degrees, and the angle between B ring and A ring generated correspondingly. From the surface electrostatic potential of the molecules, it can be seen that the minimum values increased (-41.24~-28.45 kcal/mol), especially molecule IV. This may be related to the presence of unique hydroxyl oxygen atoms on C ring, which will influence the negative molecular electrostatic potential near carbonyl oxygen atom (-28.45 kcal/mol). Both of the positive and negative values of the hydroxyl hydrogen atom and oxygen atoms of the carboxyl group reduced simultaneously, implying strong interactions with the surrounding amino acids. When the number of hydroxyl groups on B ring is two, the positive electrostatic potential value are increased significantly at the vicinity of the hydroxyl hydrogen atom, especially the molecule IV. This may be related to the strong hydrogen bonding interactions of the two-hydroxyl hydrogen atoms with amino acid Asp214, which may lead to the flow of chemical electrons.
CONCLUSION
The introduction of hydroxyl group to the flavonoid ring undoubtedly increases the hydrogen bonding interaction between the hydroxyl group and the surrounding amino acids, which may reduce the intermolecular binding energy and increase the inhibitory activity [14]. When the number of hydroxyl group on the B ring is increased to two, the inhibitory ability to S. cerevisiae α-glucosidase is quiet strong, such as quercetin[14]. However, due to the hydrophobic N-heptane alkyl and terminal hydrophilic carboxyl group appeared on the 7-position, the intermolecular interactions at the active site were affected. It was reported that the introduction of a carboxyl alkyl group into 7-OH of apigenin can enhance its binding with α-glucosidase and increase the α-glucosidase inhibitory activity [20], which is consistent with our docking results. Results showed that the electron density of the chemical bond C14-O17 of molecule II increases (~0.075 eÅ-3) while chemical bond O17-H18 decreases (~0.162 eÅ-3) at the active site. However, for molecule III, the electron density of two chemical bonds C-O of B-ring hydroxyl groups decreased (0.186 eÅ-3 and 0.164 eÅ-3), and increased on the chemical bond O-H (0.109 eÅ-3 and 0.174 eÅ-3). The similar situation was observed on molecule IV. This shows that para-hydroxyl group of the B ring not only forms an intermolecular hydrogen bonding interaction with the amino acid Asp214, but also has a strong electrostatic interaction with surrounding amino acids Glu276, Thr215 and His111. Amino acids Phe157 and Phe177 maintain the molecule at the active site by forming hydrophobic interactions with the alkyl group. When the hydroxyl group is connected to B ring, it can be seen from Fig. (7d) that the lowest positive electrostatic potentials are distributed near the hydroxyl hydrogen atom (57.13 Kcal/mol) and carboxyl hydrogen atom (52.43 Kcal/mol).
Compounds with a large number of hydroxyl groups may have more solvent interactions [55], however, from this docking analysis, it is concluded that the position and number of hydroxyl groups on the carboxyalkyl flavonoid B and C rings are important to the hypoglycemic activity. It is shown that, from the structure-activity relationship study, quercetin is the most active flavonoids (IC50=15±3μM), whose structural features include the presence of two hydroxyl groups located strategically on ring A and B, and one on ring C [14]. When carboxyalkyl group is introduced into A ring, the number of hydroxyl groups should not be an important factor, whereas their position proves to be a relevant factor, showing that the presence of three well-defined zones in the structure, a hydrophobic long alkyl and carboxyalkyl group on A ring, and a hydrophilic one on B ring, are necessary. In other words, it appears that the presence of one hydroxyl groups located strategically on ring B and the absence of polar groups on ring C are of great importance for the hypoglycemic of the carboxyalkyl flavonoids. However, further biological tests are needed to be done to validate the computational predictions.
ACKNOWLEDGEMENTS
Declared None.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
This work was supported by the Fundamental Research Funds for the Central Universities (31920190155).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.
REFERENCES
- 1.Camelo Castillo W., Boggess K., Stürmer T., Brookhart M.A., Benjamin D.K., Jr, Jonsson Funk M. Trends in glyburide compared with insulin use for gestational diabetes treatment in the United States, 2000-2011. Obstet. Gynecol. 2014;123(6):1177–1184. doi: 10.1097/AOG.0000000000000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qaseem A., Humphrey L.L., Sweet D.E., Starkey M., Shekelle P. Clinical Guidelines Committee of the American College of Physicians. Oral pharmacologic treatment of type 2 diabetes mellitus: A clinical practice guideline from the American College of Physicians. Ann. Intern. Med. 2012;156(3):218–231. doi: 10.7326/0003-4819-156-3-201202070-00011. [DOI] [PubMed] [Google Scholar]
- 3.Thiyagarajan G., Muthukumaran P., Sarath Kumar B., Muthusamy V.S., Lakshmi B.S. Selective Inhibition of PTP1B by Vitalboside A from syzygium cumini enhances insulin sensitivity and attenuates lipid accumulation via partial agonism to pparγ: In Vitro and In Silico investigation. Chem. Biol. Drug Des. 2016;88(2):302–312. doi: 10.1111/cbdd.12757. [DOI] [PubMed] [Google Scholar]
- 4.Etxeberria U., de la Garza A.L., Campión J., Martínez J.A., Milagro F.I. Antidiabetic effects of natural plant extracts via inhibition of carbohydrate hydrolysis enzymes with emphasis on pancreatic alpha amylase. Expert Opin. Ther. Targets. 2012;16(3):269–297. doi: 10.1517/14728222.2012.664134. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L., Chen Q., Li L., Kwong J.S., Jia P., Zhao P., Wang W., Zhou X., Zhang M., Sun X. Alpha-glucosidase inhibitors and hepatotoxicity in type 2 diabetes: A systematic review and meta-analysis. Sci. Rep. 2016;6:32649–32618. doi: 10.1038/srep32649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim M.A., Bester M.J., Neitz A.W.H., Gaspar A.R.M. Structural properties of bioactive peptides with α-glucosidase inhibitory activity. Chem. Biol. Drug Des. 2018;91(2):370–379. doi: 10.1111/cbdd.13105. [DOI] [PubMed] [Google Scholar]
- 7.Zeng L., Zhang G., Lin S., Gong D. Inhibitory mechanism of apigenin on α-glucosidase and synergy analysis of flavonoids. J. Agric. Food Chem. 2016;64(37):6939–6949. doi: 10.1021/acs.jafc.6b02314. [DOI] [PubMed] [Google Scholar]
- 8.Mokale S.N., Palkar A.D., Dube P.N., Sakle N.S., Miniyar P.B. Design, synthesis and in vivo screening of some novel quinazoline analogs as anti-hyperlipidemic and hypoglycemic agents. Bioorg. Med. Chem. Lett. 2016;26(2):272–276. doi: 10.1016/j.bmcl.2015.12.037. [DOI] [PubMed] [Google Scholar]
- 9.Mosihuzzman M., Naheed S., Hareem S., Talib S., Abbas G., Khan S.N., Choudhary M.I., Sener B., Tareen R.B., Israr M. Studies on α-glucosidase inhibition and anti-glycation potential of Iris loczyi and Iris unguicularis. Life Sci. 2013;92(3):187–192. doi: 10.1016/j.lfs.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 10.Zeng L., Zhang G., Liao Y., Gong D. Inhibitory mechanism of morin on α-glucosidase and its anti-glycation properties. Food Funct. 2016;7(9):3953–3963. doi: 10.1039/C6FO00680A. [DOI] [PubMed] [Google Scholar]
- 11.Yan J., Zhang G., Pan J., Wang Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014;64:213–223. doi: 10.1016/j.ijbiomac.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Gao H., Nishioka T., Kawabata J., Kasai T. Structure-activity relationships for alpha-glucosidase inhibition of baicalein, 5,6,7-trihydroxyflavone: The effect of A-ring substitution. Biosci. Biotechnol. Biochem. 2004;68(2):369–375. doi: 10.1271/bbb.68.369. [DOI] [PubMed] [Google Scholar]
- 13.Na B., Nguyen P.H., Zhao B.T., Vo Q.H., Min B.S., Woo M.H. Protein tyrosine phosphatase 1B (PTP1B) inhibitory activity and glucosidase inhibitory activity of compounds isolated from Agrimonia pilosa. Pharm. Biol. 2016;54(3):474–480. doi: 10.3109/13880209.2015.1048372. [DOI] [PubMed] [Google Scholar]
- 14.Proença C., Freitas M., Ribeiro D., Oliveira E.F.T., Sousa J.L.C., Tomé S.M., Ramos M.J., Silva A.M.S., Fernandes P.A., Fernandes E. α-Glucosidase inhibition by flavonoids: An in vitro and in silico structure-activity relationship study. J. Enzyme Inhib. Med. Chem. 2017;32(1):1216–1228. doi: 10.1080/14756366.2017.1368503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Zuo Z., Lin G. Intestinal and hepatic glucuronidation of flavonoids. Mol. Pharm. 2007;4(6):833–845. doi: 10.1021/mp700077z. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Liu D., Huang Y., Gao Y., Qian S. Biopharmaceutics classification and intestinal absorption study of apigenin. Int. J. Pharm. 2012;436(1-2):311–317. doi: 10.1016/j.ijpharm.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Kim M.K., Park K.S., Lee C., Park H.R., Choo H., Chong Y. Enhanced stability and intracellular accumulation of quercetin by protection of the chemically or metabolically susceptible hydroxyl groups with a pivaloxymethyl (POM) promoiety. J. Med. Chem. 2010;53(24):8597–8607. doi: 10.1021/jm101252m. [DOI] [PubMed] [Google Scholar]
- 18.Wen X., Walle T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab. Dispos. 2006;34(10):1786–1792. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]
- 19.Hakamata W., Nakanishi I., Masuda Y., Shimizu T., Higuchi H., Nakamura Y., Saito S., Urano S., Oku T., Ozawa T., Ikota N., Miyata N., Okuda H., Fukuhara K. Planar catechin analogues with alkyl side chains: A potent antioxidant and an alpha-glucosidase inhibitor. J. Am. Chem. Soc. 2006;128(20):6524–6525. doi: 10.1021/ja057763c. [DOI] [PubMed] [Google Scholar]
- 20.Su Z.R., Fan S.Y., Shi W.G., Zhong B.H. Discovery of xanthine oxidase inhibitors and/or α-glucosidase inhibitors by carboxyalkyl derivatization based on the flavonoid of apigenin. Bioorg. Med. Chem. Lett. 2015;25(14):2778–2781. doi: 10.1016/j.bmcl.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Zhen J., Dai Y., Villani T., Giurleo D., Simon J.E., Wu Q. Synthesis of novel flavonoid alkaloids as α-glucosidase inhibitors. Bioorg. Med. Chem. 2017;25(20):5355–5364. doi: 10.1016/j.bmc.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z., Ma S. Recent advances in synthetic α-glucosidase inhibitors. ChemMedChem. 2017;12(11):819–829. doi: 10.1002/cmdc.201700216. [DOI] [PubMed] [Google Scholar]
- 23.Leech A.R. Molecular Modelling. Essex: Longman; 1997. [Google Scholar]
- 24.Bader R.F.W. Atoms in Molecules: A Quantum Theory. Oxford, U.K.: Oxford University Press; 1990. [Google Scholar]
- 25.Guisasola E.E.B., Gutierrez L.J., Salcedo R.E., Garibotto F.M., Andujar S.A., Enriz R.D., Rodriguez A.M. Conformational transition of A beta(42) inhibited by a mimetic peptide. A molecular modeling study using QM/MM calculations and QTAIM analysis. Comput. Theor. Chem. 2016;1080:56–65. doi: 10.1016/j.comptc.2016.02.002. [DOI] [Google Scholar]
- 26.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian 09, Revision E.01. Wallingford: Gaussian Inc.; 2009. [Google Scholar]
- 27.Deeb O., Clare B.W. Comparison of AM1 and B3LYP-DFT for inhibition of MAO-A by phenylisopropylamines: A QSAR study. Chem. Biol. Drug Des. 2008;71(4):352–362. doi: 10.1111/j.1747-0285.2008.00643.x. [DOI] [PubMed] [Google Scholar]
- 28.Evidente M., Santoro E., Petrovic A.G., Cimmino A., Koshoubu J., Evidente A., Berova N., Superchi S. Absolute configurations of phytotoxic inuloxins B and C based on experimental and computational analysis of chiroptical properties. Phytochemistry. 2016;130:328–334. doi: 10.1016/j.phytochem.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Katan C., Tretiak S., Werts M.H., Bain A.J., Marsh R.J., Leonczek N., Nicolaou N., Badaeva E., Mongin O., Blanchard-Desce M. Two-photon transitions in quadrupolar and branched chromophores: experiment and theory. J. Phys. Chem. B. 2007;111(32):9468–9483. doi: 10.1021/jp071069x. [DOI] [PubMed] [Google Scholar]
- 30.Yurdakul S., Yurdakul M.F.T-I.R. FT-Raman spectra, and DFT computations of the vibrational spectra and molecular geometry of chlorzoxazone. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014;126:339–348. doi: 10.1016/j.saa.2014.02.156. [DOI] [PubMed] [Google Scholar]
- 31.Iramain M.A., Davies L., Brandán S.A. FTIR, HATR and FT-Raman studies on the anhydrous and monohydrate species of maltose in aqueous solution. Carbohydr. Res. 2016;428:41–56. doi: 10.1016/j.carres.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 32.Lu T., Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012;33(5):580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 33.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakayanathan P., Loganathan C., Iruthayaraj A., Periyasamy P., Poomani K., Periasamy V., Thayumanavan P. Biological interaction of newly synthesized astaxanthin-s-allyl cysteine biconjugate with Saccharomyces cerevisiae and mammalian α-glucosidase: In vitro kinetics and in silico docking analysis. Int. J. Biol. Macromol. 2018;118:252–262. doi: 10.1016/j.ijbiomac.2018.06.027. [DOI] [PubMed] [Google Scholar]
- 35.Jabeen F., Shehzadi S.A., Fatmi M.Q., Shaheen S., Iqbal L., Afza N., Panda S.S., Ansari F.L. Synthesis, in vitro and computational studies of 1,4-disubstituted 1,2,3-triazoles as potential α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016;26(3):1029–1038. doi: 10.1016/j.bmcl.2015.12.033. [DOI] [PubMed] [Google Scholar]
- 36.Qi Y.J., Zhao Y.M., Lu H.N., Wang X.E., Jin N.Z. Comparative analysis of the bonding modes between two antidiabetic drugs with beta-glucosidases from different species. Indian J. Pharm. Sci. 2016;78(4):525–536. doi: 10.4172/pharmaceutical-sciences.1000146. [DOI] [Google Scholar]
- 37.Yar M., Bajda M., Shahzadi L., Shahzad S.A., Ahmed M., Ashraf M., Alam U., Khan I.U., Khan A.F. Novel synthesis of dihydropyrimidines for α-glucosidase inhibition to treat type 2 diabetes: In vitro biological evaluation and in silico docking. Bioorg. Chem. 2014;54:96–104. doi: 10.1016/j.bioorg.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 38.van Bochove M.A., Swart M., Bickelhaupt F.M. Stepwise walden inversion in nucleophilic substitution at phosphorus. Phys. Chem. Chem. Phys. 2009;11(2):259–267. doi: 10.1039/B813152J. [DOI] [PubMed] [Google Scholar]
- 39.Bender A., Jenkins J.L., Scheiber J., Sukuru S.C., Glick M., Davies J.W. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009;49(1):108–119. doi: 10.1021/ci800249s. [DOI] [PubMed] [Google Scholar]
- 40.La Porta F.A., Santiago R.T., Ramalho T.C., Freitas M.P., da Cunha E.F.F. The Role of the frontier orbitals in acid-base chemistry of organic amines probed by ab initio and chemometric techniques. Int. J. Quantum Chem. 2010;110:2015–2023. doi: 10.1002/qua.22676. [DOI] [Google Scholar]
- 41.Van Bochove M.A., Bickelhaupt F.M. Nucleophilic substitution at C, Si and P: How solvation affects the shape of reaction profiles. Eur. J. Org. Chem. 2008;4:649–654. doi: 10.1002/ejoc.200700953. [DOI] [Google Scholar]
- 42.de Lima W.E.A., Pereira A.F., de Castro A.A., da Cunha E.F.F., Ramalho T.C. Flexibility in the molecular design of acetylcholinesterase reactivators: Probing representative conformations by chemometric techniques and docking/QM calculations. Lett. Drug Des. Discov. 2016;13:360–371. doi: 10.2174/1570180812666150918191550. [DOI] [Google Scholar]
- 43.Kessler J., Dračínský M., Bouř P. Parallel variable selection of molecular dynamics clusters as a tool for calculation of spectroscopic properties. J. Comput. Chem. 2013;34(5):366–371. doi: 10.1002/jcc.23143. [DOI] [PubMed] [Google Scholar]
- 44.Devipriya B., Kumaradhas P. Molecular flexibility and the electrostatic moments of curcumin and its derivatives in the active site of p300: A theoretical charge density study. Chem. Biol. Interact. 2013;204(3):153–165. doi: 10.1016/j.cbi.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Yi X., Zhang Y., Wang P., Qi J., Hu M., Zhong G. Ligands binding and molecular simulation: The potential investigation of a biosensor based on an insect odorant binding protein. Int. J. Biol. Sci. 2015;11(1):75–87. doi: 10.7150/ijbs.9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L., Li T., Shen Y., Song Y. A theoretical study of the electronic structure and charge transport properties of thieno[2,3-b]benzothiophene based derivatives. Phys. Chem. Chem. Phys. 2016;18(12):8401–8411. doi: 10.1039/C5CP07879B. [DOI] [PubMed] [Google Scholar]
- 47.Devipriya B., Kumaradhas P. Probing the effect of intermolecular interaction and understanding the electrostatic moments of anacardic acid in the active site of p300 enzyme via DFT and charge density analysis. J. Mol. Graph. Model. 2012;34:57–66. doi: 10.1016/j.jmgm.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Qi Y.J., Lu H.N., Zhao Y.M., Jin N.Z. Probing the influence of carboxyalkyl groups on the molecular flexibility and the charge density of apigenin derivatives. J. Mol. Model. 2017;23(3):70. doi: 10.1007/s00894-017-3221-3. [DOI] [PubMed] [Google Scholar]
- 49.Yearley E.J., Zhurova E.A., Zhurov V.V., Pinkerton A.A. Binding of genistein to the estrogen receptor based on an experimental electron density study. J. Am. Chem. Soc. 2007;129(48):15013–15021. doi: 10.1021/ja075211j. [DOI] [PubMed] [Google Scholar]
- 50.Srinivasan P., David Stephen A., Kumaradhas P. Effect of gold atom contact in conjugated system of one dimensional octane dithiolate based molecular wire: A theoretical charge density study. J. Mol. Struct. THEOCHEM. 2009;910:112–121. doi: 10.1016/j.theochem.2009.06.026. [DOI] [Google Scholar]
- 51.David Stephen A., Revathi M., Asthana S.N., Pawar Rajesh B., Kumaradhas P. Probing the weakest bond and the cleavage of p-chlorobenzaldehyde diperoxide energetic molecule via quantum chemical calculations and theoretical charge density analysis. Int. J. Quantum Chem. 2011;111:3741–3754. [Google Scholar]
- 52.Renuga Parameswari A., Rajalakshmi G., Kumaradhas P. A combined molecular docking and charge density analysis is a new approach for medicinal research to understand drug-receptor interaction: curcumin-AChE model. Chem. Biol. Interact. 2015;225:21–31. doi: 10.1016/j.jmgm.2011.12.003. doi: 10.1016/j.jmgm.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Ren F.D., Cao D.L., Shi W.J., Gao H.F. A theoretical prediction of the relationships between the impact sensitivity and electrostatic potential in strained cyclic explosive and application to H-bonded complex of nitrocyclohydrocarbon. J. Mol. Model. 2016;22(4):97. doi: 10.1007/s00894-016-2967-3. [DOI] [PubMed] [Google Scholar]
- 54.Selvaraju K., Jothi M., Kumaradhas P. Understanding the charge density distribution and the electrostatic properties of hexadecane molecular nanowire under electric field using DFT and AIM theory. Comput. Theor. Chem. 2012;992:9–17. doi: 10.1016/j.comptc.2012.04.019. [DOI] [Google Scholar]
- 55.Echeverría J., Opazo J., Mendoza L., Urzúa A., Wilkens M. Structure-Activity and lipophilicity relationships of selected antibacterial natural flavones and flavanones of chilean flora. Molecules. 2017;22(4):E608. doi: 10.3390/molecules22040608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s website along with the published article.
Data Availability Statement
Not applicable.