

Abstract
Introduction
Androgen Receptor (AR) plays a pivotal role in the development of male sex and contributes to prostate cancer growth. Different from other nuclear receptors that bind to the co-regulator LxxLL motif in coregulator peptide interaction, the AR Ligand Binding Domain (LBD) prefers to bind to the FxxLF motif. BUD31, a novel co-regulator with FxxLF motif, has been demonstrated to suppress wild-type and mutated AR-mediated prostate cancer growth.
Methods
To find out the interaction mechanisms of BUD31 with WT/T877A/W741L AR complex, molecular dynamics simulations were employed to study the complex BUD31 and WT/mutant ARs. The molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) results demonstrated that T877A and W741L point mutations can reduce the binding affinity between BUD31 and AR. The RMSF and dynamic cross-correlation analysis indicated that amino acid point mutations can affect the motions of loop residues in the AR structure.
Results
These results indicated that AR co-regulator binding site AF2 can serve as a target for drug discovery to solve the resistance problem.
Keywords: BUD31, Androgen receptor, AF2 binding site, interaction mechanism, molecular dynamics, co-regulator
1. INTRODUCTION
Androgen Receptor (AR) plays a crucial role in the development and progress of male reproductive tissues [1, 2]. As a member of ligand-inducible transcription factor of the Nuclear Receptor (NR) superfamily, AR shares a common structural and functional organization with other members, including an N-Terminal Domain (NTD), a DNA-binding domain (DBD) and a C-terminal Ligand-Binding Domain (LBD) containing Activation Function 2 (AF-2) [3-5]. Upon binding of the endogenous ligands, testosterone or 5α-dihydrotestosterone (DHT), AR LBD undergoes conformational changes leading to the formation of a hydrophobic co-regulator binding groove in the AF-2 of LBD [6]. This groove acts as a high-affinity binding site for a short α-helix FxxLF motif, Presenting in the AR N-terminal domain and in AR co-regulators, which can be used as a harbor to accommodate the bulky peptide side chains. AR regulates the expression of target genes through binding to the specific Androgen Response Elements (ARE) after translating into the nucleus. Meanwhile, co-regulators bind to the AF-2 to enhance AR transcription function by histone modifications, chromatin remodeling, proteasomal degradation, DNA repair and bridging of the receptor to other components of the transcription initiation process, including general transcription factors and RNA polymerase II [7-9].
It has become clear that the transcriptional activity of AR is modulated by a co-regulatory protein. Co-regulators interact with the AR to either enhance transcription or reduce the transcription of target genes but do not alter the essential transcription rate. Previous research demonstrated that AR prefers to bind aromatic-rich FxxLF motif [10-12]. Studies of the first (+1) and the fifth (+5) amino acid positions of the FxxLF motif indicate that these residues are involved in mediating high affinity and specific interactions with the AR ligand-binding domain [13]. In fact, the FxxLF-like motif exists in many AR co-regulators and plays a significant role in modulating co-regulator-mediated AR functions [14-17]. However, peptides containing a tyrosine in the +5 position of the motif were frequently identified in the screened peptides [17] by the combined phage-display screening and X-ray crystal structure analysis of the wild-type AR-DBD-LBD (WT AR-DBD-LBD) and T877A mutant AR-DBD-LBD (T877A AR-DBD-LBD) cofactor-binding grooves. An anti-AR BUD31 containing an FxxFY motif was also identified, which was found to interact tightly with AR (Fig. 1). The corresponding experimental results indicate that BUD31 can block AR transcriptional activity, the AR N-C interaction and AR-mediated cancer cell growth [18-21].
Fig. (1).

The structure of wild type AR and peptide BUD31. a: The Ribbon structure of WT AR-BUD31 complex. b: The surface of WT AR-BUD31 complex. c: The active site of WT AR.
Here we report the structural basis of FxxFY motif binding to both WT and mutant ARs. Molecular dynamics simulations were employed as a promising tool to understand the influence of point mutations on the binding of peptide BUD31 (Sequence: FDLFY) with AR. Key residues interacting with BUD31 were obtain and the important H-bonds were analyzed. Furthermore, the binding affinity was also calculated.
2. MATERIALS AND METHODS
2.1. System Preparations
The initial AR structure together with peptide BUD31 was obtained from the Protein Data Bank (PDB code: 4OED, 4OH6 and 4OKB) [22]. Crystal water molecules and heteroatom were deleted, and missing residues were complemented by preparing a ligand module in Discover studio 2.5. All of the hydrogen atoms were added using LEaP module in the AMBER 12 software [23]. The AMBER ff99 force field [24] was used as the parameter for all of the amino acid residues. The appropriate number of chloride counterions were added to maintain the electric neutrality of all the systems and then each system was immersed into a hexahedral box using TIP3P water molecules with at least 10Å distance around the complex [25].
2.2. Molecular Dynamics Simulations
To carry out MD simulations, the topology and coordinate files of the three complexes were built with LEaP module of the AMBER 12 package. Subsequently, the first minimization was performed using the steepest descent method that switched to a conjugate gradient every 2500 steps for a total of 5000 steps with 0.1 kcal/mol Å-2 restraints on the atoms of the complexes. The other two energy minimization steps were performed by only restraining the protein and further releasing all the restraints for 5000 steps of each round. After that, the system was gradually heated from 0 to 310 K over 500 ps using a Langevin thermostat with a coupling coefficient of 2.0 ps. Later on, MD equilibration of 50 ps each was performed with restraint weight 0.5 kcal/mol Å-2. Eventually, 50 ns production MD simulations were performed without any restraint on these three systems in the isothermal-isobaric (NPT) ensemble at a temperature of 310 K and a pressure of 1 atm. All MD phases were carried out in the NPT ensemble using a Berendsen barostat [26] with a target pressure of 1 bar and a pressure coupling constant of 2.0 ps. Long-range Coulombic interactions were analysed using the Particle Mesh Ewald (PME) [27, 28] summation. For the equilibrium and production phase of simulation, the SHAKE algorithm [29] was employed on all atoms covalently bonded to a hydrogen atom, allowing for an integration time step of 2 fs.
2.3. Binding Free Energy Calculations
The binding free energy and corresponding components of WT and its mutations were calculated by MM-PBSA methods [30]. At first, 5000 snapshots were taken from the last 5ns trajectory with an interval of 10 ps and the counterions and water molecules were stripped. Subsequently, MM-PBSA methods were employed to compute the difference in the binding free energy between WT and its mutations. In general, the complex approximate free energy was calculated by using the following equations:
ΔGbind = ΔEMM + ΔGsol – TΔS (1)
ΔEMM = ΔEval + ΔEele + ΔEvdw (2)
ΔGsol = ΔGPB + ΔGnp (3)
ΔGnp = γSASA + β (4)
Here, ΔGbind was considered as the sum of the change in the molecular mechanical (MM) gas-phase binding energy (ΔEMM), solvation energy (ΔGsol), and entropy term (– TΔS) (Eq. 1). The ΔEMM is calculated by (Eq. 2), where ΔEval, ΔEele and ΔEvdw represent the internal energy contribution from bonds, angles and torsions, electrostatic and van der Waals interactions, respectively, computed by using the same parameter set as used in the MD simulation. The solvation free energy
consist of two parts, ΔGPB and ΔGnp (Eq. 3). The electrostatic contribution to the solvation free energy is calculated by the Poisson-Boltzmann (PB) method using the Delphi program. The grid spacing was set to 0.5 Å. The nonpolar solvation contribution is described in (Eq. 4), where SASA is the solvent-accessible surface area and the solvation parameters, γ and β, which were set to 0.00542 kcal/mol Å-2 and 0.92 kcal/mol, respectively [31].
2.4. Simulation Analysis
In order to evaluate the correlation motion of residues caused by T877A and W741L mutations, cross-correlation analysis was employed to explore the fluctuation in the coordinates of the Cα atoms relative to their average positions from the last 5 ns of the simulations. Moreover, hydrogen bond analysis was also used to investigate the binding affinity, including bond, angle and probability. The hydrogen bond criteria include an acceptor-donor distance < 3.0 Å, and acceptor…H-donor angle > 135°. In addition, the average structure and hydrophobic surface were used to explore the binding mechanism of the protein and the peptide.
3. RESULTS
3.1. The Stability of the Studied Systems
To obtain the equilibrated and stable systems, fifty nanoseconds molecular dynamics simulations for three systems (WT-BUD31, T877A-BUD31 and W741L-BUD31) were performed. The root mean square deviation (RMSD) of backbone Cα atoms of the protein reference the structure of production dynamics were calculated and plotted in Fig. (2). In Fig. (2), the RMSD of the backbones of WT-BUD31, T877A-BUD31 and W741L-BUD31 show small fluctuations in the whole production process, indicating that the studied system reached stabilization.
Fig. (2).
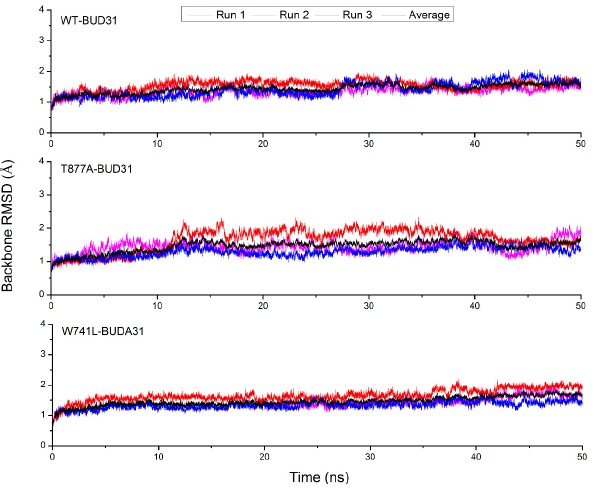
The time evolution of the RMSD values of the backone atoms of the BUD31-WT/mutant AR.
To further explore the conformation change of the protein induced by the two mutations, Root Mean Square Fluctuations (RMSF) analysis was also employed to study the stability of the systems. The structure of the AR ligand binding domain consists of 250 residues: the loop region consist of 681-695, 722-723, 759-770, 779, 798-799, 814-822, 842-851, 889-891, and 909-918, while the binding pocket includes L712, V713, V716, K717, A719, K720. F725, H729, V730, D731, Q733, M734, I737, Q738, W741, E893, 894, E897, I898, V901, and Q902. It can be seen from Fig. (3) that the flexibility of the residues 690-696, 756-761, 766-800 and 841-865, significantly increased, which was mainly concentrated in the loop region. However, the flexibility of residues for T877A-BUD31 and W741L-BUD31 decreased compared to wild type, indicating that T877A and W741L mutations reduced the flexibility of the residues, especially W741L mutation. For W741L-BUD31, it maintains the lowest flexibility among these three systems. However, at the binding site, the RMSF values remained low in all these systems, such as residues 700-750 and 890-905, which demonstrates point mutation, thereby, changing the flexibility of the Loop region, which did not change the flexibility of the active site.
Fig. (3).
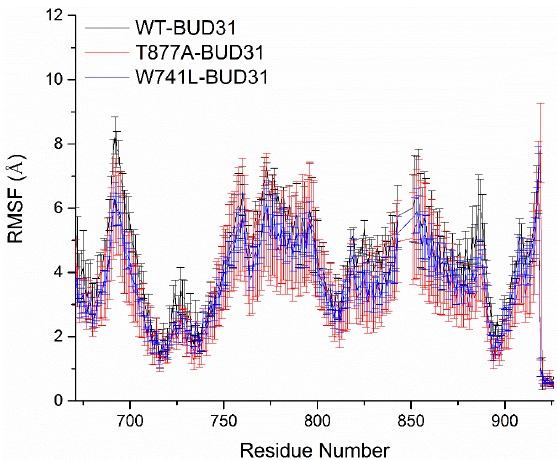
RMSF values for wild type AR and its mutants. The Root Mean Square Fluctuation (RMSF) for Cα atoms of wild type AR-BUD31 and its mutants relative to the initial structure were calculated.
3.2. Dynamic Cross-correlation Analysis
To further investigate the residue motions caused by point mutants, cross-correlation matrices of the Cα atoms fluctuations among last 10 ns were calculated and plotted in Fig. (4). Herein, highly positive regions (red and yellow) are associated with strongly correlated motions of specific residues, whereas negative regions (dark blue) are indicative of a strong anticorrelation in the specific residue movements. In Fig. (4), there are a very few highly correlated motions except for the y=x diagonal, which inflect the correlation of a certain residue with itself.
Fig. (4).

Cross-correlation matrices of the fluctuations of coordinates for Cα atoms around their mean positions during the last 10 ns of MD simulation. The extent of correlated motions and anticorrelated motions are color-coded. a: WT-BUD31. b: T877A-BUD31. c: W741L-BUD31.
Compared with WT, T877A and W741L point mutations increased the anticorrelated motions near T877 and W741 residues. This result suggests that the conformation near T877 and W741 residues changed by point mutations, while the co-regulator binding site was not affected.
3.3. MM-PBSA Calculation
In order to deeply analyze the effects of point mutations between AR and BUD31, the binding free energies and corresponding components were calculated by the MM-PBSA method. Table 1 shows that the ΔG of all three complexes are almost similar, which do not contradict with the previous report [22]. It can be seen (Table 1) that ΔEele andΔGpol are the dominant factors for regulating the binding affinity. To obtain the detailed interaction profile between AR and co-regulator BUD31, MM-PBSA method was further used to decompose the interaction energies to each residue. The contribution profile of the residues in the active site (residues within 5Å around BUD31) for ΔEele, ΔGpol and ΔG are plotted in Fig. (5). As mentioned above, electrostatic interaction, as one of the key components, on one hand, decreases the contribution of residues K717 and K720, on the other hand, it significantly increases the contribution of residues D731 and E897. For solvation free energy, point mutations decrease the contribution of residues D731, E893 and E897, but increase the contribution of residues K717 and K720. However, as a whole, the contribution values of the key residues were observed to be slightly different in the active site.
Table 1. The calculated binding free energies and corresponding components between AR and BUD31 using MM-PBSA method (unit: kcal/mol).
| Terms | WT-BUD31 | T877A -BUD31 | W741L -BUD31 |
|---|---|---|---|
| ΔEvdwa | -50.1±1.69 | -47.85±2.18 | -46.86±2.26 |
| ΔEeleb | -217.83±5.18 | -238.84±8.40 | -242.7±10.03 |
| ΔGpolc | 221.46±7.70 | 241.95±7.27 | 245.64±10.98 |
| ΔGnon-pold | 25.86±0.38 | 12.37±0.71 | 24.75±0.70 |
| ΔGbinde | -20.61±5.91 | -19.99±2.15 | -19.17±1.14 |
avan der Waals interaction energies between AR and BUD31.
bElectrostatic interaction energies between AR and BUD31.
cPolar contributions to the solvation free energy.
dNonpolar contributions to the solvation free energy.
eΔGbind = ΔEele + ΔEvdw + ΔGpol + ΔGnon-pol.
Fig. (5).
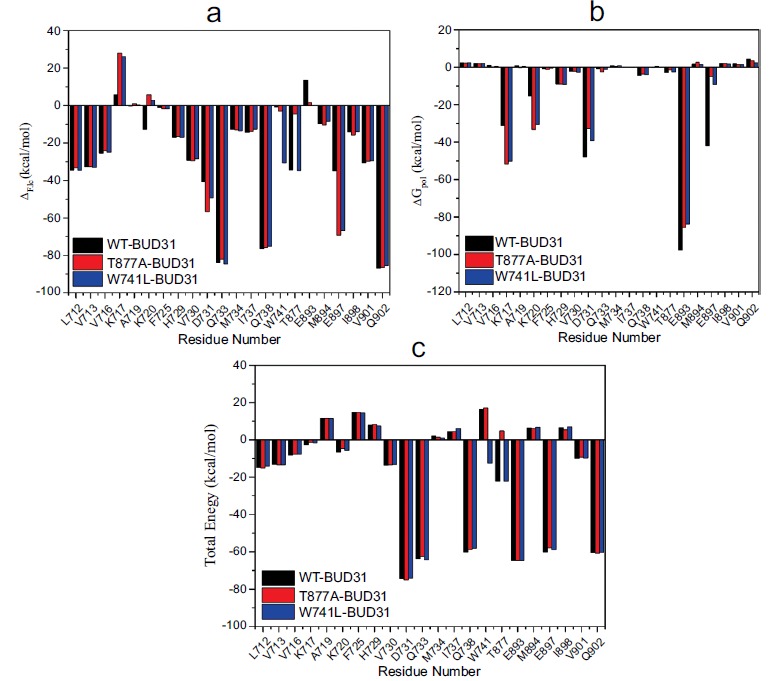
The residues contribution of AR to BUD31 binding. a: per residue contribution profile for ΔGele. b: per residue contribution profile for ΔGpol. c: per residue contribution profile for ΔG.
3.4. Hydrogen Bond Analysis
In the process of molecular dynamics simulations, BUD-31 can form hydrogen bonds with certain key residues, such as K720, Q733 and E897. All hydrogen bonds with a probability higher than 10% are listed in Table 2. From Table 2, it can be seen that in these three systems, N-H in the amino group of K720, as well as O-H in the hydroxyl group of Q733 and E897, can form a hydrogen bond with polypeptide BUD-31. Compared with WT AR systems, the probabilities of hydrogen bonds after T877A and W741L mutations decreased, but the distance increased. These results indicate that T877A and W741L point mutations reduce hydrogen bond stability between polypeptide with Q733. Nevertheless, hydrogen bond probability as a benchmark only reflects electrostatic interactions between the residues and polypeptide BUD-31, which cannot fully dominate the binding affinity between AR and polypeptide BUD-31.
Table 2. The hydrogen bonds between AR and polypeptide BUD31.
| Complex | Acceptor | Donor | Distance (Å) | Angle (。) | Probability (%) |
|---|---|---|---|---|---|
| WT | +5Y@O | K720@HZ1 | 2.80 | 156.81 | 26.00 |
| - | +5Y@O | K720@HZ2 | 2.80 | 156.66 | 19.40 |
| - | +5Y@O | K720@HZ3 | 2.80 | 155.84 | 20.63 |
| - | Q733@OE1 | +5Y@OH | 2.75 | 164.89 | 86.80 |
| - | E897@OE2 | +1F @N | 2.85 | 157.79 | 43.45 |
| - | E897@OE1 | -2Y@N | 2.86 | 153.26 | 40.00 |
| T877A | +5Y@O | K720@HZ1 | 2.83 | 155.64 | 14.70 |
| - | +5Y@O | K720@HZ2 | 2.81 | 155.58 | 15.87 |
| - | +5Y@O | K720@HZ3 | 2.82 | 155.47 | 14.33 |
| - | Q733@OE1 | +5Y@OH | 2.76 | 162.21 | 77.80 |
| - | E897@OE1 | +1F @N | 2.89 | 165.59 | 30.47 |
| - | E897@OE2 | -1I@H | 2.83 | 154.04 | 68.00 |
| - | E897@OE2 | -2Y@H1 | 2.78 | 154.63 | 25.00 |
| - | E897@OE2 | -2Y@H2 | 2.79 | 154.25 | 23.00 |
| - | E897@OE2 | -2Y@H3 | 2.79 | 154.06 | 25.00 |
| W741L | +5Y@O | K720@HZ1 | 2.81 | 155.63 | 18.93 |
| - | +5Y@O | K720@HZ2 | 2.81 | 157.00 | 18.83 |
| - | +5Y@O | K720@HZ3 | 2.80 | 155.87 | 18.07 |
| - | Q733@OE1 | +5Y@OH | 2.75 | 164.62 | 87.57 |
| - | I242@O | +4F @N | 2.89 | 155.59 | 25.33 |
| - | Y241@O | +3L@H | 2.90 | 156.44 | 25.00 |
| - | E897@OE2 | +1F@H | 2.87 | 164.27 | 33.00 |
3.5. The Structure Comparison of WT and Mutant Complexes
To reveal the contribution of the vital residues from a structural perspective, three structures with peptide and key residues in the binding pocket taken from the average MD-simulated structure are shown in Fig. (6). Besides, in order to fully understand the actual distribution of key peptide residues in AR active sites, hydrophobic surfaces between AR and polypeptide BUD-31 are plotted in Fig. (7). It can be seen that the peptide, composed of Phe+1, Leu+4, and Phe+5, binds to the hydrophobic groove formed by the helix3, 4, 5 and 12 of AR. As expected, BUD-31 is coordinated by residues K720, M734, M894 and E897, suggesting that AR can accommodate short peptides through these conserved residues.
Fig. (6).

The average structures of WT and its mutants take from the last 10 ns of the molecular dynamics simulations with the key residues of the binding pocket of the complexes. a: WT-BUD31. b: T877A-BUD31. c: W741L-BUD31.
Fig. (7).

The hydrophobic surfaces of the complexes of WT and its mutants: a: WT-BUD31. b: T877A-BUD31. c: W741L-BUD31. Herein, orange represents hydrophobic regions, whereas blue represents hydrophilic regions.
Point mutations lead to slight differences in the interactions between the peptide and AF2 site, such as binding affinity, and orientation of key residues in +4, +5 positions of the peptides. In the +4 position, although hydrogen bond contract was not formed, the orientation of this side chain was influenced by hydrophobic residues, such as L712, V713 and V716. As previously reported, +4 position of the groove is more suitable for accommodating bulky side chains [10]. It can be seen in Fig. (7) that the side chain of
The hydrogen bonds are determined by the acceptor-donor atom distance of 3.0Å and acceptor-donor angle of 135.
+4 residues were exposed to the broad groove, indicating that the surrounding hydrophobic residues that form an appropriate orientation of the peptide side chain. At the same time, T877A and W741L mutations make the phenyl group to be distant from V713, and close to M894, which may explain to some extent that the point mutation can decrease the binding affinity.
Compared with +4 position of BUD31, +5 residues are deeply embedded into the hydrophobic groove. By comparing the hydrophobic surface (Fig. 7), we could see that both the hydrophobicity and the size of the hydrophobic pocket were obviously different in these three complexes. Although all the complexes were stabilized by two hydrogen bond contracts between the phenolic hydroxyl group of +5 peptide, hydroxyl group of Q733 and the amino group of K720, the length of the bonds changed after point mutations. This result indicates that point mutations cannot change the formation of hydrogen bond interaction, which also contributes to the difference of the bond energy between a protein and a peptide.
4. DISCUSSION
BUD31 is a novel co-regulator that regulates the transcriptional activity of the AR. However, the mechanism of BUD31 interaction with AR is not clear. The present work reveals how point mutations affect the efficacy of a peptide associated with a protein. The action mechanism was explored using molecular dynamics simulation, which provids structural insights into the binding mechanism of the co-regulator BUD31 to AR AF2. Though the result of binding free energy analysis suggests that T877A and W741L point mutations can decrease the binding affinity between co-regulator BUD31 and protein, the difference in the interaction energies between BUD31 and protein is not significant. Combined with RMSF analysis, one possible explanation is that the T877A and W741L mutations were unable to disturb the scaffolds of the AR, and just increased the fluctuation of the loop regions. This view was also proved by the cross-correlation analysis. The hydrogen bond analysis suggests that T877A point mutation reduced hydrogen bond stability by influencing the formation of the hydrogen bond between polypeptide and Q733.
CONCLUSION
In conclusion, when binding to the co-regulator AR AF2 site, no matter WT or mutant protein, the binding poses of peptide BUD31 have no significant difference, the possible reason is that AF2 is a highly reserved binding site. The structural insights obtained from this study are important for us to design novel peptide drugs to overcome the resistance caused by T877A and W741L mutations.
ACKNOWLEDGEMENTS
None.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This research is sponsored by the National Natural Science Foundation of China (21205055).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Lee D.K., Chang C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: Mechanism and clinical implication. J. Clin. Endocrinol. Metab. 2003;88(9):4043–4054. doi: 10.1210/jc.2003-030261. [DOI] [PubMed] [Google Scholar]
- 2.Quigley C.A., De Bellis A., Marschke K.B., el-Awady M.K., Wilson E.M., French F.S. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr. Rev. 1995;16(3):271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 3.Tsai M.J., O’Malley B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf D.J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R.M. The nuclear receptor superfamily: The second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wärnmark A., Treuter E., Wright A.P., Gustafsson J.A. Activation functions 1 and 2 of nuclear receptors: Molecular strategies for transcriptional activation. Mol. Endocrinol. 2003;17(10):1901–1909. doi: 10.1210/me.2002-0384. [DOI] [PubMed] [Google Scholar]
- 6.Chang C., Saltzman A., Yeh S., Young W., Keller E., Lee H.J., Wang C., Mizokami A. Androgen receptor: An overview. Crit. Rev. Eukaryot. Gene Expr. 1995;5(2):97–125. doi: 10.1615/CritRevEukarGeneExpr.v5.i2.10. [DOI] [PubMed] [Google Scholar]
- 7.Shang Y., Myers M., Brown M. Formation of the androgen receptor transcription complex. Mol. Cell. 2002;9(3):601–610. doi: 10.1016/S1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 8.Heinlein C.A., Chang C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002;23(2):175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 9.Kinyamu H.K., Archer T.K. Modifying chromatin to permit steroid hormone receptor-dependent transcription. Biochim. Biophys. Acta. 2004;1677(1-3):30–45. doi: 10.1016/j.bbaexp.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Dubbink H.J., Hersmus R., Verma C.S., van der Korput H.A., Berrevoets C.A., van Tol J., Ziel-van der Made A.C., Brinkmann A.O., Pike A.C., Trapman J. Distinct recognition modes of FXXLF and LXXLL motifs by the androgen receptor. Mol. Endocrinol. 2004;18(9):2132–2150. doi: 10.1210/me.2003-0375. [DOI] [PubMed] [Google Scholar]
- 11.Steketee K., Berrevoets C.A., Dubbink H.J., Doesburg P., Hersmus R., Brinkmann A.O., Trapman J. Amino acids 3-13 and amino acids in and flanking the 23FxxLF27 motif modulate the interaction between the N-terminal and ligand-binding domain of the androgen receptor. Eur. J. Biochem. 2002;269(23):5780–5791. doi: 10.1046/j.1432-1033.2002.03276.x. [DOI] [PubMed] [Google Scholar]
- 12.He B., Minges J.T., Lee L.W., Wilson E.M. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 2002;277(12):10226–10235. doi: 10.1074/jbc.M111975200. [DOI] [PubMed] [Google Scholar]
- 13.Dubbink H.J., Hersmus R., Pike A.C., Molier M., Brinkmann A.O., Jenster G., Trapman J. Androgen receptor ligand-binding domain interaction and nuclear receptor specificity of FXXLF and LXXLL motifs as determined by L/F swapping. Mol. Endocrinol. 2006;20(8):1742–1755. doi: 10.1210/me.2005-0348. [DOI] [PubMed] [Google Scholar]
- 14.Van de Wijngaart D.J., Dubbink H.J., Molier M., de Vos C., Trapman J., Jenster G. Functional screening of FxxLF-like peptide motifs identifies SMARCD1/BAF60a as an androgen receptor cofactor that modulates TMPRSS2 expression. Mol. Endocrinol. 2009;23:1776–1786. doi: 10.1210/me.2008-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahl J., Smieško M. Endocrine disruption at the androgen receptor: Employing molecular dynamics and docking for improved virtual screening and toxicity prediction. Int. J. Mol. Sci. 2018;19(6):1784. doi: 10.3390/ijms19061784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakkiah S., Kusko R., Pan B., Guo W., Ge W., Tong W., Hong H. Structural changes due to antagonist binding in ligand binding pocket of androgen receptor elucidated through molecular dynamics simulations. Front. Pharmacol. 2018;9:492. doi: 10.3389/fphar.2018.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mammino L. Complexes of 1-[3-geranyl-2,4,6-trihydroxyphenyl]-2- methylpropan-1-one with a Cu2+ ion: A DFT study. Theor. Chem. Acc. 2019;138:15. doi: 10.1007/s00214-018-2381-2. [DOI] [Google Scholar]
- 18.Teodorico C.R., Elaine F.F. da C. Ricardo B. de A. Solvent effects on 13C and 15N shielding tensors of nitroimidazoles in the condensed phase: A sequential molecular dynamics/quantum mechanics study. J. Phys. Condens. Matter. 2004;16:6159–6170. doi: 10.1088/0953-8984/16/34/015. [DOI] [Google Scholar]
- 19.Mateus A.G., Lizandro S.S., Fernando C.P., Elaine F.F. da C., Telles C.S., Teodorico C.R. Comparing structure and dynamics of solvation of different iron oxide phases for enhanced magnetic resonance imaging. ChemistrySelect. 2017;2:10136–10142. doi: 10.1002/slct.201701705. [DOI] [Google Scholar]
- 20.Yeh S., Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA. 1996;93(11):5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu C.L., Chen Y.L., Yeh S., Ting H.J., Hu Y.C., Lin H., Wang X., Chang C. The use of phage display technique for the isolation of androgen receptor interacting peptides with (F/W)XXL(F/W) and FXXLY new signature motifs. J. Biol. Chem. 2003;278(26):23691–23698. doi: 10.1074/jbc.M211908200. [DOI] [PubMed] [Google Scholar]
- 22.Hsu C.L., Liu J.S., Wu P.L., Guan H.H., Chen Y.L., Lin A.C., Ting H.J., Pang S.T., Yeh S.D., Ma W.L., Chen C.J., Wu W.G., Chang C. Identification of a new androgen receptor (AR) co-regulator BUD31 and related peptides to suppress wild-type and mutated AR-mediated prostate cancer growth via peptide screening and X-ray structure analysis. Mol. Oncol. 2014;8(8):1575–1587. doi: 10.1016/j.molonc.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Case D.A., Darden T.A., Cheatham T.E., III, Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Götz A.W., Kolossváry I., Wong K.F., Paesani F., Vanicek J., Wolf R.M., Liu J., Wu X., Brozell S.R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M-J., Cui G., Roe D.R., Mathews D.H., Seetin M.G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A. Kollman. P.A. 2012, AMBER 12 Reference Manual. San Francisco: University of California; 2012. [Google Scholar]
- 24.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. doi: 10.1002/1096-987X(200009)21:12<1049:AID-JCC3>3.0.CO;2-F. [DOI] [Google Scholar]
- 25.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 26.26 Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 27.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 28.Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 29.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-Alkanes. J. Chem. Phys. 1977;23:327–341. [Google Scholar]
- 30.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E., III Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 31.Doree S., Kim A.S., Barry H. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994;98:1978–1988. doi: 10.1021/j100058a043. [DOI] [Google Scholar]