

Abstract
Background: There are over 44 million persons who suffer with Alzheimer’s disease (AD) worldwide, no existence of cure and only symptomatic treatments are available for it. The aim of this study is to evaluate the anti-Alzheimer potential of designed AChEI analogues using computer simulation docking studies. AChEIs are the most potential standards for treatment of AD, because they have proven efficacy. Among all AChEIs donepezil possesses lowest adverse effects, it can treat mild-moderate-severe AD and only once-daily dosing is required. Therefore, donepezil is recognized as a significant prototype for design and development of new drug molecule.
Methods
In this study the Inhibitory potential of the design compounds on acetylcholinesterase enzyme has been evaluated. Docking studies has been performed which further analyzed by in-silico pharmacokinetic evaluation through pharmacopredicta after that Interaction modes with enzyme active sites were determined. Docking studies revealed that there is a strong interaction between the active sites of AChE enzyme and analyzed compounds.
Results
As a result 26 compounds have been indicates better inhibitory activity on AChE enzyme and all the screening parameters have also been satisfied by all 26 compounds. From these 26 compounds, six compounds 17, 18, 24, 30, 36 and 56 are found to be the most potent inhibitors of this series by in-silico study through INVENTUS v 1.1 software, having highest bio-affinities i.e. - 8.51, - 7.67, - 8.30, - 7.59, - 8.71 and -7.62 kcal/mol respectively, while the standard or reference drug donepezil had binding affinity of - 6.32 kcal/mol.
Conclusion
Computer aided drug design approach has been playing an important role in the design and development of novel anti- AD drugs. With the help of structure based drug design some novel analogues of donepezil have been designed and the molecular docking studies with structure based ADME properties prediction studies is performed for prediction of AChE inhibitory activity. The binding mode of proposed compounds with target protein i.e. AChE has been evaluated and the resulting data from docking studies explains that all of the newly designed analogues had significantly high affinity towards target protein compared to donepezil as a reference ligand.
Keywords: Alzheimer’s disease, acetylcholinesterase inhibitors (AChEIs), CADD, Structure based drug design, molecular docking
1. INTRODUCTION
Discovery and Development of a new drug is a very complex, risky and costly process in terms of time, money and manpower. Generally, it is found that the drug discovery and development process takes around 10-14 years and more than 1 billion dollars capital in total [1]. General process of drug discovery and development has been given in (Fig. 1).
Fig. (1).
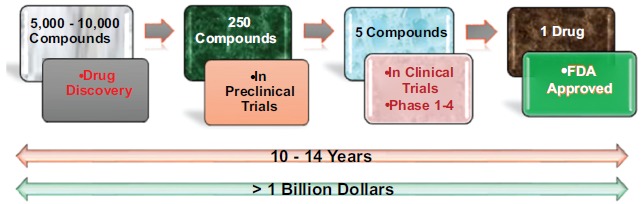
So for reducing time, cost and risk-borne factors, Computer-Aided Drug Design (CADD) method is widely used as a new drug design approach. It has been seen that by the use of CADD approaches we can reduce the cost of drug discovery and development up to 50% [4]. General principle of drug design through the modern approaches of CADD has been given in (Fig. 2).
Fig. (2).
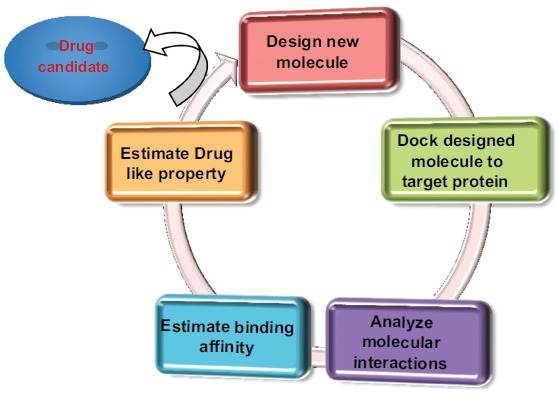
General principle for drug design through CADD [1].
Neurodegenerative diseases are one of the major challenges that modern health care system faces today and one of the most common causes of neurodegenerative diseases is Alzheimer Disease (AD) [5]. Worldwide, it accounts for 60-80% of all dementia cases [6]. Alzheimer disease is characterized by progressive loss of neurons, brain functions and cognition function.
Treatment based on the cholinergic hypothesis is the most promising and oldest one for Alzheimer’s. Besides the fact that cholinergic hypothesis elaborate only a fraction of complex neurodegeneration process in AD; acetylcholinesterase inhibitors (AChEIs) are the most potential standards for treatment of AD because they have proven efficacy [7]. In this study, donepezil is taken as a reference drug because it shows better efficacy with lesser side effects than any other AChEIs [8], reducing Amyloid-β by enhancement of α-secretase [9] but it also provides only modest clinical benefit for the treatment of AD [10]. Therefore, the aim of this study is to find out most potent drug candidates for the treatment of AD. CADD can be utilized as a powerful tool in the discovery and development of an efficient drug for AD. Through CADD we can filter out the most active predictive compounds from the large libraries of compounds, optimize the different parameters of lead compounds like pharmacokinetic parameters or increase its affinity, etc., and design the hybrid novel compounds [11].
2. MATERIALS AND METHODS
2.1. Target Protein: Screening, Selection, Identification and Visualization
The three-dimensional structures of acetylcholinesterase (AChE) target protein have been screened and identified through different databases for searching the cure of AD; the X-ray co-crystallized structures of human AChE (PDB ID - 4EY7) and donepezil have been selected and retrieved from RCSB-PDB [12]. It follows that the human enzyme is preferable as a starting model rather than Torpedo AChE enzyme for the structure based drug design, molecular modeling and docking like in-silico studies [13].
In the present study, INVENTUS v1.1 software has been used for the identification and visualization of 3D structure of target protein. The structure of 4EY7 has in total 2 chains which consist of 1084 residues with 2.3509 Å resolution. The structure is shown in Fig. (3). Energy optimization of target protein the energy minimization process has been performed through Energy Opt module of Inventus v-1.1 software and Steepest Descent (SD) and Conjugate Gradient (CG) algorithm methods have been used to minimize the potential energy of protein molecule.
Fig. (3).
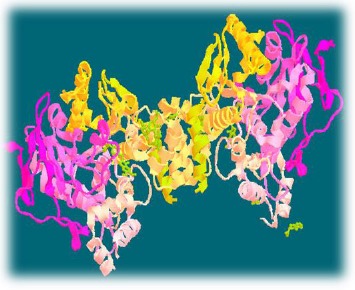
Structure of human AChE.
In this study, it has been observed that the initial energy of the protein is 9.221E+06 and final energy after minimization is –1.337E+04. The value after energy minimization tends towards negative, which indicates increases in the stability of the protein.
2.2. Active Site Analysis of Merged File (with the Standard Ligand)
In Inventus v 1.1 software, the active site detection in a protein structure is based on its geometry like volume, depth, etc. Our target protein (PDB ID: 4EY7) is a co-crystallized structure so, the binding site of donepezil has been taken as the active site for further process. The active site residues within the distance of 5 Ǻ are TYR 69, ASP 71, TRP 83, GLY 117, GLY 118, TYR 121, TYR 130, GLU 199, SER 200, TRP 277, LUE 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439, ILE 442 (Fig. 4).
Fig. (4).
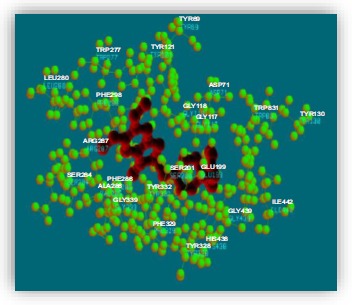
Active site analysis of merged file.
2.3. Virtual Screening (HitsGen)
Hitgen module has been used to screen 44 million compounds embedded with physico-chemical properties in Inventus. Libraries have been segmented as per target classes, disease and its nature, etc. from which the most potent compounds with physico-chemical properties of our interest have been screened out and later these compounds can be further used for docking studies.
2.4. Molecular Modelling
In molecular modeling, reference molecule is modified to develop more efficient inhibitors based on maximizing complementary interactions with the active site of the target protein through the help of structure-activity relationships (SAR) information of reference molecule [14, 15], the data obtained from high throughput screening and creating three dimensional structure based pharmacophore model by using Ligand Scout drug design software (Fig. 5).
Fig. (5).
Structure-based pharmacophore model development.
2.5. Molecular Docking
In this study, all the analogues are subjected to docking with prepared target protein by using Novo-Docker module of Inventus v1.1 software to carry out the protein analogue interaction studies and bio-affinity calculations.
2.6. Pharmacokinetic Prediction (PharmacoPredicta)
PharamacoPredicta is comprehensive predictive ADME software which is developed and validated to predict relevant pharmacokinetic characteristics of selected hit/lead compounds before proceeding ahead with synthesis and animal studies.
Following predictions based on chemical structure have been calculated in the PharmacoPredicta software system: Caco-2 (B→A), Caco-2 (A→B) permeability, absorption (FDp) classification level, efflux, blood-brain barrier permeability, protein binding and volume of distribution. A prediction confidence metric has been provided for each of these [17-19]. Results of these screening parameters are given in Tables 2 and 3.
Table 2. Caco – permeability and efflux parameters of designed compounds.
| Compound | Caco74ab | Caco74ab-Confidence | Caco74ba | Caco74ba-Confidence | Efflux | Efflux-Confidence |
|---|---|---|---|---|---|---|
| Reference | 4.90E-05 | Medium | 5.07E-05 | High | 0 | High |
| Analogue4 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue8 | 4.90E-05 | Medium | 5.73E-05 | High | 0 | High |
| Analogue11 | 4.90E-05 | Medium | 5.73E-05 | High | 1 | High |
| Analogue17 | 4.90E-05 | High | 2.21E-05 | High | 0 | High |
| Analogue18 | 4.90E-05 | High | 5.61E-05 | High | 0 | High |
| Analogue21 | 4.90E-05 | High | 5.73E-05 | High | 1 | High |
| Analogue22 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue23 | 4.90E-05 | High | 5.49E-05 | High | 0 | High |
| Analogue24 | 4.90E-05 | Medium | 4.49E-05 | High | 0 | High |
| Analogue28 | 4.90E-05 | Medium | 5.48E-05 | High | 0 | High |
| Analogue30 | 4.90E-05 | High | 1.08E-05 | High | 0 | High |
| Analogue32 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue33 | 4.90E-05 | Medium | 5.73E-05 | High | 0 | High |
| Analogue34 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue36 | 4.90E-05 | High | 8.72E-06 | High | 0 | High |
| Analogue38 | 4.90E-05 | Medium | 1.34E-05 | High | 0 | High |
| Analogue39 | 4.90E-05 | High | 4.97E-05 | High | 0 | High |
| Analogue40 | 4.90E-05 | High | 2.59E-05 | High | 0 | High |
| Analogue41 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue42 | 4.90E-05 | High | 2.10E-05 | High | 0 | High |
| Compound | Caco74ab | Caco74ab-Confidence | Caco74ba | Caco74ba-Confidence | Efflux | Efflux-Confidence |
| Analogue44 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue46 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue47 | 4.90E-05 | Medium | 1.34E-05 | High | 0 | High |
| Analogue50 | 4.90E-05 | High | 5.73E-05 | High | 0 | High |
| Analogue55 | 4.90E-05 | High | 1.44E-05 | High | 0 | High |
| Analogue56 | 4.90E-05 | High | 3.84E-05 | High | 0 | High |
Table 3. BBB, fdp, vdss parameters of designed compounds.
|
Compounds
ID |
BBB | BBB-Confidence | fdp | fdp-Confidence | Pro-bind | probind-Confidence | vdss | vdss-Confidence |
|---|---|---|---|---|---|---|---|---|
| Reference | 1 | High | High | Medium | 1 | High | 1000 | High |
| Analogue4 | 1 | High | High | High | 0 | High | 1000 | High |
| Analogue8 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue11 | 1 | High | High | High | 1 | High | 100 | High |
| Analogue17 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue18 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue21 | 1 | High | High | High | 1 | High | 100 | High |
| Analogue22 | 1 | High | High | High | 1 | High | 1000 | High |
| Analogue23 | 1 | High | High | High | 1 | High | 100 | High |
| Analogue24 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue28 | 1 | High | High | Medium | 0 | High | 10 | High |
| Analogue30 | 1 | High | High | High | 1 | High | 10 | High |
| Analogue32 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue33 | 1 | High | High | Medium | 1 | High | 10 | High |
| Analogue34 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue36 | 1 | High | High | High | 1 | High | 100 | High |
| Analogue38 | 1 | High | High | Medium | 1 | High | 1 | High |
| Analogue39 | 1 | High | High | High | 1 | High | 1000 | High |
| Analogue40 | 1 | High | High | Medium | 1 | High | 1000 | High |
| Analogue41 | 1 | High | High | Medium | 1 | High | 100 | High |
| Analogue42 | 1 | High | High | Medium | 1 | High | 1000 | High |
| Analogue44 | 1 | High | High | High | 1 | High | 1000 | High |
| Analogue46 | 1 | High | High | High | 1 | High | 1000 | High |
| Analogue47 | 1 | High | High | Medium | 1 | High | 1 | High |
| Analogue50 | 1 | High | High | Medium | 1 | High | 1000 | High |
| Analogue55 | 1 | High | High | High | 1 | High | 10 | High |
| Analogue56 | 1 | High | High | Medium | 1 | High | 100 | High |
3. RESULT AND DISCUSSION
Furthermore, we have filtered out top 26 analogues on the following basis:
A). Lipinski rule of five for drug-likeness, according to which a compound is more likely to be membrane permeable and easily absorbed by the body if it matches the following criteria:
Its molecular weight is less than 500 (M. wt. ≤500 daltons).
The compounds lipophilicity (logP) ≤ 5.
The sum of nitrogen and oxygen atoms is ≤10.
Hydrogen bond donor (H. B. D.) ≤ 5.
Hydrogen bond acceptor (H. B. A.) ≤ 10.
B). Lipinski’s rule for CNS drugs that CNS penetration is likely if:
Molecular weight ≤ 400.
Log P ≤ 5.
Hydrogen bond donor ≤ 3.
Hydrogen bond acceptor ≤ 7.
Number of rotatable bond ≤ 8 [22].
C). Better drug-receptor interaction or bio-affinity values than reference and prediction of BBB penetration
These top 26 compounds which follow Lipinski rule of five for drug-likeness, Lipinski rule for CNS drugs and gave better bio-affinity values or binding strength with target protein AChE (4EY7) than the reference drug (donepezil), are given in Table 1. Interaction pattern of these analogues with target protein is also given in Table 4. With the help of this interaction pattern, we can predict or analyze the amino acids and functional groups involved in strong interactions with our analogues. Finally from all 26 compounds binding interaction of the reference compound and top 6 designed analogues have been shown in Figs. (6 - 12).
Table 1. Docking results of reference and designed compounds.
| S. No. | Compounds | Properties |
|---|---|---|
| Ref. Donepezil |
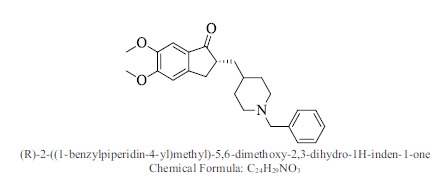 |
Bioaff.: - 6.32 M. wt.: 379.49 LogP: 4.93 H. B. D.: 0 H. B. A.: 4 |
| 4. | 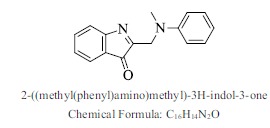 |
Bioaff.: - 6.53 M. wt.: 250.30 LogP: 3.38 H. B. D.: 0 H. B. A.: 3 |
| 8. | 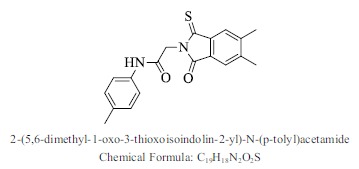 |
Bioaff.: - 6.51 M. wt.: 338.42 LogP: 3.67 H. B. D.: 1 H. B. A.: 4 |
| 11. | 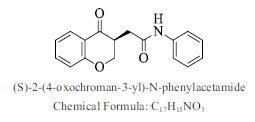 |
Bioaff.: - 6.52 M. wt.: 281.31 LogP: 3.19 H. B. D.: 1 H. B. A.: 4 |
| 17. | 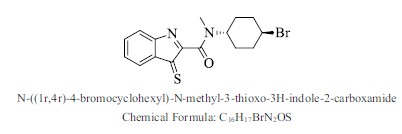 |
Bioaff.: - 8.51 M. wt.: 365.29 LogP: 4.23 H. B. D.: 0 H. B. A.: 3 |
| S. No. | Compounds | Properties |
| 18. | 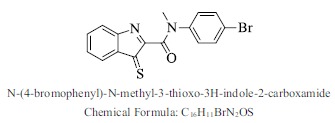 |
Bioaff.: - 7.67 M. wt.: 359.24 LogP: 3.92 H. B. D.: 0 H. B. A.: 3 |
| 21. | 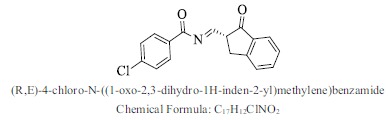 |
Bioaff.: - 6.54 M. wt.: 297.74 LogP: 3.61 H. B. D.: 0 H. B. A.: 3 |
| 22. | 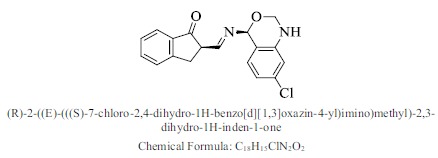 |
Bioaff.: - 6.54 M. wt.: 326.78 LogP: 3.86 H. B. D.: 1 H. B. A.: 4 |
| 23. | 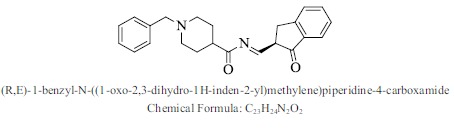 |
Bioaff.: - 6.48 M. wt.: 360.45 LogP: 4.12 H. B. D.: 0 H. B. A.: 4 |
| 24. | 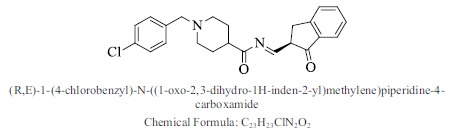 |
Bioaff.: - 8.30 M. wt.: 394.89 LogP: 4.77 H. B. D.: 0 H. B. A.: 4 |
| S. No. | Compounds | Properties |
| 28. | 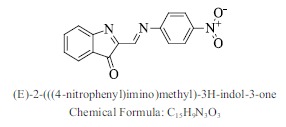 |
Bioaff.: - 6.46 M. wt.: 279.25 LogP: 3.27 H. B. D.: 0 H. B. A.: 6 |
| 30. | 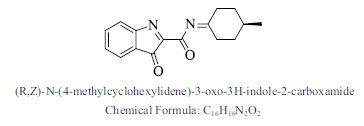 |
Bioaff.: - 7.59 M. wt.: 268.31 LogP: 3.13 H. B. D.: 0 H. B. A.: 4 |
| 32. | 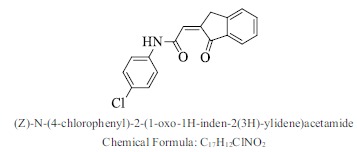 |
Bioaff.: - 6.54 M. wt.: 297.74 LogP: 3.64 H. B. D.: 1 H. B. A.: 3 |
| 33. | 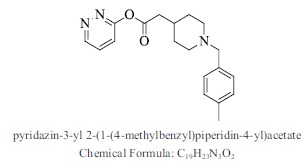 |
Bioaff.: - 6.50 M. wt.: 325.40 LogP: 3.56 H. B. D.: 0 H. B. A.: 5 |
| 34. | 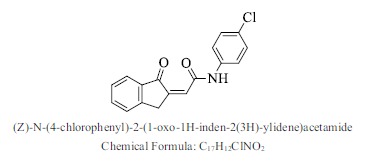 |
Bioaff.: - 6.54 M. wt.: 297.74 LogP: 3.64 H. B. D.: 1 H. B. A.: 3 |
| S. No. | Compounds | Properties |
| 36. | 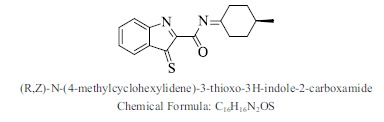 |
Bioaff.: - 8.71 M. wt.: 284.38 LogP: 3.67 H. B. D.: 0 H. B. A.: 3 |
| 38. | 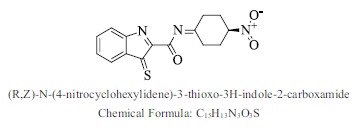 |
Bioaff.: - 6.38 M. wt.: 315.35 LogP: 2.96 H. B. D.: 0 H. B. A.: 6 |
| 39. | 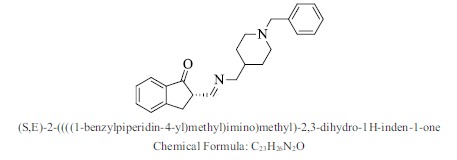 |
Bioaff.: - 6.51 M. wt.: 346.47 LogP: 4.88 H. B. D.: 0 H. B. A.: 3 |
| 40. | 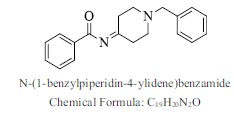 |
Bioaff.: - 6.49 M. wt.: 292.37 LogP: 4.13 H. B. D.: 0 H. B. A.: 3 |
| 41. | 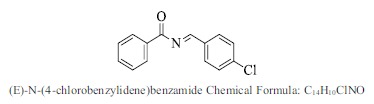 |
Bioaff.: - 6.53 M. wt.: 243.69 LogP: 3.60 H. B. D.: 0 H. B. A.: 2 |
| 42. | 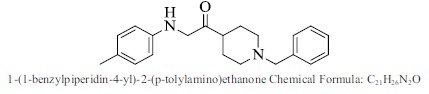 |
Bioaff.: - 6.52 M. wt.: 322.44 LogP: 4.74 H. B. D.: 1 H. B. A.: 3 |
| S. No. | Compounds | Properties |
| 44. | 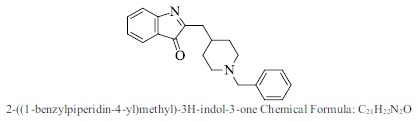 |
Bioaff.: - 6.54 M. wt.: 318.41 LogP: 4.83 H. B. D.: 0 H. B. A.: 3 |
| 46. | 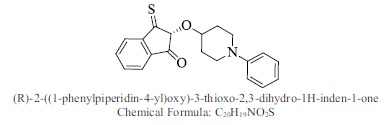 |
Bioaff.: - 6.53 M. wt.: 337.44 LogP: 4.80 H. B. D.: 0 H. B. A.: 3 |
| 47. | 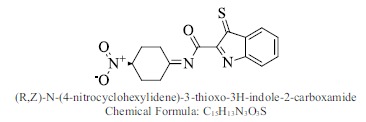 |
Bioaff.: - 6.54 M. wt.: 315.35 LogP: 2.96 H. B. D.: 0 H. B. A.: 6 |
| 50. | 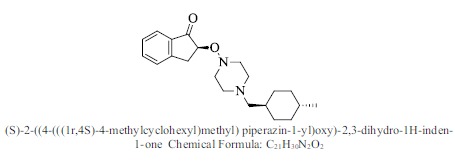 |
Bioaff.: - 6.37 M. wt.: 342.48 LogP: 4.88 H. B. D.: 0 H. B. A.: 4 |
| 55. | 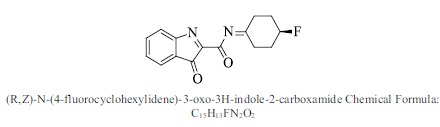 |
Bioaff.: - 6.51 M. wt.: 272.27 LogP: 3.12 H. B. D.: 0 H. B. A.: 4 |
| 56. | 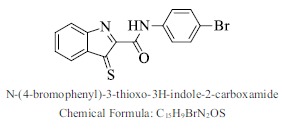 |
Bioaff.: - 7.62 M. wt.: 345.21 LogP: 3.89 H. B. D.: 1 H. B. A.: 3 |
Finally, all the analogues have been prepared in-silico with the help of ChemDraw software [16], in the consideration of Lipinski rule of five.
Table 4. Illustration of amino acid residues involved in drug-receptor interaction.
| Ana. No. | Residues Involved in Different Types of Interactions | |
|---|---|---|
| Reference Donepezil |
Active site residues: TYR 69, ASP 71, THR 72, LEU 73, GLY 79, MET 82, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLU 199, SER 200, TRP 277, LUE 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, TRP 430, HIS 438, GLY 439, TYR 440, ILE 442. Hydrophobic interaction: GLY 118, TYR 121, GLU 199, VAL 285, PHE 286, TYR 328, PHE 329, TYR 332, HIS 438. Electrostatic interaction: TYR 69, TRP 83, GLY 118, TYR 121, GLU 199, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439, TYR 440. |
|
| 4 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, TYR 121, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, PRO 437, HIS 438, GLY 439, TYR 440. Hydrophobic interaction: TYR 121, PHE 286, ARG 287, TYR 328, PHE 329, TYR 332. Electrostatic interaction: ASP 71, TRP 83, TYR 121, TRP 277, PHE 286, ARG 287, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. |
|
| 8 | Active site residues: TYR 69, ASP 71, LEU 73, MET 82, TRP 83, ASN 84, TYR 116, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLY 123, ALA 124, LEU 127, TYR 130, GLU 199, SER 200, TRP 277, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, VAL 331, TYR 332, HIS 438, GLY 439, ILE 442. Hydrophobic interaction: TYR 121, PHE 286, ARG 287, TYR 328, PHE 329, TYR 332. Electrostatic interaction: ASP 71, TRP 83, TYR 121, TRP 277, PHE 286, ARG 287, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. |
|
| 11 | Active site residues: TYR 69, ASP 71, LEU 73, TRP 83, ASN 84, TYR 116, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLU 199, SER 200, TRP 277, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. Hydrophobic interaction: TRP 83, GLY 117, GLY 118, TYR 121, SER 122, TYR 328, PHE 329, TYR 332, HIS 438. Electrostatic interaction: TYR 69, TRP 83, GLY 117, GLY 118, TYR 121, SER 122, GLU 199, SER 200, TRP 277, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 17 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, THR 80, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLU 199, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. Hydrophobic interaction: TYR 69, TYR 121, PHE 286, PHE 288, PHE 329, TYR 332. Electrostatic interaction: TYR 69, ASP 71, TRP 83, GLY 118, TYR 121, SER 122, SER 200, TRP 277, PHE 286, TYR 328, PHE 329, TYR 332. |
|
| 18 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, TYR 121, SER 122, TRP 277, LEU 280, GLU 283, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438. Hydrophobic interaction: TYR 69, TYR 121, TRP 277, TYR 332. Electrostatic interaction: ASP 71, TYR 121, TRP 277, VAL 285, TYR 328, PHE 329, TYR 332. |
|
| 21 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, THR 80, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLY 123, TYR 130, SER 200, TRP 277, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, TRP 430, HIS 438. Hydrophobic interaction: GLY 118, TYR 121, PHE 288, TYR 328, TYR 332. Electrostatic interaction: GLY 117, GLY 118, GLY 119, TYR 121, SER 122, SER 200, TRP 277, PHE 286, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 22 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, THR 80, TRP 83, ASN 84, GLY 118, TYR 121, SER 122, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438. Hydrophobic interaction: TYR 69, ASP 71, TYR 121, TYR 332. Electrostatic interaction: ASP 71, TRP 83, TYR 121, TRP 277, SER 284, VAL 285, PHE 286, PHE 288, PHE 329, TYR 332. |
|
| 23 | Active site residues: TYR 69, ASP 71, LEU 73, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TYR 130, GLU 199, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439, TYR 440, ILE 442. Hydrophobic interaction: ASP 71, GLY 118, TYR 121, SER 122, GLU 199, SER 284, VAL 285, PHE 286, TYR 332, HIS 438. Electrostatic interaction: TRP 83, GLY 118, TYR 121, SER 122, GLU 199, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, PHE 329, TYR 332, HIS 438, GLY 439, ILE 442. |
|
| Ana. No. | Residues Involved in Different Types of Interactions | |
| 24 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, ASN 84, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLY 123, SER 200, TRP 277, HIS 278, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. Hydrophobic interaction: TYR 69, GLY 118, TYR 121, SER 122, TRP 277, PHE 329, TYR 332. Electrostatic interaction: TYR 69, ASP 71, TRP 83, GLY 118, TYR 121, TRP 277, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332. |
|
| 28 | Active site residues: TYR 69, ASP 71, GLY 79, THR 80, MET 82, TRP 83, TYR 121, SER 122, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. Hydrophobic interaction: TRP 83, TYR 121, SER 122, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332. Electrostatic interaction: ASP 71, TRP 83, TYR 121, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, TYR 332. |
|
| 30 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, ASN 84, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TYR 130, GLU 199, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, TRP 430, HIS 438, GLY 439. Hydrophobic interaction: ASP 71, TYR 121, SER 284, TYR 328, TYR 332. Electrostatic interaction: TRP 83, GLY 118, TYR 121, GLU 199, SER 200, TRP 277, VAL 285, PHE 286, ARG 287, PHE 329, TYR 332. |
|
| 32 | Active site residues: TYR 69, ASP 71, TRP 83, GLY 118, TYR 121, SER 122, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439. Hydrophobic interaction: TRP 83, TYR 121, PHE 329, TYR 332, HIS 438. Electrostatic interaction: TRP 83, TYR 121, TRP 277, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 33 | Active site residues: ASP 71, THR 80, TRP 83, TYR 116, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, VAL 129, TYR 130, GLU 199, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, TRP 430, HIS 438, GLY 439, ILE 442. Hydrophobic interaction: TYR 130, TRP 277, VAL 285, PHE 286, ARG 287, TYR 328, TYR 332. Electrostatic interaction: TRP 83, GLY 117, GLY 118, TYR 121, TYR 130, GLU 199, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439, ILE 442. |
|
| 34 | Active site residues: TYR 69, ASP 71, LEU 73, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439. Hydrophobic interaction: GLY 118, TYR 121, TRP 277, PHE 329, TYR 332, HIS 438. Electrostatic interaction: TYR 69, GLY 118, TYR 121, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332. |
|
| 36 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. Hydrophobic interaction: TRP 83, TYR 121, TYR 328, PHE 329, TYR 332. Electrostatic interaction: TYR 69, ASP 71, TRP 83, GLY 118, TYR 121, TRP 277, VAL 285, ARG 287, TYR 328, PHE 329, TYR 332. |
|
| 38 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, SER 200, TRP 277, HIS 278, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. Hydrophobic interaction: TYR 69, TYR 121, TRP 277, PHE 286, PHE 329, TYR 332. Electrostatic interaction: TYR 69, ASP 71, GLY 118, TYR 121, TRP 277, PHE 288, PHE 329, TYR 332. |
|
| 39 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, TRP 83, ASN 84, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLY 123, TYR 130, GLU 199, SER 200, TRP 277, HIS 278, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439, ILE442. Hydrophobic interaction: TYR 121, GLU 199, SER 200, TYR 332. Electrostatic interaction: TYR 69, TRP 83, GLY 118, TYR 121, TYR 130, GLU 199, SER 200, TRP 277, PHE 329, TYR 332, HIS 438, GLY 439, ILE442. |
|
| 40 | Active site residues: TYR 69, ASP 71,THR 72, LEU 73, GLY 79, THR 80, TRP 83, TYR 121, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, TRP 430, HIS 438, GLY 439. Hydrophobic interaction: TYR 69, ASP 71, TYR 121, TYR 328, TYR 332. Electrostatic interaction: TYR 69, ASP 71,LEU 73, TRP 83, TYR 121, TRP 277, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| Ana. No. | Residues Involved in Different Types of Interactions | |
| 41 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TRP 277, HIS 278, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, VAL 331, TYR 332, HIS 438, GLY 439,TYR 440. Hydrophobic interaction: TYR 69, TRP 83, TYR 121, TYR 332. Electrostatic interaction: TYR 69, ASP 71, LEU 73, TRP 83, GLY 118, TYR 121, TRP 277, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 42 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, GLY 79, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, TYR 130, GLU 199, SER 200, ALA 201, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439, TYR 440, ILE442. Hydrophobic interaction: TYR 69, TRP 83, TYR 121, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. Electrostatic interaction: TYR 69, ASP 71, TRP 83, GLY 118, TYR 121, GLU 199, SER 200, TRP 277, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 44 | Active site residues: TYR 69, ASP 71, LEU 73, THR 80, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TYR 130, GLU 199, SER 200, ALA 201, TRP 277, HIS 278, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, VAL 331, TYR 332, TRP 430, HIS 438. Hydrophobic interaction: TRP 83, GLY 118, TYR 121, SER 284, TYR 332. Electrostatic interaction: TYR 69, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 200, TRP 277, VAL 285, PHE 286, PHE 288, TYR 328, PHE 329, TYR 332. |
|
| 46 | Active site residues: TYR 69, ASP 71, LEU 73, THR 80, TRP 83, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TYR 130, GLU 199, SER 200, ALA 201, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439, ILE442. Hydrophobic interaction: TRP 83, TYR 121, TRP 277, PHE 329, TYR 332. Electrostatic interaction: ASP 71, TRP 83, TYR 121, GLU 199, SER 200, TRP 277, SER 284, VAL 285, PHE 286, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. |
|
| 47 | Active site residues: TYR 69, ASP 71, THR 80, TRP 83, ASN 84, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, TYR 130, GLU 199, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438, GLY 439, ILE442. Hydrophobic interaction: TYR 121, SER 284, PHE 286, TYR 332. Electrostatic interaction: TRP 83, TYR 121, GLU 199, SER 200, TRP 277, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438. |
|
| 50 | Active site residues: TYR 69, ASP 71, LEU 73, TRP 83, TYR 116, GLY 117, GLY 118, GLY 119, TYR 121, SER 122, GLY 123, ALA 124, LEU 127, TYR 130, GLU 199, SER 200, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439, ILE442. Hydrophobic interaction: GLY 117, GLY 118, TYR 121, GLY 123, TRP 277, PHE 329, TYR 332. Electrostatic interaction: TYR 69, TRP 83, GLY 117, GLY 118, TYR 121, SER 122, GLY 123, LEU 127, TYR 130, TRP 277, VAL 285, PHE 286, PHE 288, TYR 328, PHE 329, TYR 332. |
|
| 55 | Active site residues: TYR 69, ASP 71, THR 72, LEU 73, TRP 83, GLY 118, TYR 121, SER 122, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, PHE 288, TYR 328, PHE 329, TYR 332, GLY 333, HIS 438. Hydrophobic interaction: TYR 69, ASP 71, TYR 121, TRP 277, TYR 332. Electrostatic interaction: TYR 69, TYR 121, TRP 277, TYR 328, PHE 329, TYR 332. |
|
| 56 | Active site residues: TYR 69, ASP 71, LEU 73, TRP 83, GLY 118, TYR 121, SER 200, TRP 277, LEU 280, SER 284, VAL 285, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332, HIS 438, GLY 439. Hydrophobic interaction: ASP 71, TYR 121, TRP 277, PHE 286, ARG 287, PHE 288, TYR 328, PHE 329, TYR 332. Electrostatic interaction: TYR 69, TRP 83, TYR 121, TRP 277, SER 284, VAL 285, PHE 286, ARG 287, TYR 328, PHE 329, TYR 332. |
|
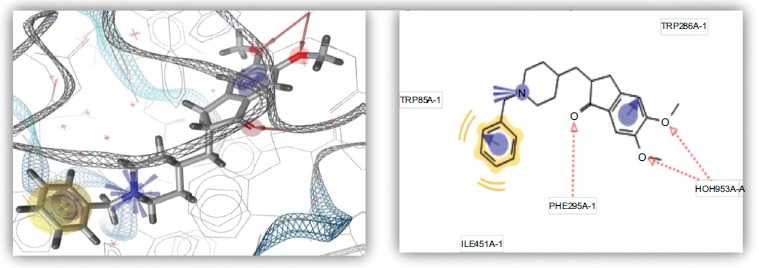
Fig. (6).

Binding mode of reference compound with AChE.
Fig. (12).
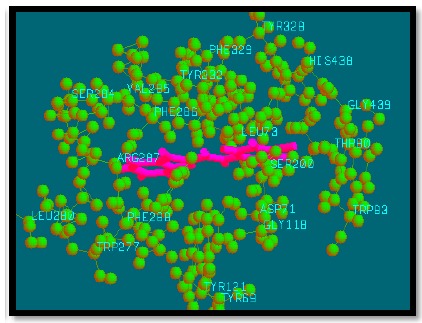
Binding mode of analogue 56 with AChE.
After molecular docking, the ADME (structure model only) properties of the all designed analogues have been analyzed by using pharmacopredicta module of inventus. The key features of this module, which have been used in our study, are described below:
3.1. ADME (Structure Based)
Human Absorption, FDp (%)
Results are classified as:
Low (0 – 33% absorbed)
Medium (33 – 64% absorbed)
-
High (67 – 100% absorbed)
Caco - 2 Permeability (A → B or apical to basolateral) Peff at pH 7.4 (cm/s).
Caco - 2 Permeability (B → A) or basolateral to apical) Peff at pH 7.4 (cm/s).
Efflux at pH 7.4 (0 if ≤ 5.3, 1 if >5.30).
Blood-brain barrier permeability (0 if no penetration, 1 if penetration).
Protein binding (0 if ≤ 85% or 1 if >85%).
Volume of Distribution at steady state, VDSS (liters).
All of these ADME (structure based) parameters of designed compounds have been calculated by using Pharmacopredicta module of INVENTUS v 1.1 software and values of these predictions are given in Tables 2 and 3. Graphical representations of ADME (structure only) properties prediction of reference and top 6 analogues have been given in (Figs. 13-17).
Fig. (13).
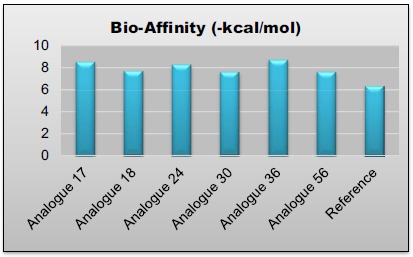
Graphical representation of Bio-affinity values of lead compounds.
Fig. (17).
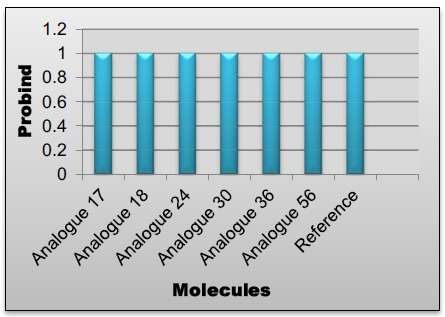
Protein binding values of lead compounds.
Finally, as a result we have found out top 26 compounds which show good bio-affinity as compared to reference drug, passed BBB penetration, Lipinski rule of five for drug likeness and also passed Lipinski rule for CNS drugs. From these 26 compounds, six compounds 17, 18, 24, 30, 36 and 56 were found to be the most potent inhibitors of this series by in-silico study through INVENTUS v 1.1 software, having highest bio-affinities i.e. – 8.51, - 7.67, - 8.30, - 7.59, - 8.71 and -7.62 kcal/mol while the standard or reference drug donepezil had binding affinity of - 6.32 kcal/mol. It has been perceived that if we follow the traditional synthetic route for drug design and development process, it is very tedious, costly, time consuming and risky process and the chances of failure in the last stages of development are also very high. So by the use of these in-silico approaches, we can filter the most appropriate compounds for further synthetic process, thus saving time, money, manpower with very less chances of failure in the last stages of drug discovery and development because our final filtered compounds had showed better in-silico results as compared to reference drug. Therefore, they could be used in clinical trials to test their efficacy and for the future drug design research and optimization for producing more efficacious drug analogues of AChEIs.
CONCLUSION
Computer aided drug design aids in enormous research of novel drugs candidates for the treatment of various diseases. It is playing an increasingly important role in design and development of novel anti-AD drugs, which significantly saves the limited resources and accelerates the drug development process.
Some novel derivatives of (R)-2-((1-benzylpiperidin-4-yl)methyl)-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one have been designed and the molecular docking studies with structure based ADME properties prediction studies is performed for prediction of AChE inhibitory activity. The binding mode of proposed compounds with target protein i.e. AChE has been evaluated and the resulting data from docking studies explains that all of the newly designed analogues had significantly high affinity towards target protein compared to donepezil as a reference ligand.
The compounds with higher bio-affinity values, passed BBB permeation prediction and also follow Lipinski rule of five criteria were analogues 4, 8, 11, 17, 18, 21, 22, 23, 24, 28, 30, 32, 33, 34, 36, 38, 39, 40, 41, 42, 44, 46, 47, 50, 55 and 56. Six compounds 17, 18, 24, 30, 36 and 56 were proved to be most potent inhibitors of this series by in-silico study with highest bio-affinities i.e. - 8.51, - 7.67, - 8.30, - 7.59, - 8.71 and - 7.62 kcal/mol, respectively while the reference drug donepezil had binding affinity of - 6.32 kcal/mol. The structure based pharmacokinetic properties of all designed compounds also have been predicted by using pharmacopredicta module of inventus v 1.1 software and it were found that most of the properties of final leads were similar as reference. The obtained result indicates that before synthesis, biological activity testing and clinical trials of new analogues, one can use molecular mechanics and analogue based drug designing methods for qualitative assessment of binding affinities for speeding up drug discovery process by eliminating less potent compounds from synthesis. Furthermore, the compounds 17, 18, 24, 30, 36 and 56 were found as the most suitable analogues in the study and speculated to show better results than donepezil in laboratory as well. Therefore, these compounds could be used in clinical trials to test their effectiveness for social benefits, as a pattern for future design, optimization and investigation to produce more effective analogues.
Fig. (7).
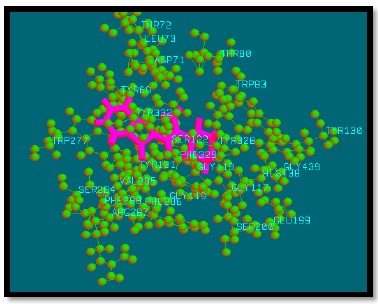
Binding mode of analogue 17 with AChE.
Fig. (8).

Binding mode of analogue 18 with AChE.
Fig. (9).

Binding mode of analogue 24 with AChE.
Fig. (10).

Binding mode of analogue 30 with AChE.
Fig. (11).
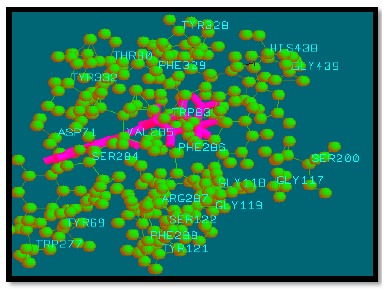
Binding mode of analogue 36 with AChE.
Fig. (14).
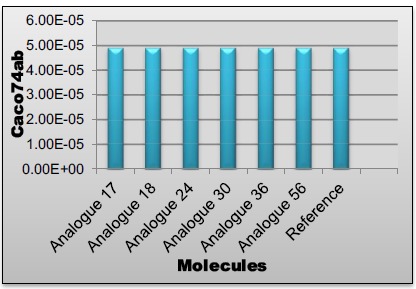
Caco74ab values of lead compounds.
Fig. (15).
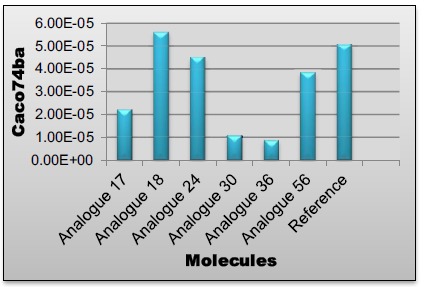
Caco74ba values of lead compounds.
Fig. (16).
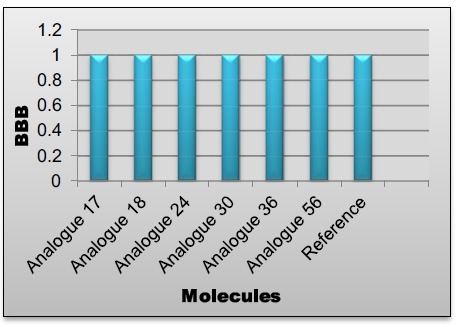
BBB values of lead compounds.
ACKNOWLEDGEMENTS
Declared none.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Daina A., Blatter M.C., Baillie Gerritsen V., Palagi P.M., Marek D., Xenarios I. Drug Design Workshop: A Web-Based Educational Tool To Introduce Computer-Aided Drug Design to the General Public. J. Chem. Educ. 2017;94(3):335–344. doi: 10.1021/acs.jchemed.6b00596. [DOI] [Google Scholar]
- 2. Medical chemistry project. (Accessed from: chemistryproject.info/-images/processgraphic.jpg )
- 3. We speak science. (Accessed from: https://wespeakscience.com/ wpcontent/uploads-/2017/07/Drug-Development-750x400.jpg )
- 4.Xiang M., Cao Y., Fan W., Chen L., Mo Y. Computer-aided drug design: Lead discovery and optimization. Comb. Chem. High Throughput Screen. 2012;15(4):328–337. doi: 10.2174/138620712799361825. [DOI] [PubMed] [Google Scholar]
- 5.Ghanemi A. Alzheimer’s disease therapies: Selected advances and future perspectives. Alexandria Journal of Medicine. 2015;51(1):1–3. doi: 10.1016/j.ajme.2014.09.006. [DOI] [Google Scholar]
- 6.Gustavsson A., Green C., Jones R.W., Förstl H., Simsek D., de Reydet de Vulpillieres F., Luthman S., Adlard N., Bhattacharyya S., Wimo A. Current issues and future research priorities for health economic modelling across the full continuum of Alzheimer’s disease. Alzheimers Dement. 2017;13(3):312–321. doi: 10.1016/j.jalz.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Zeb M.W., Riaz A., Szigeti K. Donepezil: A review of pharmacological characteristics and role in the management of alzheimer disease. Clinical Medicine Insights: Geriatrics. 2017;10:1–14. [Google Scholar]
- 8.Gouri K.S., Dani K.J., Chowdary K.P. Evaluation of drug treatment options for Alzheimer’s disease - a review of earlier studies. World J. Pharm. Pharm. Sci. 2017;6(4):177–182. doi: 10.20959/wjpps20174-8726. [DOI] [Google Scholar]
- 9.Sugimoto H. The new approach in development of anti-Alzheimer’s disease drugs via the cholinergic hypothesis. Chem. Biol. Interact. 2008;175(1-3):204–208. doi: 10.1016/j.cbi.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 10.Christensen D.D. Higher-dose (23 mg/day) donepezil formulation for the treatment of patients with moderate-to-severe Alzheimer’s disease. Postgrad. Med. 2012;124(6):110–116. doi: 10.3810/pgm.2012.11.2589. [DOI] [PubMed] [Google Scholar]
- 11.Zeng H., Wu X. Alzheimer’s disease drug development based on Computer-Aided Drug Design. Eur. J. Med. Chem. 2016;121:851–863. doi: 10.1016/j.ejmech.2015.08.039. [DOI] [PubMed] [Google Scholar]
- 12.PDB. http: // www.rcsb.org/pdb/
- 13.Cheung J., Rudolph M.J., Burshteyn F., Cassidy M.S., Gary E.N., Love J., Franklin M.C., Height J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012;55(22):10282–10286. doi: 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- 14.Sağlık B.N., Ilgın S., Özkay Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016;124:1026–1040. doi: 10.1016/j.ejmech.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 15.Sugimoto H., Ogura H., Arai Y., Limura Y., Yamanishi Y. Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharmacol. 2002;89(1):7–20. doi: 10.1254/jjp.89.7. [DOI] [PubMed] [Google Scholar]
- 16. Chemdraw Software (Accessed from: https://en.wikipedia.org/wiki/ChemDraw )
- 17. Novodocker (Accessed from: http://www.novoinformatics.com/ moleculediscovery-centre.html )
- 18. http://www.novoinformatics.com/ inventus.php
- 19. Inventus (Accessed from: http://www.novoinformatics.com/ )
- 20.Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today. Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 22.Pajouhesh H., Lenz G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]