

Abstract
Nitric oxide (NO) is a short-lived, endogenously produced, signaling molecule which plays multiple roles in mammalian physiology. Underproduction of NO is associated with several pathological processes; hence a broad range of NO donors have emerged as potential therapeutics for cardiovascular and respiratory disorders, wound healing, the immune response to infection, and cancer. However, short half-lives, chemical reactivity, rapid systemic clearance, and cytotoxicity have hindered the clinical development of most low molecular weight NO donors. Hence, for controlled NO delivery, there has been extensive effort to design novel NO-releasing biomaterials for tumor targeting. This review covers the effects of NO in cancer biology, NO releasing moieties which can be used for NO delivery, and current advances in the design of NO releasing biomaterials focusing on their applications for tumor therapy.
Keywords: Cancer therapy, Controlled delivery, Nanoparticles, Nitric oxide donors, Nitric oxide releasing biomaterials, Nitric oxide
1. INTRODUCTION
Although the therapeutic applications of glyceryl trinitrate (GTN) have been established for over 165 years, until the 1980s little was known about GTN’s physiological mechanism of action [1, 2]. Then the pioneering research of Furchgott, Ignarro, Murad and Moncada identified nitric oxide (NO) as the endothelium-derived relaxing factor [3, 4]. NO is a multifunctional free radical, with a molecular weight of only 30 Da, which has an unshared electron in its outer shell. As a simple
diatomic free radical, NO is a short-lived molecule with a half-life (t1/2) between 0.1 to 5s in aqueous solutions, and in vivo its levels are continuously modulated by the nitric oxide synthases (NOS) [5]. Its t1/2 in aqueous solutions is approximately nine fold shorter compared to hydrophobic solvents and this is mainly due to the autoxidation reaction which occurs in the presence of oxygen [6]. Due to NO’s lipophilicity it can readily diffuse through cell membranes and, under physiological conditions, its diffusion constant is similar to molecular oxygen [7, 8].
As a signaling molecule NO regulates key physiological processes, such as gene regulation, vasorelaxation, vascular permeability, bronchodilation, platelet aggregation, angiogenesis, neuronal communication, hormone secretion, inflammation, gastrointestinal mobility and wound healing [9, 10]. The over or under-production of NO causes or contributes to several pathophysiological conditions including cancer [11]. Over the past decades there have been extensive efforts to investigate the effects of NO on cancer biology, however, the findings are controversial. Overall NO has been termed a double edged sword, as it has both tumoricidal and tumor promoting effects. The concentration and duration of its presence at a particular site are thought to play a prominent role in cancer biology [12].
Inhaled NO is now recognized as an invaluable tool for decreasing pulmonary inflammation, neonatal pulmonary hypertension, and for heart and lung surgery. Beyond these applications it has limited, if any, other clinical value due to its low water solubility, instability and inconvenient handling of authentic aqueous solutions of NO [13-15]. Hence, there has been increasing interest in the development of NO releasing compounds to generate NO in situ.
For cancer therapy the utility of authentic aqueous solutions of NO, and most currently available NO donor agents (consisting of low molecular weight molecules), is highly limited due to their low half-lives, instability under physiological conditions, rapid systemic clearance, unspecific NO release and NO-independent toxicities. With the emergence of nanotechnology, the application of intelligent biomaterials to the controlled delivery of NO has emerged as a unique strategy for tumor targeting.
Herein we discuss the role of NO in cancer biology, and the therapeutic use of NO releasing functional groups, with a focus on the six main groups: organic nitrates, organic nitrites, metal complexes, sydnonimines, diazeniumdiolates, and S-nitrosothiols. We also review current advances in the design of NO releasing biomaterials and their applications for tumor therapy.
2. THE BIOLOGICAL ACTIONS OF NO
The biological actions of NO can occur via direct or indirect chemical reactions. The best characterized example is of NO directly binding to metal complexes of different proteins to form metal nitrosyl complexes, regulating their biological activity [16]. For example, NO reacts with soluble guanylate cyclase (sGC) at its haem moiety; upon binding the enzyme is activated, enabling the catalytic transformation of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). Downstream of this event kinase-media- ted signal transduction proteins responsive to cGMP are activated, such as protein kinase G (PKG). This has been recognized as the principal pathway by which NO mediates many physiolo- gical processes including: smooth muscle relaxa- tion, neurotransmission, inhibition of platelet aggregation and adhesion [17]. For the classical mechanism suggested for vasodilatory effects of NO, PKG sequestrates calcium (Ca2+) into intracellular stores via stimulation of the sarco-endoplas- mic reticulum Ca2+ ATPase (SERCA), thereby decreasing cytoplasmic Ca2+ concentration which in- hibits the cell’s contractile apparatus [18] through interactions with myosin Fig. 1. NO-mediated vasodilation also occurs via inhibition of Ca2+-dependent K+ channels [19]. Inhibition of such channels, which are specifically important in cerebral vasodilatation [20], results in depletion of cellular calcium and therefore relaxes the vascular muscle.
Fig. (1).
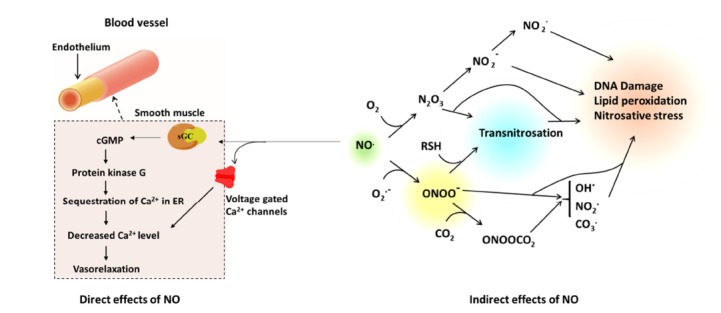
The direct and indirect effects of NO in cell signaling. The interaction of NO with O2 and O2.- results in species such as N2O3 and ONOO-, which modulate some of the biological activities of NO such as DNA damage and lipid peroxidation.
In addition to metal binding, NO may also undergo chemical reactions with a variety of endogenous radical species, producing reactive nitrogen species, which are responsible for additional signaling roles and certain types of NO-mediated toxicity in vivo. For instance, the reaction of NO with O2 or the superoxide anion (O2•-) produces nitrogen dioxide (NO2) and peroxynitrite (ONOO-) respectively [21]. These reactive species act as potent oxidizing and nitrating agents [21] which can result in changes to DNA, lipid peroxidation, and protein modifications via nitration, nitrosative and oxidative reactions Fig. 2 [22].
Fig. (2).
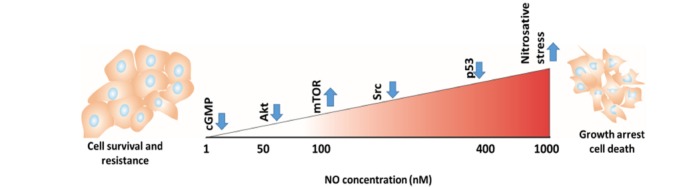
The effect of sustained levels of NO on signal transduction mechanisms in cancer cells. These data were generated by treating cancer cells with diethylenetriamine NONOate (DETA/NO) for 24 h, then the activation of specific pathways was evaluated [30-32].
3. NO IN CANCER BIOLOGY
The role of NO in tumor therapy is diverse. Depending on both the concentration and duration of NO action within the tumor cells, it can affect cancer initiation, enhance cell progression, tumor blood flow, angiogenesis, metastasis, and apoptosis, cell death and tumor suppression (for reviews see [9, 23, 24]). The targeted release of NO may also provide opportunities for cancer therapy and enhance the efficacy of chemo- and radiotherapy, provided that the appropriate concentration of NO reaches the tumor [24].
Vannini et al. have recently categorized the concentration of NO in tissue as low (50-100 nM), intermediate (100-400 nM) or high (400-1000 nM) [25]. At low and intermediate concentrations, NO normally stimulates cancer cell progression, prevents apoptosis and enhances angiogenesis and metastasis via various signaling pathways which are crucial for tumor cell survival, such as the extracellular signal-regulated kinase (ERK), Akt, mammalian target of rapamycin (mTOR), Ras and epidermal growth factor receptor (EGFR) pathways [23]. Higher concentrations of NO are recognized as having an anti-tumor effects by inducing apoptosis and sensitizing tumors to chemo- and radiotherapy [24], however the exact mechanisms responsible for establishing different NO activities are unknown. It appears that there is a gradation of NO effectiveness in activating these different pathways [26-28] (Fig. 3. For example, stimulation of MCF-7 breast cancer cells, macrophages, or endothelial cells with activation of PKG, increased ERK-P and Akt-P expression at low nM levels of NO (10-60 nM). Increased hypoxia-induced factor 1 alpha (HIF-1α) expression was then detected at approximately 100 nM NO [29, 30], while p53 expression was associated with higher levels of NO that led to apoptosis [31]. In addition, NO is known as a physiological regulator of mitochondrial respiration [32]. At both low and high concentrations it inhibits mitochondrial respiration by different mechanisms [32]. At low concentrations NO inhibits cytochrome c oxidase in competition with oxygen, and this process is fully reversible [33]. Probably by nitrosating or oxidising thiol centers of proteins and reacting with the iron-sulfur centers, at high concentrations NO inhibits the respiratory chain complexes I, II and V, as well as creatine kinase [32]. This results in depolarization of the mitochondrial membrane and induces swelling and calcium release [34].
Fig. (3).
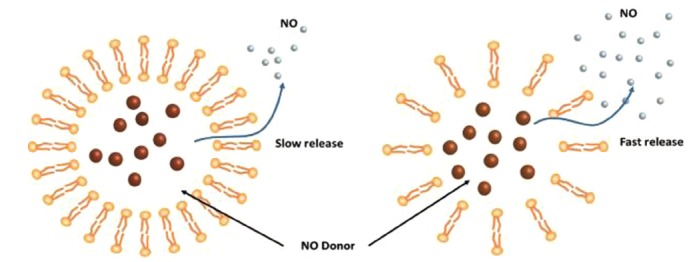
The encapsulation of N-diazeniumdiolates in liposomal structures. The stability of the NO donors was significantly enhanced when encapsulated in the liposomes. In addition, NO release from the liposomes could be regulated by altering the NO donor molecular structure, or the phospholipid composition of the liposome (independently or in combination) [137]. By changing the composition of the phospholipid layer from highly dense (left) to loosely packed (right), the stability of the NO releasing liposome dramatically decreased.
Due to the complexity of the signaling pathways involved, NO administration may result in different effects on cancer. For example, S-nitroso-N-acetyl-DL-penicillamine (SNAP) inhibited cell proliferation in umbilical vein endothelial cells [35], while in mouse clonal osteogenic cells it stimulated cell proliferation [36]. In addition, the expression of NOS has been investigated in several in vitro and in vivo cancer studies (for review see [25]). In animal models, depending on the tumor microenvironment and the tumor type, NOS overexpression resulted in tumorigenic or anti-tumor effects [37-39]. Similarly, it has both pro- and anti-metastatic effects [40]. In metastasis, epithelial cells lose normal cell-cell adhesion and gain mesenchymal markers which promote cell migration and invasion, a process known as epithelial to mesenchymal transition (EMT) [41]. Emerging evidence has shown that the NF‐κB family of transcription factors are pivotal regulators of both promoting and maintaining an invasive phenotype [42]. Several studies have shown that a high level of NO, delivered via NO donors such as diethylenetriamine NONOate (DETA/NO), inhibits the EMT phenotype in metastatic cancer cell lines by dysregulation of NF‐κB [43, 44]. However others have reported that NO facilitated the induction of MET and tumor invasion [45].
4. DRUG DELIVERY SYSTEMS AND EFF- ECTS OF NO ON EPR BASED ANTICAN- CER-DRUG DELIVERY
In cancer therapy, biomacromolecules larger than 40 kDa have been developed as drug carriers to promote tumor targeting. The macromolecules that are traditionally used for drug delivery systems (DDS) include; liposomes, polymeric nanoparticles, micelles, dendrimers and inorganic nanoparticles made of iron oxide, gold, quantum dots or metal oxide frameworks [46-48].
The tumor vasculature is structurally and functionally abnormal [49]. The vessels spread chaotically with abnormally varied caliber, and the smooth muscle layer is not correctly formed. Hence vascular walls contain wide fenestrations [50-53]. Furthermore, tumor tissues have ineffici- ent lymphatic drainage [54]. A significant number of vessels make shunts between arterial and venous ends, effectively stealing blood from regions of the tumor tissue and inducing localized hypoxia [53, 55, 56].
This leaky tumor vasculature, slow venous return and poor lymphatic clearance lead to the accumulation of macromolecules within the tumor, and this phenomenon was termed the enhanced permeability and retention (EPR) effect of macromolecules (larger than 7 nm) by Maeda and colleagues [57].
Despite the remarkable advances in DDS based on EPR, a limited number of nanocarriers have reached the clinic so far [58]. In this context, one major challenge is the lack of an EPR effect for macromolecules in the hypoxic area of tumors, particularly the necrotic regions of primary and metastatic cancers [59, 60]. Therefore extensive efforts have been made to increase the EPR effect in tumor tissue [61].
Several factors affecting vascular permeability have been investigated, including: vascular endo- thelial growth factors, bradykinin, prostaglandins and NO [62], as any intervention which enhances tumor blood supply should increase the EPR effect of macromolecules. For example, the NOS inhibitor Nω-monomethyl-l-arginine (NMMG), was shown to suppress vascular permeability and hence the EPR effect [63], while NO donors have been found to potentiate the EPR effect. Examples of potentiation include the local administration of the NO donor GTN directly into tumor sites, which increased the concentration of PEG-conjugated zinc protoporphyrin IX anticancer NPs, indicating an elevated EPR effect and thereby improved delivery of the drug to the tumor [64]. Similarly, infusion of isosorbide dinitrate (ISDN) into the local feeding artery of tumours enhanced the site-specific delivery of anticancer NPs in humans [61].
EPR enhancement can also be achived by elevating blood pressure, as this will increase tumor blood flow [59], and several clinical studies have used NO donors such as GTN to improve the responsiveness of tumors to chemo and/or radiotherapy (Table 1). On the other hand, the systemic administration of unstable NO donors may result in a significant decrease in systemic blood pressure thereby decreasing the EPR effect [59]. Therefore, the utility of NO donors to enhance the EPR effect in solid tumors, or to reach a tumorcidal level of NO, is highly dependent of the stability of the NO donors.
Table 1. Example clinical studies which used NO donors to improve the effects of chemo and/or radiotherapy.
| Drug | NO Donor | Type of the Study | Cancer Type | Results | References |
|---|---|---|---|---|---|
| Cisplatin and irinotecan |
Isosorbide mononitrate (ISMN) |
Phase II clinical trial | Non-small cell lung cancer | ISMN did not improve the treatment outcome of the chemotherapy | [174] |
| 5-FU and radiation | GTN patch | Phase I clinical trial | Lung cancer | The patches were well-tolerated and significantly enhanced the efficacy of the treatment. |
[175] |
| Combination of vinorelbine and cisplatin |
GTN | Phase II clinical trial | Non-small cell lung cancer | Improve overall response in patients with stage IIIB/IV NSCLC patients | [176] |
| Vinorelbine, cisplatin and concurrent radiotherapy | GTN patch | Phase II clinical trial | Non-small cell lung cancer | Increase in the overall survival of compared patient after 2 years | [177, 178] |
| Prostate-specific antigen (PSA) |
GTN patch | Phase II clinical trial | Prostate cancer | Significantly (p<0.001) longer doubling PSA time (31.8 months) | [179] |
| Transcatheter arterial chemoembolization | GTN | Clinical trial | Liver cancer | GTN improved the delivery of tumor-targeted therapy via enhanced permeability and retention. | [180] |
In addition, as mentioned above, NO acts differently over a range of concentrations within the tumor, and therefore it must be delivered at a designated concentration to tumor cells. Hence, the development of stable and tuned NO releasing compounds are required for applications towards tumor therapy. In the following sections we initially review the NO donor moieties which can be used for controlled delivery of NO, and then focus on recent developments in the controled delivery of NO using NO releasing macromole- cules for tumor therapy.
5. MAJOR CLASSES OF NO DONORS
There are at least 16 families of NO precursor and direct NO donors with remarkably varied chemical reactivities and NO-release kinetics. However, organic nitrates, organic nitrites, metal complexes, sydnonimines, diazeniumdiolates, and S-nitrosothiols are the major groups used in experimental and clinical applications.
5.1. L-Arginine and N-Hydroxyguanidine
NO is synthesized via conversion of L-arginine to NO and citrulline by the NOS family (Scheme 1). There are three different NOS isoforms in mammalian organisms: neuronal (nNOS), inducible (iNOS), and endothelial NOS (eNOS). The isoforms have similar mechanisms for NO synthesis however, their distribution in different cell types, and their mechanism of activation, are different [65]. nNOS and eNOS are mainly expressed in neuronal and endothelial cells respectively, but iNOS is expressed in both endothelial as well as nonvascular cell types such as macrophages, fibroblasts, and hepatocytes [66]. A major functional difference between NOS isoforms is that the activity of iNOS is initiated in response to inflammatory mediators and the expression of iNOS in healthy states is absent. Therefore, the activity of iNOS is subject to protein transcription, and the expression of iNOS has been shown to be a detrimental player in the development of disease. Whereas eNOS is constitutively active and regulates physiological processes such as vasodilation [66].
Scheme 1.
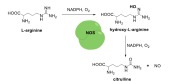
The mechanism of NO biosynthesis.
Although it is not an actual NO donor, L-arginine administration has been widely used to increase NO generation. For example, it displays beneficial properties in stroke [67, 68], angiogen- esis [69] and wound healing [70]. The biosyn- thesis of NO is an oxygen dependent pathway, and so analogues of the intermediate generated during NO synthesis, the N-hydroxyguanidines such as ω-hydroxy-L-arginine (NHA) [2], have been used as an alternative for L-arginine delivery [71]. This family is particularly important in the treatment of hypoxic conditions such as stroke, as converting NHA to NO can occur at low oxygen concentra- tions [72].
5.2. Organic Nitrates
Organic nitrates (RONO2) are nitric acid esters of alcohols. They are the oldest class of NO donors and currently the most commonly used NO donor drugs [73]. Organic nitrates can be synthe- sized by esterification of the corresponding alco- hols, or by the reaction of alkyl halides with AgNO3 (Scheme 2) [2]. GTN, isosorbide mononitrate (ISMN), pentaerythritol tetranitrate (PETN) and nicorandil are the most wildly used exampes in clinical studies. Most organic nitrates are highly stable, and the mechanism by which they release NO is not fully understood. However, it has been suggested that the NO formation can happen via both enzyme-dependent and non-enzymatic pathways [2]. Several enzymes such as NADPH-dependent cytochrome P450 [74, 75] and certain isoenzymes of the glutathione-S-transferase (GST) family [76] have been recognized in the bio-activation of organic nitrates. It has been shown that thiols significantly affect both in vitro
Scheme 2.
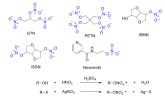
Some examples of the most commonly used organic nitrates and their general mechanism of synthesis. The nitrate group is highlighted (blue). (The color version of the figure is available in the electronic copy of the article).
and in vivo responses to GTN by promoting its NO release [77, 78] however these reactions are much slower than enzymatic activation. Therefore the dependency on enzymatic reaction to liberate NO is a major drawback to the clinical use of these drugs, as tolerance develops over time [79].
The organic nitrates have several established clinical applications. GTN is used as a cheap and effective drug for the rapid reversal of pain associated with acute angina [80, 81], and also occasionally for heart failure [82]. ISMN, which is a slower NO-releasing member of the organic nitrate family, has been used in the treatment of chronic angina [83]. As well as providing rapid and sustained dilation of veins, reducing cardiac afterload work, it causes dilation of cardiac vessels, which restores oxygen supply to ischemic cardiac muscle.
5.3. Organic Nitrites
Analogous to nitrates, organic nitrites are esters of alcohols and nitrous acid. They are mainly synthesized via reacting alcohols with nitrous acid or other nitrosating agents, as well as by the reaction of alcohols with gaseous NO (Scheme 3) [2]. The nitrosyl nitrogen atoms in organic nitrites are highly electronegative, which results in electron deficiency, hence they are highly prone to nucleophilic attack, which results in trans-nitrosation between the organic nitrite and the nucleophile [84]. Butyl nitrite (BN), isobutyl nitrite (ISBN), tert-butyl nitrite (TBN), amyl nitrite (AMN), and isoamyl nitrite (IAMN) have been used as vasodilators in the clinic.
Scheme 3.
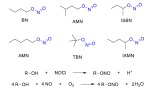
Chemical structures of common organic nitrites and their mechanism of synthesis. The nitrite group is highlighted (blue). (The color version of the figure is available in the electronic copy of the article).
It has been shown that organic nitrites activate NO signalling pathways, relaxing pulmonary vessels and decreasing blood pressure [85]. The release of NO from organic nitrites can occur through S-nitrosothiol formation due to the very fast trans-nitrosation with sulfhydryl groups [84]. Despite the higher potency of organic nitrites (as they are more reactive and less dependent on enzymatic activation) they are not as commonly used as organic nitrates due to their cytotoxicity [2], however AMN and BN have frequently been used in the treatment of angina pectoris [86]. The advantage of the organic nitrites is that they are less susceptible to inducing drug tolerance in comparison to organic nitrates such as GTN [87]. However a lack of selectivity and bioavailability, as well as cytotoxicity and carcinogenicity, restrict the applications of this class of NO donor [86].
5.4. Metal Complexes
NO is a strong ligand in metal complexes, and its binding constant is normally much higher than those of CO and O2 [88]. Numerous metal centers (primarily iron) react with NO to give adducts (Scheme 4), and the primary mechanism by which NO regulates many signaling pathways is through binding to metal centers, such as the haem group or iron-sulfur clusters of proteins. NO, as a ligand in metal complexes, has various oxidation states from M-NO- to M-NO+ and the oxidation state determines the reactivity of NO in the complex [89]. For example NO in sodium nitroprusside (SNP, Na2[Fe(CN)5NO]) exhibits significant NO+ character and iron has an oxidation state of Fe3+. The NO ligand is therefore subject to nucleophilic attack, hence the rate of NO release from SNP can be affected by thiols and other potential nucleophiles in the environment.
Scheme 4.

Example structures of NO releasing metal complexes, and the mechanism of SNP synthesis.
SNP was the first metal-NO compound discovered over 150 years ago, and has been in widespread clinical use for 40 years [90]. SNP is used as an arterial and venous vasodilator in cardiac surgery, hypertensive crisis, heart failure, vascular surgery, and pediatric surgery [91]. However SNP degradation is accompanied by five equivalents of cyanide release, which results in “cyanide toxicity” [91]. This dose limiting toxicity of cyanide release has been well-documented in several clinical cases and animal studies and precludes long term administration [92, 93].
In addition to iron, ruthenium (Ru) also has a high affinity for NO, and this has resulted in the development of a family of photoactive NO-releasing Ru complexes [94]. By modification of the π-bonding of other ligands, the affinity of Ru for NO can be modified, thereby regulating NO release from the complex [95]. Several photoactive Ru nitrosyls have been tested in in vitro and in vivo studies for controlled NO delivery [96]. However, efficient NO release requires high power UV light (300-400 nm) which is harmful to tissues [94] and this hurdle has prevented the translation of this family of NO donors into clinical use.
5.5. Diazeniumdiolates
The diazeniumdiolates (also known as ‘NONOates’) consist of a diolate group attached to a primary or secondary amine or polyamine. In solid form they are stable, but decompose sponta- neously in solution at physiological pH and tem- perature to generate two equivalents of NO per diolate [97]. They can be simply synthesized by exposing a primary or secondary amine to NO under high pressure [98], and depending on the structure of parent amine, temperature, and pH, their decomposition rates vary from seconds to hours [99]. Unlike most other NO donors, the NO release of diazeniumdiolates is not catalyzed by cellular metabolites or enzymes, and follows first-order kinetics. Hence the duration of action of the drugs can be directly predicted from their rates of decomposition in vitro. In addition, independent NO release explains the lack of tolerance experi- enced with these compounds [100]. Some of the most studied NONOates are methylamine hexa- methylene methylamine NONOate (MAHMA/ NO), diethylamine NONOate (DEA/NO), sper- mine NONOate (SPER/NO), proli NONOate (PROLI/NO) and diethylenetriamine NONOate (DETA/NO) (Scheme 5). No drug from this class of NO donors has advanced to clinical stages, however, they have frequently been tested in various experimental models of cardiovascular disease [97] mainly as supplementary treatments for vital cardiovascular operations such as balloon angioplasty and bypass, to prevent thrombosis and neointimal formation [101-105].
Scheme 5.
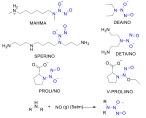
Chemical structures of representative NONOates and their mechanism of synthesis. The diazeniumdiolate moiety is highlighted (blue). (The color version of the figure is available in the electronic copy of the article).
5.6. Sydnonimines
Sydnonimines are another class of NO releasing agents which, due to the concomitant generation of O2•- and NO, are recognized as an ONOO- donors [106, 107]. In general, sydnonimines are synthe- sized by α-cyanoalkylation of dialkylhydrazines followed by nitrosation under acidic conditions (Scheme 6). 3-morpholinosydnonimine (SIN-1) and its precursor molsidomine (N-ethoxycarbonyl-3-morpholinosydnonimine) are the most exten-
Scheme 6.
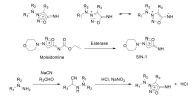
Chemical structures of sydnonimines and their mechanism of synthesis.
sively studied compounds of this class. N-acyl derivatives of sydnonimines in solid form are stable and in the absence of light can be kept at room temperature. Sydnonimines can be degraded by both enzymatic and nonenzymatic processes at physiological pH. Nonenzymatic hydrolysis is accelerated by alkaline pH, the presence of O2, and light. The enzymatic deacetylation and conversion of molsidomine to SIN-1 occurs primarily in the liver, SIN-1 then spontaneously decomposes to NO and O2•- in the blood [108, 109]. As ONOO- is highly reactive, it is believed that many of the actions of sydnonimines are mediated via cGMP-independent pathways, and possibly activation of K+ channels [110]. One of the main advantages of molsidomines as NO donors is the absence of tolerance. Hence, in the early 1980s, sydnonimines were widely studied as alternatives to organic nitrates in stable angina, and in the treatment of coronary heart disease [109]. However, they failed to show clear benefit in many large-scale clinical trials [111].
5.7. S-nitrosothiols
S-nitrosothiols (SNTs), also known as thionitrites, are an important class of NO donor which are mainly synthesized by the reaction of a thiol with a nitrite. SNTs, with certain exceptions, are very unstable compounds in aqueous solution, especially the primary and secondary SNTs [112]. Degradation of SNTs involves both homolytic and heterolytic cleavage of the S-NO bond [113] which results in the corresponding disulfide and NO, NO+ or NO- (Scheme 7). Homolytic cleavage is a two-step reaction which starts by cleavage of S-NO bond to make the thiyl radical (RS•), followed by reaction of two RS• radicals to form a disulfide (RSSR) [114]. One of the main reasons why tertiary SNTs are more stable than primary and secondary analogues could be due to the easier dimerization of the less sterically hindered RS•. The degradation of SNTs may also occur through trans-nitrosation to other nucleophilic species, such as thiols, without the appearance of NO as a free entity [113].
Scheme 7.
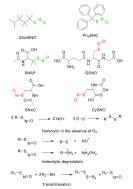
Chemical structures of common SNTs, and the mechanisms of SNT decomposition. NO releasing moieties are highlighted (tertiary SNT, green; primary SNT, red). (The color version of the figure is available in the electronic copy of the article).
The instability of SNTs in vivo is more pronounced, as their degradation rate is accelerated by vitamin C, trace amounts of metal ions, thiols such as GSH, and oxygen, as well as by enzymatic cleavage.
S-nitrosoglutathione (GSNO), N-acetylpenicill- amine (SNAP), trityl S-nitrosothiol, (Ph3SNO) and tert-butyl S-nitrosothiol (tButSNO) are the most commonly studied SNTs [114], alongside the recently developed tert-dodecane S-nitrosothiol (tDodSNO) [115] (Scheme 7).
5.8. Other NO-Releasing Moieties
N-nitrosamine, N-hydroxy nitrosamine, nitrosi- mine, furoxan and oximes are other functional groups which can serve as NO releasing moieties. However the majority of studies still rely upon the six main classes of NO donors.
One of the most important drugs which con- tains the N-nitrosamine group is N-methyl-N-nitrosourea streptozocin (STZ), which has antitumor as well as diabetogenic and carcinogenic activities [2]. NO release by STZ in the pancreatic β cell is known as one of the central mecha- nisms for these properties [116]. β cells of the pancreas are sensitive to NO and ROS as they possess low levels of ROS scavenging enzymes [117], and overproduction of NO damages the DNA content of the cells. N-hydroxy-N-nitrosamines are potent anti-hypertensive and anti-coagulative agents which are heat-stable in solution. Cupferron, alanosine, and dopastin are the most studied compounds in this class of NO donor, which have mainly been investigated as anticancer drugs [118].
Furoxans generally are stable compounds against thermal degradation, acids and electrophiles but not toward bases and nucleophiles [2]. Their deg- radation and NO release take place in the presence of thiols. They currently have no clinical application, but in animal studies, some furoxans such as C92-4609, and C93-4759 displayed tolerance-resistant vasodilation [119, 120].
6. Hybrid NO-releasing anticancer drugs
Due to the diverse activity of NO in bio- logical processes, as well as the complexity of NO delivery to tumor tissue, low molecular weight NO releasing compounds have made limited progress for tumor treatment so far. In particular, short half-lives, non-specific NO release, and rapid systemic clearance have hindered the clinical development of most NO donor compounds as anticancer drugs. However, there has been an extensive effort to develop hybrid NO donor drugs by attachment of NO donor moieties to currently available anti- cancer drugs to maintain the pharmacological activity of the parent drug, while gaining benefit from the biological actions of NO [121]. To get the desired biological activity (such as anticancer action) from NO release in the hybrid, the ratio of the parent drug to NO equivalents released can be varied [122]. The most commonly used NO releasing moieties for the design of hybrid drugs are S-nitrosothiols, diazeniumdiolates, furoxans, and organic nitrates.
NO releasing versions of NSAIDs have been used for cancer therapy. For example, several NO releasing hybrids of aspirin have been designed using furoxan [123], nitrate and S-nitrosothiol moieties [124]. NCX4040, synthesized by esterifi- cation of the carboxyl group of aspirin with 4-hydroxybenzyl nitrate (Scheme 8) showed 250-6,000-fold greater efficacy at inhibiting the growth of a panel of cancer cell lines [125]. NO releasing topoisomerase inhibitors have also been synthe- sized by conjugation of the anticancer drug doxorubicin (Dox) with a phenylsulfonyl furoxan moiety (NO-DOXO-1). These Dox analogs were
Scheme 8.
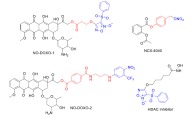
Example structures of hybrid NO releasing anticancer drugs. The parent drug (black) can be modified by NO releasing moieties (blue) and linker groups (red). (The color version of the figure is available in the electronic copy of the article).
shown to accumulate in Dox resistant human colon cancer cells (HT29-dx), and resulted in high cytotoxicity [126]. The same group has recently designed a novel light responsive NO releasing Dox to overcome multidrug resistance using a derivative of 4-nitro-3-(trifluoromethyl)aniline (NO-DOXO-2) [127].
The acetylation status of histones regulates levels of gene expression, and overexpression of histone deacetylases (HDAC) is oberved in nume- rous types of cancer. Hence, HDAC inhibitors have been suggested as a promising agents for cancer therapy [128]. The antiproliferative activity of a NO releasing HDAC inhibitor (Scheme 8) has been reported as a superior version of vorinostat, a HDAC inhibitor drug, against the human erythro- leukemia (HEL) cell line [129].
7. NO-RELEASING MACROMOLECULES
As mentioned above, so far very few low molecular weight NO releasing drugs have been used for cancer therapy whose main mechanism of action is through NO donation. Hence, in response to the need for controlled NO delivery, there has been extensive effort to design macromolecular NO-releasing vehicles [130-132]. In the following section we review the biomaterials which have been used for controlled delivery of NO particularly for cancer therapy (a few recent studies are summarized in Table 2).
Table 2. A summary of recent examples of NO releasing NPs developed for cancer therapy, with or without combination chemotherapy.
| Functional Group | Particle | Study-Cancer Type |
Combined
Anticancer Agent |
Stimulus for NO Release | References |
|---|---|---|---|---|---|
| NONOate | Modified silica NP | In vitro- ovarian cancer | None | None | [181] |
| NONOate | Modified silica NP | In vitro - non-small cell lung cancer | Cisplatin | None | [182] |
| S-nitrosothiol | Micelle | In vitro and in vivo - breast cancer | Doxorubicin loaded NPs | None | [144] |
| S-nitrosothiol | Polymeric NP | In vitro- ovarian cancer | Doxorubicin | Vis light | [169] |
| Roussin's Black Salt | Metal silica coated NP | In vitro - breast cancer | Doxorubicin | NIR-ray | [170] |
| Nitrobenzene | Polymeric NP |
In vitro – cervical cancer |
None | UV-ray | [127] |
| S-nitrosothiol | Silica NP | In vitro - cervical cancer | Radiotherapy | X-ray | [171] |
| L-arginine | Silica NP | In vitro and in vivo - pancreatic cancer | None | Ultrasound | [172] |
| N-nitrosamine | Nano-sandwich | In vitro and in vivo - breast cancer | Doxorubicin | UV-ray | [183] |
7.1. Encapsulation of Gaseous NO or Low Molecular Weight NO Donors
One of the initial strategies was direct encapsulation of NO gas as an active therapeutic. Perfluorocarbon (PFC) microbubbles are emulsions of synthetic hydrophobic fluorinated hydrocarbons and surfactants which can store large amounts of O2 and NO [133]. These emulsions are biologically inactive and can be safely injected into the vascular system [134]. It has been shown that PFC long-circulating particles can provide a physiologically significant pool of endogenous plasma NO, which can be used as a pharmacological tool for various cardiovascular complications associated with NO imbalance [135]. Polymeric carriers such as poly (vinyl alcohol) shelled NO microbubbles [136] and liposome encapsulations of gaseous NO [137] have also been investigated as NO delivery systems. Nevertheless, NO gas encapsulations are unstable and gaining control over their NO release is difficult.
Another strategy for controlled NO delivery is via the encapsulation of NO donor drugs. Here the potential advantage is that the NO donor remains trapped within the particle, avoiding unwanted cytotoxicity. For example a ruthenium nitrosyl complex was embedded in a poly-lactic-co-glycolic acid (PLGA) matrix, from which only NO release was observed with no leakage of the remaining metal fragment [138, 139].
Unlike encapsulation of gaseous NO, NO release from encapsulated NO donors is sensitive to environment, and the presence of water, vitamin C and GSH may affect the rate of NO release from the particle [130]. Recently Schoenfisch’s group has reported encapsulation of a N-diazeniumdio- late in liposomal structures. The NO release of the system could be precisely controlled by varying the composition of the lipid layer [140]. PROLI/NO, DEA/NO, PAPA/NO, or SPER/NO have been encapsulated in such phospholipid bilayers to form different NO-releasing liposomes [140]. The encapsulation process significantly increased the t1/2 of the NO donors, for example, t1/2 of SPER/NO increased from 37 min in its free form to around 2 days when encapsulated within the liposome. The physical barrier against proton diffusion/exchange provided by the lipid bilayer was demonstrated to be the main mechanism for the prolonged NO release properties of the liposomes. Interestingly, the NO-release kinetics of different liposomes with identical NO donors was dependent on the compactness of the lipid chains of each individual liposome, which is a property set upon liposome formation [140]. Therefore, by manipulation of the structure of lipid bilayer, the water permeability of the liposomes and hence the kinetics of NO release could be tuned, with tighter packing of the lipid chains decreasing the N-diazeniumdiolate NO donor decomposition rate Fig. (3) [140, 141].
Recently we have designed a photoactive NO releasing nanoparticle (NP) by encapsulation of the S-nitrosothiol tDodSNO into a co-polymer of styrene and maleic acid (SMA) to afford SMA-tDodSNO. The encapsulation imparted water solubility to the highly hydrophobic tDodSNO, and protected it from degradation reactions with glutathione [142, 143]. In the absence of photoactivation, the S-nitrosothiol group within the NPs had a t1/2 of 104 h, while photoactivation dropped this to 3.5 min. The NPs acted as a photo-switchable NO donor and induced localized vasodilation in aortic rings, and vascular hyperpermeability in mesenteric beds. When SMA-tDodSNO was co-adminis- tered with SMA-Dox (SMA NPs loaded with doxorubicin) it significantly enhanced its anticancer properties [144].
In addition, it has been shown that encapsulation of low molecular weight NONOates in liposomes improves the tumor targeting of such NO donors due to the EPR effects of the NPs [145], and administration of such NO releasing NPs with anticancer drugs increases their anticancer efficacy [144, 145].
7.2. Polymeric Organic and Inorganic NO Releasing Scaffolds
In addition, to improve the control of NO release in biological media, a wide-variety of NO donor materials such as macromolecule scaffolds, polymeric NPs, micelles, dendrimers, sol-gel derived silica NPs and surface modified metal/metal oxide NPs have been examined as NO delivery systems [130, 146]. These polymeric NO donors have been examined as treatments for a large number of pathological conditions ranging from cardiovascular diseases, wound healing, neuropathy and stroke to cancer [131, 147].
As an example of this approach, analysis of the tumor microenvironment of solid tumors has revealed that a significant part of the tumor mass is made of immune cells. Hence, tumor immune cells have been characterized as suitable targets for tumor therapy [148]. In immune cells iNOS produces high concentrations of NO, so it is possible to achieve a higher concentration of NO in tumor tissue by delivering arginine to the immune cells of the tumor [149]. Therefore there has been increasing interest in the application of poly-arginine for tumor targeting.
Kudo et al. have designed polyion complex micelles from a poly(ethylene glycol)-block-poly(l-arginine) block copolymer (PEG-b-PArg) and chondroitin sulfate for systemic anticancer immunotherapy [149] Fig. 4. Without activation the NPs did not generate NO when exposed to iNOS in vitro. After trypsin pretreatment and hydrolysis of the polymeric Arg to monomeric arginine, iNOS exposure then resulted in NO generation. Therefore the activation of the NPs required enzymatic degradation to monomeric arginine in the lysosomes of immune or cancer cells in tumor tissue. Hence, the systemic administration of the NPs did not induce any tangible adverse effects. However they did significantly suppressed tumor growth rate in a dose-dependent manner [149].
Fig. (4).
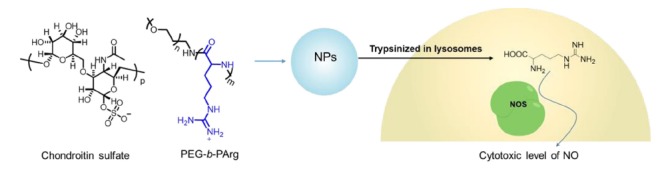
Schematic illustration of polyionic NPs made from PEG-b-PArg and chondroitin sulfate for systemic anticancer immunotherapy [145]. The L-arginine moiety is highlighted (blue). (The color version of the figure is available in the electronic copy of the article).
The pivotal role of albumin in the circulation of NO in the plasma has been well established [150]. Human serum albumin (HSA) has one site of S-nitrosation at the Cys-34 thiol, which can carry NO equivalents in the circulatory system in the form of an S-nitrosothiol [150]. S-nitrosated HSA (HSASNO) has significantly superior stability compared with low molecular weight SNTs [151]. In numerous studies HSA has been used as a long-acting and safe NO donor for several pathological conditions including cancer (this has been recently reviewed [151]). As it only has one S-NO group HSASNO produced low levels of NO (nM range) when administered at physiological levels mimicking HSA in blood, therefore under pathological conditions it acts as a cytoprotective agent as it produces low concentrations of NO. To improve its NO release properties HSA has been chemically modified by attachment of linkers and then binding to low molecular weight SNTs to form poly-HSASNO. Here two HSA molecules were linked together using the amino acid linker (GGGGS)2 to form a HAS-dimer which had a significantly longer circulation time than the mono-
meric form of HSA [152]. It has been shown that the HSA-dimer had an enhanced accumulation in solid tumors via an increased EPR effect [153] Fig. 5 . Nitrosation of the HSA-dimer resulted in NO releasing NPs (SNO-HSA-dimer), which also further enhanced its EPR effect in tumors [153], as well as other anti-cancer loaded NPs. In xenograft mice with B16 or C26 tumors, the SNO-HSA-dimer increased tumor accumulation of the anti-cancer agents N-(2-hydroxypropyl) methacrylamide (HPMA)-zinc protoporphyrin (ZnPP) and PEGylated liposomal doxorubicin (Doxil) compared with only NnPP or Doxil treated mice (by a factor of 3-4 in C26 tumors and 6 in B16 tumors), and thereby enhanced the anticancer efficacy of these drugs Fig. 6 , and increased survival of the animals [154]. The administration of SNO-HSA-dimer itself was found to be safe in the animals, as it did not affect blood pressure or heart rate.
Fig. (5).
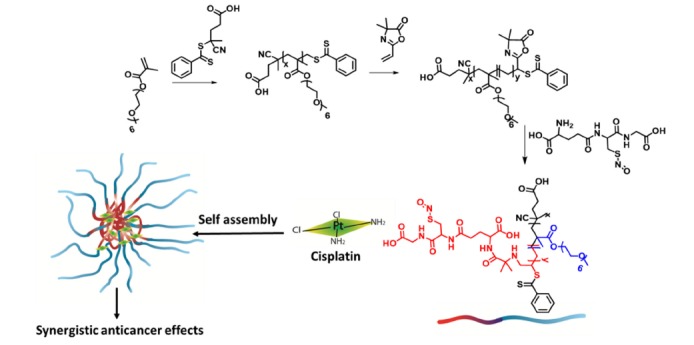
The synthesis of NO-releasing micelles(black/blue) using GSNO (red) for synergistic cytotoxicity against cancer cells [146]. (The color version of the figure is available in the electronic copy of the article).
Fig. (6).
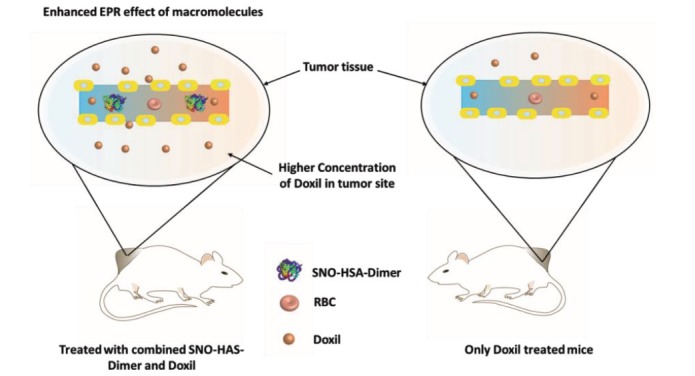
The NO releasing NP SNO-HSA-dimer enhanced the EPR effect of macromolecules in tumor tissue, and hence augmented the anti-tumor effects of the Dox loaded NP Doxil.
In 2002 the first microparticle formed from an NO releasing polymer was reported by the Meyerhoff group [155]. The group deployed methyl methacrylate as an amine bearing monomer, and utilized 1,6-hexanedioldimethacrylate as a reactive cross-linker to form polymeric microbeads. The secondary amine groups were then converted to diazeniumdiolates to form NO releasing particles with a size of 100-200 µm. The particles had t1/2 values for NO release ranging from 30 to 60 min when suspended in PBS [155]. Following this initial work there has been an extensive effort to develop tunable release polymers for the controlled delivery of NO. For example GSNO was covalently attached to a diblock copolymer made from the polymerization of oligoethylene glycol-methacry- late (OEG-MA) and (4-cyanopentanoic acid)-4-dithiobenzoate to afford an NO releasing polymer, which in aqueous environments self-assembled and made nano-sized micelles. The polymeric NPs improved the stability of GSNO (3.5 fold) without affecting the efficacy of intracellular delivery. Additionally, in the presence of ascorbic acid, the t1/2 of the NPs was significantly longer than the GSNO alone, demonstrating protection from S-nitrosothiol degradation pathways. When neuroblastoma cells were pre-treated with these NO releasing NPs they were significantly more sensitive to cisplatin treatment, with a 5-fold shift in IC50.
The use of dendrimers for controlled NO delivery was first described by the Schoenfisch group [156]. They used the commercially available generation 3 and 5 polypropylenimine dendrimers, and reacted then with high pressure gaseous NO (5 atm) to form NONOate dendrimers. NONOate
dendrimers generated from primary amines were not effective in providing sustained release of NO. However secondary amine NONOate dendrimers showed a high storage capacity for NO, and the release durations were significantly longer compared to small molecule alkyl secondary amine NONOates; however, the t1/2 of most of the NO releasing dendrimers were less than 2 h [156]. In another study from the same group, generation 4 polyamidoamine (PAMAM) dendrimers were functionalized with either SNAP or N-acetyl-L-cysteine to yield thiol terminated dendrimers, then converted to SNT terminated dendrimers [157]. Similarly to low molecular weight SNTs, the kinetics of NO release were found to be highly sensitive to Cu+ and photoactivation. The SNAP-dendrimer was particularly effective, showing 62% inhibition of platelet aggregation, which was almost 3 times greater than SNAP alone [157].
Gold nanoparticles (GNPs) have been widely used as non-toxic carriers in drug and gene delivery systems [158]. The inert and non-toxic gold core [159] and facile synthesis of GNPs make them very popular agents for drug delivery [158]. Rothrock et al. initially used GNPs for the controlled delivery of NO [160]. The synthesis strategy is shown in Fig. 7 , approximately 2 nm diameter GNPs were made by reduction of tetrachloroaurate (AuCl4-) with sodium citrate or sodium borohydride in the presence of hexanethiol ligands. Then the ligands were exchanged with bromoalkane thiols, followed by reaction with ethylenediamine, butylamine or hexanediamine. When the GNPs (functionalized with a primary or secondary amine) were exposed to high pressure gaseous NO, the final NO-releasing NPs were generated [160]. Theoretically an advantage of these NO releasing NPs is the ability to control both the amount of loaded NO, and the kinetics of NO release, by modification of the amount and/or structure of amine groups in the NPs [130]. However, due to the poor NO storage capacity and low solubility in aqueous media, the application of this class of NO-releasing NPs is limited [130].
Fig. (7).
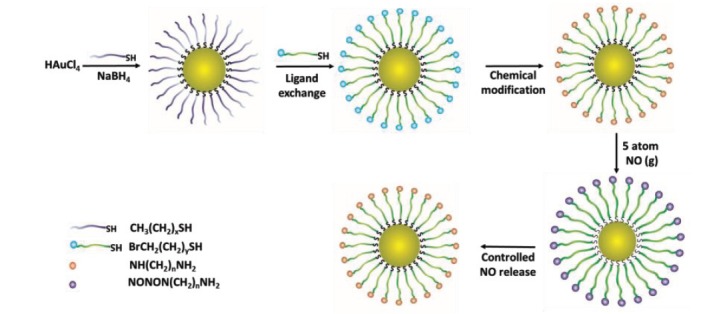
NO releasing gold NPs. HAuCl4 was reduced by sodium borohydride in the presence of an alkylthiol, and the ligand then exchanged and chemically modified to make the NO releasing NPs [154].
Zeolites are aluminosilicate minerals which contain a variety of pore sizes and shapes [161], while metal-organic frameworks (MOFs) are a polymeric material consisting of metal ions bridged by organic ligands [162]. These classes of highly porous materials have been widely used for ion exchange, catalysis, and gas adsorption [161, 162]. The chemical characteristics of these materials, especially nano-scaled systems, make them potentially suitable for medical applications including the delivery of NO [163]. In zeolites, NO is chemisorbed to the cationic centers of the crystals [130], while in MOFs it can chemisorb to either the cationic centers or the organic structure of the framework. MOFs are well known for their high porosity and large surface areas, which gives them the capability of storing and delivering gases such as NO via physiosorbing (physically trapping) within their pores [164, 165]. The physiochemical properties of MOFs (such as the level of porosity) can remarkably affect their control over NO delivery [166-168]. In general, the biomedical application of most zeolites and MOFs is restricted due to their low water solubility and instability [130].
More recently, stimuli responsive NO delivery has been utilized to control NO release on demand and in tumor tissue. For example, light responsive NO releasing NPs were made from the encapsulation of N,N′-di-sec-butyl-N,N′-dinitroso-1,4-phe- nylenediamine (BNN6), a NO releasing molecule, Dox and a mPEG-PLGA copolymer Fig. 8 [169]. Upon the irradiation by UV light the NO content of BNN6 was released, and the generated NO gas broke the nanoparticle shell and led to the release of Dox. Hence, the release of both NO and Dox was responsive to UV irradiation. Incubation of OVCAR-8/ADR cells with the NPs resulted in a higher intracellular concentration of Dox compared to the equivalent treatment with free Dox, potentially due to NO release from the NPs inhibiting MDR [169]. In addition, other sources of energies (e.g. NIR, X rays and ultrasound) have been used to tune the NO release and anticancer properties of the NPs [170-172] as summarized in Table .
Fig. (8).
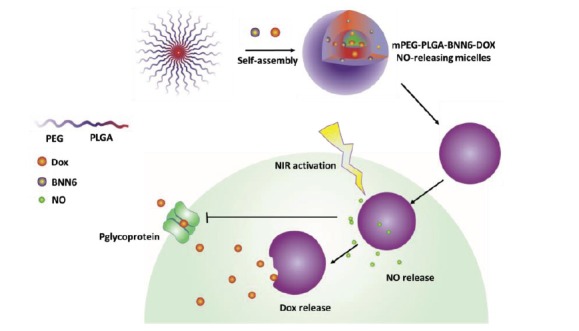
BNN6 is a NO releasing micelle which contains Dox. Upon photo-irradiation NO was released from BNN6, the nanoparticle shell broken, and Dox then released. In addition, the released NO significantly inhibited MDR (p-glycoprotein) and thereby resistance to Dox.
CONCLUSION AND FUTURE PERSPECTIVES
As detailed above, NO plays a key role in tumor biology and therapy, and there are several approaches that have been utilized to induce antitumor activities, or improve the efficacy of chemotherapy and radiotherapy, from NO releasing compounds. So far at least 16 families of NO precursor and NO donor functional groups have been developed. However only organic nitrates and SNP have clinical applications, predominantly for cardiovascular disease. Hence the development of stable and tuned NO donor compounds is a priority for drug discovery programs. Nanotechnology has revolutionized the NO delivery field, and the level of interest in NO releasing NPs has exploded over last decade. By protecting polymers carrying NO releasing functional groups from hydrolysis, researchers have developed stable and controllable NO donors with superior biological functions, paving the way for their therapeutic applications towards a wide range of diseases including cancer.
However, the controlled delivery of cytotoxic levels of NO to tumor tissues remains a major challenge. Therefore, similarly to the hybrid NO donor drugs, investigations into NO releasing NPs have recently shifted to using them as an approach to strengthen the efficacy of existing drugs. Here NO delivery can inhibit MDR and potentiate the efficacy of chemotherapeutic drugs (recently reviewed [173-183]). Furthermore, the use of NO releasing nanocarriers can sensitize hypoxic cells to radiation, circumventing radiation resistance, which is a major hurdle in treating solid tumors [127].
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- AMN
= Amyl nitrite
- AuCl4-
= Tetrachloroaurate
- BNN6
= N,N′-di-sec-butyl-N,N′-dinitroso-1,4-phenylenediamine
- cGMP
= Cyclic guanosine monophosphate
- Cyt c
= Cytochrome c
- DDS
= Drug delivery systems
- DEA/NO
= Diethylamine NONOate
- Dox
= DETA
- /NO
Diethylenetriamine NONOate Doxorubicin
- Doxil
= Liposomal doxorubicin
- EGFR
= Epidermal growth factor receptor
- eNOS
= Endothelial NOS
- EPR
= Enhanced permeability and retention
- GNPs
= Gold nanoparticles
- GSH
= Glutathione
- GSNO
= S-nitrosoglutathione
- GTN
= Glyceryl trinitrate
- HSA
= Human serum albumin
- HSASNO
= S-nitrosated human serum albumin
- IAMN
= Isoamyl nitrite
- IC50
= Inhibitory Concentration (50%)
- iNOS
= Inducible NOS
- ISBN
= Isobutyl nitrite
- ISDN
= Isosorbide dinitrate
- ISMN
= Isosorbide mononitrate
- MA/NO
= Methylamine hexamethylene methylamine NONOate
- MDR
= Multi drug resistance
- MOFs
= Metal-organic frameworks
- NADPH
= Nicotinamide adenine dinucleotide phosphate
- nNOS
= Neuronal NOS
- NO
= Nitric oxide
- NOS
= Nitric oxide synthases
- NP
= Nanoparticle
- OEG-MA
= Oligoethylene glycol-methacrylate
- PAMAM
= Polyamidoamine
- PETN
= Pentaerythritol tetranitrate
- PFC
= Perfluorocarbon
- Ph3SNO
= Trityl S-nitrosothiol
- PKG
= Protein kinase G
- PLGA
= Poly-lactic-co-glycolic acid
- PROLI/NO
= Proli NONOate
- RNS
= Reactive nitrogen species
- ROS
= Reactive oxygen species
- RSH
= Thiol
- SIN-1
= 3-Morpholinosydnonimine
- SMA
= Styrene maleic acid
- SNAP
= S-nitroso-N-acetyl-DL-penicillamine
- SNP
= Sodium nitroprusside
- SNT
= S-nitrosothiol
- SPER/NO
= Spermine NONOate
- STZ
= Streptozocin
- t1/2
= Half-life
- TBME
= Tert-butyl methyl ether
- tButONO
= Tert-butyl nitrite
- tButSNO
= Tert-butyl S-nitrosothiol
- tDodSNO
= Tert-dodecane S-nitrosothiol
- VEGF
= Vascular endothelial growth factor
- ZnPP
= Zinc protoporphyrin
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by (Arabian Gulf university research grant E003-PI-04/17) to KG. GIG was supported by a Laurenson Award from the Otago Medical Research Foundation.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Duong H.T., Kamarudin Z.M., Erlich R.B.F et al. Intracellular nitric oxide delivery from stable NO-polymeric nanoparticle carriers. Chem. Commun. 2013;49(39):4190–4192. doi: 10.1039/c2cc37181b. [DOI] [PubMed] [Google Scholar]
- 2.Wang P.G., Xian M., Tang X., et al. Nitric oxide donors: chemical activities and biological applications. Chem. Rev. 2002;102(4):1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro L.J. Nitric oxide: a unique endogenous signaling molecule in vascular biology (Nobel lecture). Angew. Chem. Int. Ed. 1999;38(13‐14):1882–1892. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 4.Moncada S., Palmer R.M., Higgs E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 5.Ignarro L.J. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu. Rev. Pharmacol. Toxicol. 1990;30(1):535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]
- 6.Shaw A.W., Vosper A.J. Solubility of nitric oxide in aqueous and nonaqueous solvents. J. Chem. Soc., Faraday Trans. I. 1977;73:1239–1244. [Google Scholar]
- 7.Malinski T., Taha Z., Grunfeld S., Patton S., Kapturczak M., Tomboulian P. Diffusion of nitric oxide in the aorta wall monitored in situ by porphyrinic microsensors. Biochem. Biophys. Res. Commun. 1993;193(3):1076–1082. doi: 10.1006/bbrc.1993.1735. [DOI] [PubMed] [Google Scholar]
- 8.Moller M.N., Denicola A. Diffusion of nitric oxide and oxygen in lipoproteins and membranes studied by pyrene fluorescence quenching. Free Radic. Biol. Med. 2018;128:137–143. doi: 10.1016/j.freeradbiomed.2018.04.553. [DOI] [PubMed] [Google Scholar]
- 9.Choudhari S.K., Chaudhary M., Bagde S., Gadbail A.R., Joshi V. Nitric oxide and cancer: a review. World J. Surg. Oncol. 2013;11(1):118. doi: 10.1186/1477-7819-11-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lincoln J., Hoyle C.H., Burnstock G. Nitric oxide in health and disease: Burnstock. Cambridge: Cambridge University Press; 1997. [Google Scholar]
- 11.Wink D.A., Vodovotz Y., Laval J., Laval F., Dewhirst M.W., Mitchell J.B. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19(5):711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 12.Wink D.A., Mitchell J.B. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998;25(4):434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 13.Maruyama K., Zhang E., Maruyama J. Clinical application of inhaled nitric oxide. In: Yoshikawa T., Naito Y., editors. Gas Biology Research in Clinical Practice. Basel, Switzerland: Karger Publishers; 2011. pp. 43–55. [Google Scholar]
- 14.Wu H.W., Li Z.G., Liu G., Lu G.Z., Liang H.Y. Effect of nitric oxide inhalation for the treatment of neonatal pulmonary hypertension. Eur Rev Med Pharmaco. 2016;20(21):4607–4611. [PubMed] [Google Scholar]
- 15.Troncy E., Francoeur M., Blaise G. Inhaled nitric oxide: clinical applications, indications, and toxicology. Can. J. Anaesth. 1997;44(9):973–988. doi: 10.1007/BF03011970. [DOI] [PubMed] [Google Scholar]
- 16.Cooper C.E. Nitric oxide and iron proteins. Biochim. Biophys. Acta. 1999;1411(2):290–309. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 17.Martin E., Davis K., Bian K., Lee Y., Murad F. Cellular signaling with nitric oxide and cyclic guanosine monophosphate. Semin. Perinatol. 2000;24(1):2–6. doi: 10.1016/s0146-0005(00)80045-2. [DOI] [PubMed] [Google Scholar]
- 18.Cohen R.A., Weisbrod R.M., Gericke M., Yaghoubi M., Bierl C., Bolotina V.M. Mechanism of nitric oxide-induced vasodilatation: refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ. Res. 1999;84(2):210–219. doi: 10.1161/01.res.84.2.210. [DOI] [PubMed] [Google Scholar]
- 19.Paterno R., Heistad D.D., Faraci F.M. Functional activity of Ca2+-dependent K+ channels is increased in basilar artery during chronic hypertension. Am J Physiol Heart Circ. 1997;272(3):H1287–H91. doi: 10.1152/ajpheart.1997.272.3.H1287. [DOI] [PubMed] [Google Scholar]
- 20.Paternò R., Faraci F.M., Heistad D.D. Role of Ca2+-dependent K+ channels in cerebral vasodilatation induced by increases in cyclic GMP and cyclic AMP in the rat. Stroke. 1996;27(9):1603–1608. doi: 10.1161/01.str.27.9.1603. [DOI] [PubMed] [Google Scholar]
- 21.Blough N.V., Zafiriou O.C. Reaction of superoxide with nitric oxide to form peroxonitrite in alkaline aqueous solution. Inorg. Chem. 1985;24(22):3502–3504. [Google Scholar]
- 22.Rubbo H., Radi R., Trujillo M., et al. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994;269(42):26066–26075. [PubMed] [Google Scholar]
- 23.Fukumura D., Kashiwagi S., Jain R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer. 2006;6(7):521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 24.Hirst D., Robson T. Targeting nitric oxide for cancer therapy. J. Pharm. Pharmacol. 2007;59(1):3–13. doi: 10.1211/jpp.59.1.0002. [DOI] [PubMed] [Google Scholar]
- 25.Vannini F., Kashfi K., Nath N. The dual role of iNOS in cancer. Redox Biol. 2015;6(Suppl. C):334–343. doi: 10.1016/j.redox.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Z., Fu J., Zhang Y. Nitric oxide donor-based cancer therapy: advances and prospects. J. Med. Chem. 2017;60(18):7617–7635. doi: 10.1021/acs.jmedchem.6b01672. [DOI] [PubMed] [Google Scholar]
- 27.Predonzani A., Calì B., Agnellini A.H., Molon B. Spotlights on immunological effects of reactive nitrogen species: when inflammation says nitric oxide. World J. Exp. Med. 2015;5(2):64. doi: 10.5493/wjem.v5.i2.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wink D.A., Hines H.B., Cheng R.Y., et al. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011;89(6):873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas D.D., Espey M.G., Ridnour L.A., et al. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc. Natl. Acad. Sci. USA. 2004;101(24):8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ridnour L.A., Isenberg J.S., Espey M.G., Thomas D.D., Roberts D.D., Wink D.A. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. USA. 2005;102(37):13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wink D.A., Hines H.B., Cheng R.Y., et al. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011;89(6):873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta. 1999;1411(2):351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 33.Brown G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369(2-3):136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 34.Bal-Price A., Brown G.C. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J. Neurochem. 2000;75(4):1455–1464. doi: 10.1046/j.1471-4159.2000.0751455.x. [DOI] [PubMed] [Google Scholar]
- 35.Heller R., Polack T., Gräbner R., Till U. Nitric oxide inhibits proliferation of human endothelial cells via a mechanism independent of cGMP. Atherosclerosis. 1999;144(1):49–57. doi: 10.1016/s0021-9150(99)00041-6. [DOI] [PubMed] [Google Scholar]
- 36.Kanamaru Y., Takada T., Saura R., Mizuno K. Effect of nitric oxide on mouse clonal osteogenic cell, MC3T3-E1, proliferation in vitro. Kobe J. Med. Sci. 2001;47(1):1–12. [PubMed] [Google Scholar]
- 37.Xie K., Huang S., Dong Z., et al. Transfection with the inducible nitric oxide synthase gene suppresses tumorigenicity and abrogates metastasis by K-1735 murine melanoma cells. J. Exp. Med. 1995;181(4):1333–1343. doi: 10.1084/jem.181.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lala P.K., Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001;2(3):149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhang R., Ma A., Urbanski S.J., McCafferty D-M. Induction of inducible nitric oxide synthase: a protective mechanism in colitis-induced adenocarcinoma. Carcinogenesis. 2006;28(5):1122–1130. doi: 10.1093/carcin/bgl224. [DOI] [PubMed] [Google Scholar]
- 40.Cheng H., Wang L., Mollica M., Re A.T., Wu S., Zuo L. Nitric oxide in cancer metastasis. Cancer Lett. 2014;353(1):1–7. doi: 10.1016/j.canlet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min C., Eddy S.F., Sherr D.H., Sonenshein G.E. NF‐κB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008;104(3):733–744. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 43.Bonavida B., Baritaki S. Inhibition of epithelial-to-mesenchymal transition (EMT) in cancer by nitric oxide: pivotal roles of nitrosylation of NF-κB, YY1 and Snail. For. Immunopathol. Dis. Therap. 2012;3(2):125–133. doi: 10.1615/ForumImmunDisTher.2012006065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan X., Wang X., Lei W., et al. Nitric oxide suppresses transforming growth factor‐β1-induced epithelial-to-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology. 2009;50(5):1577–1587. doi: 10.1002/hep.23156. [DOI] [PubMed] [Google Scholar]
- 45.Powan P., Chanvorachote P. Nitric oxide mediates cell aggregation and mesenchymal to epithelial transition in anoikis-resistant lung cancer cells. Mol. Cell. Biochem. 2014;393(1-2):237–245. doi: 10.1007/s11010-014-2066-7. [DOI] [PubMed] [Google Scholar]
- 46.Jain K.K. Drug Delivery Systems. Switzerland: Springer Science and Business Media; 2008. [Google Scholar]
- 47.Kumari A., Yadav S.K., Yadav S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces. 2010;75(1):1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Hughes G.A. Nanostructure-mediated drug delivery. Nanomedicine (Lond.) 2017;1(1):22–30. doi: 10.1016/j.nano.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Jain R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 50.Vaupel P., Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9(Suppl. 5):4–9. doi: 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- 51.Secomb T.W., Hsu R., Park E.Y., Dewhirst M.W. Green’s function methods for analysis of oxygen delivery to tissue by microvascular networks. Ann. Biomed. Eng. 2004;32(11):1519–1529. doi: 10.1114/b:abme.0000049036.08817.44. [DOI] [PubMed] [Google Scholar]
- 52.Dewhirst M., Ong E., Braun R., et al. Quantification of longitudinal tissue pO2 gradients in window chamber tumours: impact on tumour hypoxia. Br. J. Cancer. 1999;79(11-12):1717. doi: 10.1038/sj.bjc.6690273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewhirst M.W., Cao Y., Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer. 2008;8(6):425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 55.Vaupel P., Thews O., Hoeckel M. Treatment resistance of solid tumors. Med. Oncol. 2001;18(4):243–259. doi: 10.1385/MO:18:4:243. [DOI] [PubMed] [Google Scholar]
- 56.Gacche R.N. Compensatory angiogenesis and tumor refractoriness. Oncogenesis. 2015;4:e153. doi: 10.1038/oncsis.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maki S., Konno T., Maeda H. Image enhancement in computerized tomography for sensitive diagnosis of liver cancer and semiquantitation of tumor selective drug targeting with oily contrast medium. Cancer. 1985;56(4):751–757. doi: 10.1002/1097-0142(19850815)56:4<751::aid-cncr2820560409>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 58.Taurin S., Nehoff H., Greish K. Anticancer nanomedicine and tumor vascular permeability; where is the missing link? J. Control. Release. 2012;164(3):265–275. doi: 10.1016/j.jconrel.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 59.Maeda H., Wu J., Sawa T., Matsumura Y., Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release. 2000;65(1):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 60.Fang J., Nakamura H., Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011;63(3):136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 61.Iyer A.K., Khaled G., Fang J., Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today. 2006;11(17):812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Wu J., Akaike T., Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998;58(1):159–165. [PubMed] [Google Scholar]
- 63.Akaike T., Horie H., Noguchi Y., et al. Excessive production of nitric oxide in rat solid tumor and its implication in rapid tumor growth. Cancer. 1996;77(8):1598–1604. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1598::AID-CNCR27>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 64.Seki T., Fang J., Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100(12):2426–2430. doi: 10.1111/j.1349-7006.2009.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vannini F., Kashfi K., Nath N. The dual role of iNOS in cancer. Redox Biol. 2015;6:334–343. doi: 10.1016/j.redox.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lind M., Hayes A., Caprnda M., et al. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017;93:370–375. doi: 10.1016/j.biopha.2017.06.036. [DOI] [PubMed] [Google Scholar]
- 67.Kubota M., Sakakihara Y., Mori M., Yamagata T., Momoi-Yoshida M. Beneficial effect of L-arginine for stroke-like episode in MELAS. Brain Dev. 2004;26(7):481–483. doi: 10.1016/j.braindev.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 68.Finsterer J., Zarrouk-Mahjoub S. A beneficial effect of l-arginine for stroke-like episodes is currently unsupported. Mol. Genet. Metab. Rep. 2018;15:67. doi: 10.1016/j.ymgmr.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howell K., Costello C.M., Sands M., Dooley I., McLoughlin P. L-Arginine promotes angiogenesis in the chronically hypoxic lung: a novel mechanism ameliorating pulmonary hypertension. Am J Physiol-Lung C. 2009;296(6):1042–1050. doi: 10.1152/ajplung.90327.2008. [DOI] [PubMed] [Google Scholar]
- 70.Barbul A., Lazarou S.A., Efron D.T., Wasserkrug H.L., Efron G. Arginine enhances wound healing and lymphocyte immune responses in humans. Surgery. 1990;108(2):331–336. [PubMed] [Google Scholar]
- 71.Clement B., Schade D., Kotthaus J. N-ω- hydroxy-L-arginine derivatives for the treatment of diseases. 2016 [Google Scholar]
- 72.Reid K.M., Tsung A., Kaizu T., et al. Liver I/R injury is improved by the arginase inhibitor, N ω-hydroxy-nor-L-arginine (nor-NOHA). Am J Physiol-Gastr L. 2007;292(2):512–517. doi: 10.1152/ajpgi.00227.2006. [DOI] [PubMed] [Google Scholar]
- 73.Münzel T., Steven S., Daiber A. Organic nitrates: update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vascul. Pharmacol. 2014;63(3):105–113. doi: 10.1016/j.vph.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 74.Schroder H. Cytochrome P-450 mediates bioactivation of organic nitrates. J. Pharmacol. Exp. Ther. 1992;262(1):298–302. [PubMed] [Google Scholar]
- 75.McDonald B.J., Bennett B.M. Biotransformation of glyceryl trinitrate by rat aortic cytochrome P450. Biochem. Pharmacol. 1993;45(1):268–270. doi: 10.1016/0006-2952(93)90403-j. [DOI] [PubMed] [Google Scholar]
- 76.Kenkare S.R., Han C., Benet L.Z. Correlation of the response to nitroglycerin in rabbit aorta with the activity of the mu class glutathione S-transferase. Biochem. Pharmacol. 1994;48(12):2231–2235. doi: 10.1016/0006-2952(94)00415-3. [DOI] [PubMed] [Google Scholar]
- 77.Loscalzo J. N-Acetylcysteine potentiates inhibition of platelet aggregation by nitroglycerin. J. Clin. Invest. 1985;76(2):703–708. doi: 10.1172/JCI112024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hutter J., Schmidt M., Rittler J. Effects of sulfhydryl-containing compounds on nitroglycerin-induced coronary dilatation in isolated working rat hearts. Eur. J. Pharmacol. 1988;156(2):215–222. doi: 10.1016/0014-2999(88)90324-x. [DOI] [PubMed] [Google Scholar]
- 79.Ignarro L.J., Napoli C., Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide. Circ. Res. 2002;90(1):21–28. doi: 10.1161/hh0102.102330. [DOI] [PubMed] [Google Scholar]
- 80.Thompson A. Counselling in practice: Glyceryl trinitrate for acute angina. Australian Pharmacist. 2016;35(1):46. [Google Scholar]
- 81.Ahlner J., Andersson R., Torfgård K., Axelsson K. Organic nitrate esters: clinical use and mechanisms of actions. Pharmacol. Rev. 1991;43(3):351–423. [PubMed] [Google Scholar]
- 82.Gardiner S., Compton A., Kemp P., Bennett T. Regional and cardiac haemodynamic responses to glyceryl trinitrate, acetylcholine, bradykinin and endothelin‐1 in conscious rats: effects of NG‐nitro‐l‐arginine methyl ester. Br. J. Pharmacol. 1990;101(3):632–639. doi: 10.1111/j.1476-5381.1990.tb14132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Akhras F., Jackson G. Efficacy of nifedipine and isosorbide mononitrate in combination with atenolol in stable angina. Lancet. 1991;338(8774):1036–1039. doi: 10.1016/0140-6736(91)91900-f. [DOI] [PubMed] [Google Scholar]
- 84.Goldstein S., Czapski G. Mechanism of the nitrosation of thiols and amines by oxygenated NO solutions: the nature of the nitrosating intermediates. J. Am. Chem. Soc. 1996;118(14):3419–3425. [Google Scholar]
- 85.Cederqvist B., Persson M.G., Gustafsson L.E. Direct demonstration of NO formation in vivo from organic nitrites and nitrates, and correlation to effects on blood pressure and to in vitro effects. Biochem. Pharmacol. 1994;47(6):1047–1053. doi: 10.1016/0006-2952(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 86.Omar S.A., Artime E., Webb A.J. A comparison of organic and inorganic nitrates/nitrites. Nitric Oxide. 2012;26(4):229–240. doi: 10.1016/j.niox.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 87.Bauer J.A., Nolan T., Fung H.L. Vascular and hemodynamic differences between organic nitrates and nitrites. J. Pharmacol. Exp. Ther. 1997;280(1):326–331. [PubMed] [Google Scholar]
- 88.Williams R. Nitric oxide in biology: its role as a ligand. Chem. Soc. Rev. 1996;25(2):77–83. [Google Scholar]
- 89.Lim M.H., Lippard S.J. Metal-based turn-on fluorescent probes for sensing nitric oxide. Acc. Chem. Res. 2007;40(1):41–51. doi: 10.1021/ar950149t. [DOI] [PubMed] [Google Scholar]
- 90.Tinker J.H., Michenfelder J.D. Sodium nitroprusside: pharmacology, toxicology and therapeutics. Anesthesiology. 1976;45(3):340–354. [PubMed] [Google Scholar]
- 91.Hottinger D.G., Beebe D.S., Kozhimannil T., Prielipp R.C., Belani K.G. Sodium nitroprusside in 2014: a clinical concepts review. J. Anaesthesiol. Clin. Pharmacol. 2014;30(4):462–471. doi: 10.4103/0970-9185.142799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amaranath L., Kellermeyer W.F. Tachyphylaxis to sodium nitroprusside. Anesthesiology. 1976;44(4):345–348. doi: 10.1097/00000542-197604000-00016. [DOI] [PubMed] [Google Scholar]
- 93.Perschau R.A., Modell J.H., Bright R.W., Shirley P.D. Suspected sodium nitroprusside-induced cyanide intoxication. Anesth. Analg. 1977;56(4):533–537. doi: 10.1213/00000539-197707000-00015. [DOI] [PubMed] [Google Scholar]
- 94.Fry N.L., Mascharak P.K. Photoactive ruthenium nitrosyls as NO donors: how to sensitize them toward visible light. Acc. Chem. Res. 2011;44(4):289–298. doi: 10.1021/ar100155t. [DOI] [PubMed] [Google Scholar]
- 95.Bezerra C.W., da Silva S.C., Gambardella M.T., et al. Water π-donation in trans-tetraammineruthenium (II): effect on coordinated-water properties induced by a trans NO ligand. Inorg. Chem. 1999;38(25):5660–5667. [Google Scholar]
- 96.Mascharak P.K. Recent progress in photoinduced no delivery with designed ruthenium nitrosyl complexes. Adv. Inorg. Chem. 2015;67:145–170. [Google Scholar]
- 97.Miller M., Megson I. Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007;151(3):305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morley D., Keefer L.K. Nitric oxide/nucleophile complexes: a unique class of nitric oxide-based vasodilators. J. Cardiovasc. Pharmacol. 1993;22:S3–S9. [PubMed] [Google Scholar]
- 99.Hrabie J.A., Klose J.R., Wink D.A., Keefer L.K. New nitric oxide-releasing zwitterions derived from polyamines. J. Org. Chem. 1993;58(6):1472–1476. [Google Scholar]
- 100.Brilli R.J., Krafte-Jacobs B., Smith D.J., et al. Intratracheal instillation of a novel NO/nucleophile adduct selectively reduces pulmonary hypertension. J. Appl. Physiol. 1997;83(6):1968–1975. doi: 10.1152/jappl.1997.83.6.1968. [DOI] [PubMed] [Google Scholar]
- 101.Lavery K.S., Rhodes C., Mcgraw A., Eppihimer M.J. Anti-thrombotic technologies for medical devices. Adv. Drug Deliv. Rev. 2017;112:2–11. doi: 10.1016/j.addr.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 102.Krausz A, Friedman AJ. Nitric oxide as a surgical adjuvant. Fut Sci OA. 2015;1(1) doi: 10.4155/fso.15.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diodati J.G., Quyyumi A.A., Hussain N., Keefer L.K. Complexes of nitric oxide with nucleophiles as agents for the controlled biological release of nitric oxide: antiplatelet effect. Thromb. Haemost. 1993;70(4):654–658. [PubMed] [Google Scholar]
- 104.Maragos C.M., Morley D., Wink D.A., et al. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem. 1991;34(11):3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 105.Pearce C.G., Najjar S.F., Kapadia M.R., et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic. Biol. Med. 2008;44(1):73–81. doi: 10.1016/j.freeradbiomed.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hermann M., Kapiotis S., Hofbauer R., et al. Salicylate inhibits LDL oxidation initiated by superoxide/nitric oxide radicals. FEBS Lett. 1999;445(1):212–214. doi: 10.1016/s0014-5793(99)00043-5. [DOI] [PubMed] [Google Scholar]
- 107.Blaylock M.G., Cuthbertson B.H., Galley H.F., Ferguson N.R., Webster N.R. The effect of nitric oxide and peroxynitrite on apoptosis in human polymorphonuclear leukocytes. Free Radic. Biol. Med. 1998;25(6):748–752. doi: 10.1016/s0891-5849(98)00108-7. [DOI] [PubMed] [Google Scholar]
- 108.Feelisch M., Ostrowski J., Noack E. On the mechanism of NO release from sydnonimines. J. Cardiovasc. Pharmacol. Ther. 1989;14:S13–S22. [PubMed] [Google Scholar]
- 109.Molsidomine R.J. Blood Vessels. 1990;27(2-5):282–294. doi: 10.1159/000158820. [DOI] [PubMed] [Google Scholar]
- 110.Peng W., Hoidal J.R., Farrukh I.S. Regulation of Ca(2+)-activated K+ channels in pulmonary vascular smooth muscle cells: role of nitric oxide. J. Appl. Physiol. 1996;81(3):1264–1272. doi: 10.1152/jappl.1996.81.3.1264. [DOI] [PubMed] [Google Scholar]
- 111.Megson I.L., Webb D.J. Nitric oxide donor drugs: current status and future trends. Expert Opin. Investig. Drugs. 2002;11(5):587–601. doi: 10.1517/13543784.11.5.587. [DOI] [PubMed] [Google Scholar]
- 112.Al-Sa’doni H., Ferro A. S-nitrosothiols as nitric oxide-donors: chemistry, biology and possible future therapeutic applications. Curr. Med. Chem. 2004;11(20):2679–2690. doi: 10.2174/0929867043364397. [DOI] [PubMed] [Google Scholar]
- 113.Williams D.L.H. The chemistry of S-nitrosothiols. Acc. Chem. Res. 1999;32(10):869–876. [Google Scholar]
- 114.Zhang C., Biggs T.D., Devarie-Baez N.O., Shuang S., Dong C., Xian M. S-Nitrosothiols: chemistry and reactions. Chem. Commun. 2017;53(82):11266–11277. doi: 10.1039/c7cc06574d. [DOI] [PubMed] [Google Scholar]
- 115.Giles N.M., Kumari S., Gang B.P., Yuen C.W., Billaud E.M., Giles G.I. The molecular design of s‐nitrosothiols as photodynamic agents for controlled nitric oxide release. Chem. Biol. Drug Des. 2012;80(3):471–478. doi: 10.1111/j.1747-0285.2012.01420.x. [DOI] [PubMed] [Google Scholar]
- 116.Adeghate E., Parvez S.H. Nitric oxide and neuronal and pancreatic beta cell death. Toxicology. 2000;153(1-3):143–156. doi: 10.1016/s0300-483x(00)00310-3. [DOI] [PubMed] [Google Scholar]
- 117.Spinas G.A. The dual role of nitric oxide in islet β-cells. Physiology (Bethesda) 1999;14(2):49–54. doi: 10.1152/physiologyonline.1999.14.2.49. [DOI] [PubMed] [Google Scholar]
- 118.McGill A.D., Zhang W., Wittbrodt J., Wang J., Schlegel H.B., Wang P.G. Para-Substituted N-nitroso-N-oxyben-zenamine ammonium salts: a new class of redox-sensitive nitric oxide releasing compounds. Bioorg. Med. Chem. 2000;8(2):405–412. doi: 10.1016/s0968-0896(99)00300-4. [DOI] [PubMed] [Google Scholar]
- 119.Hecker M., Vorhoff W., Bara A.T., Mordvintcev P.I., Busse R. Characterization of furoxans as a new class of tolerance-resistant nitrovasodilators. Naunyn Schmiedebergs Arch. Pharmacol. 1995;351(4):426–432. doi: 10.1007/BF00169084. [DOI] [PubMed] [Google Scholar]
- 120.Bohn H., Brendel J., Martorana P.A., Schonafinger K. Cardiovascular actions of the furoxan CAS 1609, a novel nitric oxide donor. Br. J. Pharmacol. 1995;114(8):1605–1612. doi: 10.1111/j.1476-5381.1995.tb14946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serafim R.A., Pernichelle F.G., Ferreira E.I. The latest advances in the discovery of nitric oxide hybrid drug compounds. Expert Opin. Drug Discov. 2017;12(9):941–953. doi: 10.1080/17460441.2017.1344400. [DOI] [PubMed] [Google Scholar]
- 122.Bandarage U.K., Chen L., Fang X., et al. Nitrosothiol esters of diclofenac: synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs. J. Med. Chem. 2000;43(21):4005–4016. doi: 10.1021/jm000178w. [DOI] [PubMed] [Google Scholar]
- 123.Turnbull C.M., Cena C., Fruttero R., Gasco A., Rossi A.G., Megson I.L. Mechanism of action of novel NO-releasing furoxan derivatives of aspirin in human platelets. Br. J. Pharmacol. 2006;148(4):517–526. doi: 10.1038/sj.bjp.0706743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tesei A., Zoli W., Fabbri F., et al. NCX 4040, an NO-donating acetylsalicylic acid derivative: efficacy and mechanisms of action in cancer cells. Nitric Oxide. 2008;19(2):225–236. doi: 10.1016/j.niox.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 125.Kashfi K., Rigas B. Molecular targets of nitric-oxide-donating aspirin in cancer. Biochem. Soc. Trans. 2005;33(Pt 4):701–704. doi: 10.1042/BST0330701. [DOI] [PubMed] [Google Scholar]
- 126.Chegaev K., Riganti C., Lazzarato L., et al. Nitric oxide donor doxorubicins accumulate into doxorubicin-resistant human colon cancer cells inducing cytotoxicity. ACS Med. Chem. Lett. 2011;2(7):494–497. doi: 10.1021/ml100302t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chegaev K., Fraix A., Gazzano E., et al. Light-regulated NO release as a novel strategy to overcome doxorubicin multidrug resistance. ACS Med. Chem. Lett. 2017;8(3):361–365. doi: 10.1021/acsmedchemlett.7b00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lane A.A., Chabner B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009;27(32):5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 129.Duan W., Li J., Inks E.S., et al. Design, synthesis, and antitumor evaluation of novel histone deacetylase inhibitors equipped with a phenylsulfonylfuroxan module as a nitric oxide donor. J. Med. Chem. 2015;58(10):4325–4338. doi: 10.1021/acs.jmedchem.5b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Riccio D.A., Schoenfisch M.H. Nitric oxide release: part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012;41(10):3731–3741. doi: 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carpenter A.W., Schoenfisch M.H. Nitric oxide release: part II. Therapeutic applications. Chem. Soc. Rev. 2012;41(10):3742–3752. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coneski P.N., Schoenfisch M.H. Nitric oxide release: part III. Measurement and reporting. Chem. Soc. Rev. 2012;41(10):3753–3758. doi: 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wilcox D.T., Glick P.L., Karamanoukian H.L., Leach C., Morin F.C., Fuhrman B.P. Perfluorocarbon-associated gas exchange improves pulmonary mechanics, oxygenation, ventilation, and allows nitric oxide delivery in the hypoplastic lung congenital diaphragmatic hernia lamb model. Crit. Care Med. 1995;23(11):1858–1863. doi: 10.1097/00003246-199511000-00012. [DOI] [PubMed] [Google Scholar]
- 134.Tao Z., Ghoroghchian P.P. Microparticle, nanoparticle, and stem cell-based oxygen carriers as advanced blood substitutes. Trends Biotechnol. 2014;32(9):466–473. doi: 10.1016/j.tibtech.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 135.Rafikova O., Sokolova E., Rafikov R., Nudler E. Control of plasma nitric oxide bioactivity by perfluorocarbons. Circulation. 2004;110(23):3573–3580. doi: 10.1161/01.CIR.0000148782.37563.F8. [DOI] [PubMed] [Google Scholar]
- 136.Cavalieri F., Finelli I., Tortora M., et al. Polymer microbubbles as diagnostic and therapeutic gas delivery device. Chem. Mater. 2008;20(10):3254–3258. [Google Scholar]
- 137.Huang S-L., Kee P.H., Kim H., et al. Nitric oxide-loaded echogenic liposomes for nitric oxide delivery and inhibition of intimal hyperplasia. Am. J. Cardiol. 2009;54(7):652–659. doi: 10.1016/j.jacc.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gomes A.J., Barbougli P.A., Espreafico E.M., Tfouni E. -[Ru(NO)(NH3)4(py)](BF4)3.H2O encapsulated in PLGA microparticles for delivery of nitric oxide to B16-F10 cells: Cytotoxicity and phototoxicity. J. Inorg. Biochem. 2008;102(4):757–766. doi: 10.1016/j.jinorgbio.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 139.Bohlender C., Landfester K., Crespy D., Schiller A. Unconventional non‐aqueous emulsions for the encapsulation of a phototriggerable NO‐donor complex in polymer nanoparticles. Part. Part. Syst. Charact. 2013;30(2):138–142. [Google Scholar]
- 140.Suchyta D.J., Schoenfisch M.H. Controlled release of nitric oxide from liposomes. ACS Biomater. Sci. Eng. 2017;3(9):2136–2143. doi: 10.1021/acsbiomaterials.7b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Suchyta D.J., Schoenfisch M.H. Encapsulation of N-diazeniumdiolates within liposomes for enhanced nitric oxide donor stability and delivery. Mol. Pharm. 2015;12(10):3569–3574. doi: 10.1021/acs.molpharmaceut.5b00248. [DOI] [PubMed] [Google Scholar]
- 142.Alimoradi H., Barzegar-Fallah A., Sammut I.A., Greish K., Giles G. Encapsulation of tDodSNO generates a nitric oxide releasing nanoparticle. Free Radic. Biol. Med. 2019;130:297–305. doi: 10.1016/j.freeradbiomed.2018.10.433. [DOI] [PubMed] [Google Scholar]
- 143.Alimoradi H., Barzegar-Fallah A., Sammut I.A., Griesh K., Giles G.I. Data characterizing the biophysical and nitric oxide release properties of the tDodSNO - styrene maleic anhydride nanoparticle SMA-tDodSNO. Data Brief. 2018;21:1771–1775. doi: 10.1016/j.dib.2018.10.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Alimoradi H., Greish K., Barzegar-Fallah A., Alshaibani L., Pittalà V. Nitric oxide-releasing nanoparticles improve doxorubicin anticancer activity. Int. J. Nanomedicine. 2018;13:7771. doi: 10.2147/IJN.S187089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tahara Y., Yoshikawa T., Sato H., et al. Encapsulation of a nitric oxide donor into a liposome to boost the enhanced permeation and retention (EPR) effect. MedChemComm. 2017;8(2):415–421. doi: 10.1039/c6md00614k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Quinn J.F., Whittaker M.R., Davis T.P. Delivering nitric oxide with nanoparticles. J. Control. Release. 2015;205:190–205. doi: 10.1016/j.jconrel.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 147.de Mel A., Murad F., Seifalian A.M. Nitric oxide: a guardian for vascular grafts? Chem. Rev. 2011;111(9):5742–5767. doi: 10.1021/cr200008n. [DOI] [PubMed] [Google Scholar]
- 148.Gajewski T.F., Schreiber H., Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013;14(10):1014. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kudo S., Nagasaki Y. A novel nitric oxide-based anticancer therapeutics by macrophage-targeted poly (l-arginine)-based nanoparticles. J. Control. Release. 2015;217:256–262. doi: 10.1016/j.jconrel.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 150.Stamler J.S., Jaraki O., Osborne J., et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. USA. 1992;89(16):7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ishima Y. Albumin-based nitric oxide traffic system for the treatment of intractable cancers. Biol. Pharm. Bull. 2017;40(2):128–134. doi: 10.1248/bpb.b16-00867. [DOI] [PubMed] [Google Scholar]
- 152.Matsushita S., Chuang V.T.G., Kanazawa M., et al. Recombinant human serum albumin dimer has high blood circulation activity and low vascular permeability in comparison with native human serum albumin. Pharm. Res. 2006;23(5):882–891. doi: 10.1007/s11095-006-9933-1. [DOI] [PubMed] [Google Scholar]
- 153.Ishima Y., Chen D., Fang J., et al. S-Nitrosated human serum albumin dimer is not only a novel anti-tumor drug but also a potentiator for anti-tumor drugs with augmented EPR effects. Bioconjug. Chem. 2012;23(2):264–271. doi: 10.1021/bc2005363. [DOI] [PubMed] [Google Scholar]
- 154.Kinoshita R., Ishima Y., Ikeda M., et al. S-Nitrosated human serum albumin dimer as novel nano-EPR enhancer applied to macromolecular anti-tumor drugs such as micelles and liposomes. J. Control. Release. 2015;217:1–9. doi: 10.1016/j.jconrel.2015.08.036. [DOI] [PubMed] [Google Scholar]
- 155.Parzuchowski P.G., Frost M.C., Meyerhoff M.E. Synthesis and characterization of polymethacrylate-based nitric oxide donors. J. Am. Chem. Soc. 2002;124(41):12182–12191. doi: 10.1021/ja020268l. [DOI] [PubMed] [Google Scholar]
- 156.Stasko N.A., Schoenfisch M.H. Dendrimers as a scaffold for nitric oxide release. J. Am. Chem. Soc. 2006;128(25):8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 157.Stasko N.A., Fischer T.H., Schoenfisch M.H. S-nitrosothiol-modified dendrimers as nitric oxide delivery vehicles. Biomacromolecules. 2008;9(3):834–841. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ghosh P., Han G., De M., Kim C.K., Rotello V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008;60(11):1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 159.Connor E.E., Mwamuka J., Gole A., Murphy C.J., Wyatt M.D. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1(3):325–327. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 160.Rothrock A.R., Donkers R.L., Schoenfisch M.H. Synthesis of nitric oxide-releasing gold nanoparticles. J. Am. Chem. Soc. 2005;127(26):9362–9363. doi: 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- 161.Pavelic K., Hadzija M. Medical applications of zeolites. In: Scott M., editor. Auerbach, Kathleen A. Carrado, Prabir KD. Handbook of Zeolite Science and Technology. 1st ed. New York, Florida, USA: CRC press; 2003. pp. 1143–1174. [Google Scholar]
- 162.James S.L. Metal-organic frameworks. Chem. Soc. Rev. 2003;32(5):276–288. doi: 10.1039/b200393g. [DOI] [PubMed] [Google Scholar]
- 163.Hinks N.J., McKinlay A.C., Xiao B., Wheatley P.S., Morris R.E. Metal organic frameworks as NO delivery materials for biological applications. Microporous Mesoporous Mater. 2010;129(3):330–334. [Google Scholar]
- 164.Xiao B., Wheatley P.S., Zhao X., et al. High-capacity hydrogen and nitric oxide adsorption and storage in a metal- organic framework. J. Am. Chem. Soc. 2007;129(5):1203–1209. doi: 10.1021/ja066098k. [DOI] [PubMed] [Google Scholar]
- 165.Nguyen J.G., Tanabe K.K., Cohen S.M. Postsynthetic diazeniumdiolate formation and NO release from MOFs. CrystEngComm. 2010;12(8):2335–2338. [Google Scholar]
- 166.McKinlay A.C., Xiao B., Wragg D.S., Wheatley P.S., Megson I.L., Morris R.E. Exceptional behavior over the whole adsorption-storage-delivery cycle for NO in porous metal organic frameworks. J. Am. Chem. Soc. 2008;130(31):10440–10444. doi: 10.1021/ja801997r. [DOI] [PubMed] [Google Scholar]
- 167.Diring S., Wang D.O., Kim C., et al. Localized cell stimulation by nitric oxide using a photoactive porous coordination polymer platform. Nat. Commun. 2013;4:2684. doi: 10.1038/ncomms3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McKinlay A., Eubank J., Wuttke S., et al. Nitric oxide adsorption and delivery in flexible MIL-88 (Fe) metal-organic frameworks. Chem. Mater. 2013;25(9):1592–1599. [Google Scholar]
- 169.Fan J., He Q., Liu Y., et al. Light-responsive biodegradable nanomedicine overcomes multidrug resistance via NO-enhanced chemosensitization. ACS Appl. Mater. Interfaces. 2016;8(22):13804–13811. doi: 10.1021/acsami.6b03737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang X., Tian G., Yin W., et al. Controllable generation of nitric oxide by near‐infrared‐sensitized upconversion nanoparticles for tumor therapy. Adv. Funct. Mater. 2015;25(20):3049–3056. [Google Scholar]
- 171.Fan W., Bu W., Zhang Z., et al. X‐ray radiation‐controlled NO‐release for on demand depth independent hypoxic radiosensitization. Angew. Chem. Int. Ed. Engl. 2015;54(47):14026–14030. doi: 10.1002/anie.201504536. [DOI] [PubMed] [Google Scholar]
- 172.Zhang K., Xu H., Jia X., et al. Ultrasound-triggered nitric oxide release platform based on energy transformation for targeted inhibition of pancreatic tumor. ACS Nano. 2016;10(12):10816–10828. doi: 10.1021/acsnano.6b04921. [DOI] [PubMed] [Google Scholar]
- 173.Kim J., Yung B.C., Kim W.J., Chen X. Combination of nitric oxide and drug delivery systems: tools for overcoming drug resistance in chemotherapy. J. Control. Release. 2017;263:223–230. doi: 10.1016/j.jconrel.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Han J.Y., Nam B.H., Kim H.Y., Yoon S.J., Kim H.T., Lee J.S. A randomized phase II study of irinotecan plus cisplatin versus irinotecan plus capecitabine with or without isosorbide-5-mononitrate in advanced non-small-cell lung cancer. Ann. Oncol. 2012;23(11):2925–2930. doi: 10.1093/annonc/mds122. [DOI] [PubMed] [Google Scholar]
- 175.Illum H., Wang D.H., Dowell J.E., et al. Phase I dose escalation trial of nitroglycerin in addition to 5-fluorouracil and radiation therapy for neoadjuvant treatment of operable rectal cancer. Surgery. 2015;158(2):460–465. doi: 10.1016/j.surg.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 176.Yasuda H., Yamaya M., Nakayama K., et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006;24(4):688–694. doi: 10.1200/JCO.2005.04.0436. [DOI] [PubMed] [Google Scholar]
- 177.Arrieta O., Blake M., de la Mata-Moya M.D., et al. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014;111(2):311–315. doi: 10.1016/j.radonc.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 178.Reinmuth N., Meyer A., Hartwigsen D., et al. Randomized, double-blind phase II study to compare nitroglycerin plus oral vinorelbine plus cisplatin with oral vinorelbine plus cisplatin alone in patients with stage IIIB/IV non-small cell lung cancer (NSCLC). Lung Cancer. 2014;83(3):363–368. doi: 10.1016/j.lungcan.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 179.Siemens D.R., Heaton J.P., Adams M.A., Kawakami J., Graham C.H. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology. 2009;74(4):878–883. doi: 10.1016/j.urology.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 180.Liu Y.S., Chuang M.T., Tsai Y.S., Tsai H.M., Lin X.Z. Nitroglycerine use in transcatheter arterial (chemo) embolization in patients with hepatocellular carcinoma and dual-energy CT assessment of Lipiodol retention. Eur. Radiol. 2012;22(10):2193–2200. doi: 10.1007/s00330-012-2484-4. [DOI] [PubMed] [Google Scholar]
- 181.Stevens E.V., Carpenter A.W., Shin J.H., Liu J., Der C.J., Schoenfisch M.H. Nitric oxide-releasing silica nanoparticle inhibition of ovarian cancer cell growth. Mol. Pharm. 2010;7(3):775–785. doi: 10.1021/mp9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Munaweera I., Shi Y., Koneru B., et al. Nitric oxide- and cisplatin-releasing silica nanoparticles for use against non-small cell lung cancer. J. Inorg. Biochem. 2015;153:23–31. doi: 10.1016/j.jinorgbio.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 183.Fan J., He Q., Liu Y., et al. Light-responsive biodegradable nanomedicine overcomes multidrug resistance via no-enhanced chemosensitization. ACS Appl. Mater. Interfaces. 2016;8(22):13804–13811. doi: 10.1021/acsami.6b03737. [DOI] [PMC free article] [PubMed] [Google Scholar]