

ABSTRACT
Astrocytes in the central nervous system (CNS) provide supportive neural functions and mediate inflammatory responses from microglia. Increasing evidence supports their critical roles in regulating brain homoeostasis in response to pro-inflammatory factors such as cytokines and pathogen/damage-associated molecular pattern molecules in infectious and neurodegenerative diseases. However, the underlying mechanisms of the trans-cellular communication are still unclear. Extracellular vesicles (EVs) can transfer a large diversity of molecules such as lipids, nucleic acids and proteins for cellular communications. The purpose of this study is to characterize the EVs cargo proteins derived from human primary astrocytes (ADEVs) under both physiological and pathophysiological conditions. ADEVs were isolated from human primary astrocytes after vehicle (CTL) or interleukin-1β (IL-1β) pre-treatment. Label-free quantitative proteomic profiling revealed a notable up-regulation of proteins including actin-associated molecules, integrins and major histocompatibility complex in IL-1β-ADEVs compared to CTL-ADEVs, which were involved in cellular metabolism and organization, cellular communication and inflammatory response. When fluorescently labelled ADEVs were added into primary cultured mouse cortical neurons, we found a significantly increased neuronal uptake of IL-1β-ADEVs compared to CTL-ADEVs. We further confirmed it is likely due to the enrichment of surface proteins in IL-1β-ADEVs, as IL-1β-ADEVs uptake by neurons was partially suppressed by a specific integrin inhibitor. Additionally, treatment of neurons with IL-1β-ADEVs also reduced neurite outgrowth, branching and neuronal firing. These findings provide insight for the molecular mechanism of the ADEVs’ effects on neural uptake, neural differentiation and maturation, and its alteration in inflammatory conditions.
KEYWORDS: Astrocytes, extracellular vesicles, extracellular matrix, exosome, IL-1β, inflammatory diseases, neurodegenerative diseases, proteomics
Introduction
Astrocytes are one of the most abundant cell types in the central nervous system (CNS) and are essential for brain homoeostasis [1]. They perform many vital roles, such as providing neurotrophic support, promoting synapse formation and plasticity, regulating the neurotransmitter clearance and maintenance of the blood–brain barrier [2]. In addition, astrocytes are major immune reactive cells in the CNS and mediate inflammatory response to brain injury and infection [3]. Reactive astrocytes release diverse molecules including inflammatory cytokines, chemokines and neurotoxic substances that exacerbate inflammation and affect neuronal functions [3]. Recently, accumulating evidence has suggested the neurotoxic role of astrocytes in CNS disorders, such as Alzheimer’s, Parkinson’s and Huntington’s diseases and amyotrophic lateral sclerosis (ALS) [4]. Nevertheless, trans-cellular communication of astrocytes to mediate such diverse biological functions is poorly investigated.
Interleukin-1β (IL-1β) is a classic pro-inflammatory cytokine and regulates the inflammatory response to brain injury. It is primarily produced by microglia and astrocytes in the CNS and one of the critical regulators for induction of reactive astrocyte phenotype [5]. IL-1β stimulation of astrocytes initiates nuclear factor kappa B (NFκB) signalling, resulting in increased expression of neurotoxins, such as glutamate and nitric oxide, ensuring neurodegeneration [6,7]. In addition, IL-1β also up-regulates Fas ligand in reactive astrocytes to induce neuronal apoptosis [8]. It remains largely unknown how glial molecules are transmitted to neurons. Previous evidence suggested several mechanisms might be involved in this process, including exocytosis, diffusion and active/passive transport [9]. Recent advancements indicate that extracellular vesicles (EVs) might play an important role in glia-neuron communication during neurodegenerative states or inflammation [10].
EVs are heterogeneous cell-derived membranous vesicles that are commonly grouped into three types including exosomes (50–150 nm), microvesicles (100–1000 nm) and apoptotic bodies (up to 5 μm) based on their size and mode of biogenesis [11]. They are released from almost all cell types of CNS including astrocytes and carry diverse intracellular cargos of proteins, lipids and nucleic acids [12]. EVs are now considered as common vehicles of intercellular communication for various physiological processes and pathological functions in the periphery and the CNS [13]. In particular, astrocytes release human immunodeficiency virus 1 protein Nef in EVs to mediate neurotoxicity, and synapsin 1 to promote neurite outgrowth [14,15]. Astrocyte-derived EVs (ADEVs) are identified as regulators of the peripheral leukocyte response to IL-1β-injected inflammatory brain lesions by targeting peroxisome proliferator–activated receptor α (PPARα) in leukocytes and activating NFκB signalling for chemotaxis [16]. Thus, molecular and functional characterization of ADEVs in inflammatory condition is necessary to clarify their roles in the progression of diseases. Our study demonstrates the comprehensive characterization of the protein composition of ADEVs with control and IL-1β treatment, and their neurophysiological roles in terms of neuronal ADEV uptake, neurite outgrowth and neuronal firing in vitro. Our findings suggest a characteristic signature of ADEVs under the inflammatory conditions.
Materials and methods
Isolation and cultivation of primary human astrocytes
Human astrocytes were isolated from conceptual brain tissues (between 96 and 132 days gestation) as previously described [8]. Deidentified brain tissue samples were obtained from the Birth Defects Laboratory at the University of Washington in Seattle in full compliance with the ethical guidelines of the National Institutes of Health (NIH), Universities of Washington and North Texas Health Science Centre. The conceptual tissues without known birth defects were used in this study. The tissue was carefully cleaned and passed through a 250 μM Nitex bag to mechanically create a single cell suspension. Cells were washed with HBSS through several low speed centrifugation/trituration steps and cultured initially at a density of 50 × 106 cells/150 cm2 at 37°C and 5% CO2 in astrocyte media (ASM) [Dulbecco’s modified Eagle medium (DMEM) with F12 nutrient mixture (Thermo Fisher Scientific, Waltham, MA), 10% foetal bovine serum (FBS, Atlas Biologicals, Fort Collins, CO), 5 mg/mL penicillin, 5 mg/mL streptomycin, 10 mg/mL neomycin (Sigma-Aldrich, St. Louis, MO) and 250 ng/mL amphotericin B (Sigma-Aldrich)]. Adherent cells were passaged every 7–10 days with 0.05% trypsin-EDTA (Sigma-Aldrich) and cultured at 20 × 106 cells/150 cm2 to enhance the purity of replicating astroglial cells until the preparations were >99% pure as measured by immunocytochemistry staining for glial fibrillary acidic protein (GFAP).
Characterization of human activated astrocytes
Metabolic activity assay
Astrocytes were plated in the 48-well culture treated plates (Corning, Corning, NY) at 1.5 × 105 per well and permitted to adhere overnight at 37°C, 5% CO2. Astrocytes were activated for 24 h IL-1β (1 or 20 ng/mL, R&D Systems, Minneapolis, MN) in ASM. Each biological donor was treated in three replicates per condition (control & IL-1β). After 24 h culture, supernatants were collected for ELISA. Cells were then treated with 400 µM glutamate for 24 h for glutamate clearance assay described below. Finally, cultures were incubated with 5% 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) in ASM for 30–45 min at 37°C, 5% CO2. Cells metabolized MTT to purple formazan crystals, which were dissolved in dimethyl sulphoxide (DMSO), and absorbance at 490 nm was measured in an M5 microplate reader (Molecular Devices, Sunnyvale, CA). Metabolic activity in treated wells was normalized to control, untreated wells (ratio).
CCL2 and CXCL8 ELISA
The supernatants collected above were assayed by CCL2 and CXCL8 ELISA (R&D Systems) according to the manufacturer’s instructions. Protein concentrations were normalized to the metabolic activity ratio of corresponding well replicates.
Immunocytochemistry (ICC)
Astrocytes were plated in the 48-well tissue culture treated plates at 1 × 105 cells per well and incubated overnight at 37°C and 5% CO2. Astrocytes were activated with IL-1β (1 ng/mL) for 24 h. Following fixation with acetone: methanol at −20°C for 20 min, astrocytes were rehydrated with blocking buffer [2% bovine serum albumin (BSA), 0.1% Triton X-100 in phosphate buffered solution (PBS)] for 30 min. Cultures were then labelled overnight at 4°C with human GFAP (rabbit, 1:700, Agilent, Santa Clara, CA), and vimentin (mouse, 1:200, monoclonal antibody, clone V9, Thermo Fisher Scientific) in blocking buffer. Alexa Fluor® secondary antibodies were conjugated to their target species at room temperature for 1.5 h in blocking buffer. Anti-rabbit-594 Alexa Fluor® secondary antibodies were used to detect GFAP, and anti-mouse 488 Alexa Fluor® secondary antibodies were used to detect vimentin (Thermo Fisher Scientific). Cells were washed three times in PBS and subsequently labelled with the nucleic acid stain 4ʹ,6-diamidino-2-phenylindole, dihydrochloride (DAPI, 1:800, Thermo Fisher Scientific) for three min at room temperature. Micrographs were taken at 200× original magnification on an Eclipse Ti-300 (Nikon Instruments, Melville, NY) using a black and white Luca R (Andor, model 604, Belfast, UK) and then pseudo-coloured to the appropriate wavelength with NIS-Elements software (Nikon Instruments).
Glutamate clearance assay
The ability of astrocytes to clear glutamate was assayed using the Amplex® Red Glutamic Acid/Glutamate Oxidase Assay Kit (Thermo Fisher Scientific). Following collection of supernatants for ELISA, control and IL-1β-activated astrocytes were treated with 400 μM glutamic acid in phenol-free ASM. After 24 h the concentration of glutamate was determined according to the manufacturer’s instructions. Glutamate clearance was calculated by comparing the glutamate concentration in treated wells to that of a well without cells (no clearance). Glutamate clearance was normalized to the metabolic activity ratio of corresponding well replicates.
Activation of astrocytes and isolation of ADEVs
Early passage (P2-P5) astrocytes were split into two groups and re-plated in 75-cm2 flasks at a density of 1 × 107 cells/flask. Normal FBS (Peak Serum, Wellington, CO) was ultra-centrifuged at 100,000 × g for 18 h at 4°C (WX 80, Sorval) to pellet exosomes and other EVs as previously described [17]. Exosome depleted FBS was then stored at −20°C until experimental use. For the IL-1β stimulated group, astrocytes were activated with 1 ng/mL IL-1β for 24 h [18]. For the control group, the same volume of vehicle was added. After activation, astrocytes were gently washed three times with warm PBS and cultured with fresh DMEM/F12 supplemented with 10% EV-depleted FBS and 1% antibiotic mixture for four days. Prior to 30 min before collecting the conditioned medium, 5 mM adenosine triphosphate (ATP) was added into both control and IL-1β stimulated astrocytes to ensure the complete EV release [19]. ADEVs were isolated by differential centrifugation as previously described [20]. Briefly, the medium was centrifuged at 300 × g for 10 min at room temperature to remove floating cells, 2,000 × g for 10 min at 4°C to remove cell debris, 10,000 × g for 30 min 4°C to remove microvesicles and apoptotic bodies. The remaining supernatant was then ultra-centrifuged at 100,000 × g overnight at 4°C in a 41Ti rotor (Beckman Coulter, Brea, CA) to obtain the ADEV pellets. The final pellets were resuspended in 1mL cold PBS and loaded on qEV original size exclusion chromatography columns (Izon Science, Christchurch, New Zealand). 12–13 fractions of 500 μL each were harvested following the manufacturer’s instructions, and then each fraction was analyzed by nanoparticle tracking analysis (NTA) to pool EV-enriched fractions (f8-11).
Nanoparticle tracking analysis (NTA) of ADEVs
EV concentration and size distribution were characterized by NTA with a NanoSight NS300 instrument (Malvern, Worcestershire, UK) and corresponding software version NTA3.1. ADEVs were pre-diluted in PBS to achieve a concentration within 107–108 range for optimal analysis. For each sample, 600 μL of diluted EVs were injected into the sample-carrier cell, and the cell was cleaned with ethanol between detections. Four videos of 30 s were acquired per sample with the parameters setting: detection level 5, camera level 13–15. The mean size and EV concentration (particles/mL) were calculated by integrating the data from four records.
EV sample preparation for mass spectrometry
EV-enriched fractions were incubated with 1% Triton X-100 (with proteasome inhibitors) to extract protein content. Sonication was then performed to rupture the EV membrane and allow the formation of homogeneous protein suspension. Protein of EVs was quantified by Pierce BCA assay kit (Thermo Fisher Scientific). For each EV preparation (n = 5 each for control and IL-1β induced ADEV), the equal amount of isolated EVs (50 μg) were acetone precipitated as described previously [21]. The final EV protein pellets were resuspended in 1× Laemmli sample buffer (Bio-Rad, Hercules, CA) and sonicated at 100% amplitude for eight min, and heated at 95°C for 10 min.
In-gel digestion and LC-MS/MS analysis
The whole EV lysate was run for 20 min on a 4–20% SDS-PAGE system to separate proteins from lower molecular weight contaminants, and the entire protein region of the gel was excised and subjected to in-gel trypsin digestion after reduction with dithiothreitol and alkylation with iodoacetamide. Peptides eluted from the gel were lyophilized and re-suspended in 25–50 µL of 5% acetonitrile (0.1% (v/v) TFA) with 2 pmol ADH digest. A 1–2 µL injection was loaded by a Waters NanoAcquity UPLC in 5% acetonitrile (0.1% formic acid) at 4 µL/min for 4 min onto a 100 µm I.D. fused-silica pre-column packed with 2 cm of 5 µm (200 Å) Magic C18AQ (Bruker-Michrom, Billerica, MA). Peptides were eluted at 300 nL/min from a 75 µm I.D. gravity-pulled analytical column packed with 25 cm of 3 µm (100 Å) Magic C18AQ particles using a linear gradient from 5–35% of mobile phase B (acetonitrile + 0.1% formic acid) in mobile phase A (water + 0.1% formic acid) over 60 min. Ions were introduced by positive electrospray ionization via liquid junction at 1.4–1.6 kV into a Q Exactive hybrid mass spectrometer (Thermo Scientific). Mass spectra were acquired over m/z 300–1750 at 70,000 resolution (m/z 200) with an AGC target of 1 × 106, and data-dependent acquisition selected the top 10 most abundant precursor ions for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.6 Da, max fill time of 110 ms, and AGC target of 1e5. Peptides were fragmented by a normalized collisional energy of 27, and fragment spectra acquired at a resolution of 17,500 (m/z 200).
Proteomic data analysis
Raw data files were peak processed with Proteome Discoverer (version 2.1, Thermo Scientific) followed by identification using Mascot Server (version 2.2, Matrix Science, Boston, MA) against the Human (Swissprot) FASTA file (downloaded 12/2017). Search parameters included Trypsin/P specificity, up to two missed cleavages, a fixed modification of carbamidomethyl cysteine, and variable modifications of oxidized methionine, pyroglutamic acid for Q, and N-terminal acetylation. Assignments were made using a 10 ppm mass tolerance for the precursor and 0.05 Da mass tolerance for the fragments. All non-filtered search results were processed by Scaffold (version 4.8.7, Proteome Software, Portland, OR) utilizing the Trans-Proteomic Pipeline (Institute for Systems Biology) with threshold values set at 95% for peptides (0.2% false-discovery rate) and 99% for proteins (two peptide minimum), and quantitative comparisons were made using the label-free intensity-based absolute quantification (iBAQ) method with all samples normalized by total ion current for the run.
Bioinformatics analysis
A total of 875 proteins identified in CTL-ADEVs and IL-1β-ADEVs were input to FunRich software (3.1.3) for GO enrichment analysis [22]. The gene symbols retrieved from UniProtKB accession numbers were mapped to cellular components (CC), molecular functions (MF) and biological processes (BP) items using default statistical parameters (threshold: count 2, ease 0.1). The protein comparison between CTL-ADEVs and IL-1β-ADEVs was conducted using iBAQ method as previously described [23]. Briefly, precursor ion intensities of all peptides matching each protein were divided by the theoretical number of peptides derived from in silico tryptic digestion. The fold change of each protein was calculated by taking the logarithm of the ratio of individual protein iBAQ values normalized to the average of the corresponding iBAQ values in the control EVs (n = 5). The 0 iBAQ values were replaced with 1. For calculating the statistical significance, individual protein iBAQ values were analyzed by student’s t-test. A volcano plot was generated using Graphpad prism 6. The significant expression threshold was defined as a criteria of p value <0.05 (−log10 (p value) > 1.3) and fold change > 2 (log2 FC >1 or < −1). We defined differentially expressed proteins (DEPs) as either significantly expressed proteins or uniquely present proteins in IL-1β-ADEVs. The heatmap of significantly expressed proteins in IL-1β-ADEVs was generated using multiExperiment Viewer software (MEV, 4.8.1). Ingenuity Pathway Analysis (IPA), a web-based software application, was used to analyze, integrate, and categorize the DEPs data according to the software manual guidelines. A standard IPA core analysis was performed including canonical pathways, disease and function annotation and upstream regulator.
ADEV labelling with fluorescent dye
ADEVs isolated from pooled astrocyte-derived conditioned medium of five donors were labelled with fluorescent dye PKH26 (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, 20 μg EV pellets were diluted in 250 μL diluent C. PKH26 dye (1 µL, final concentration 2 μM) was added to another 250 μL diluent C, and the two solutions were mixed gently by pipetting for 30 s. The labelling reaction was incubated at 37°C for 10 min, and then stopped by adding an equal volume (500 μL) of 1% BSA.
The labelled EVs (total 1 mL) were next loaded to the qEV column for size exclusion and unbounded dye removal. EV enriched fractions (f8-11) were pooled and further concentrated to final volume 100 μL using 10 kDa filter (Amicon Ultra-0.5, Merck Millipore, Burlington, MA) according to the manufacturer’s protocols. For the negative control, the same procedure was performed in the initial experiments but without EVs (dye only) to ensure lack of fluorescence in the absence of EV. Protein concentrations of labelled EVs were measured with PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific).
Primary neuron culture
Murine primary cortical neurons were isolated from embryos of CD-1 mice at day 16.5 (Jackson Laboratory, Bar Harbour, ME). Briefly, cortex of the embryonic mice was dissected in Hibernate-E medium (Gibco Invitrogen) at 4°C. Tissues were cut into 1 mm3 pieces, incubated with 0.25% trypsin-EDTA solution (Gibco Invitrogen) for 15 min at 37°C and dissociated into single cells by gentle trituration. Neurons were then plated on poly-D-lysine (PDL, Sigma-Aldrich) pre-coated regular 24-well plates (Corning) or the multi-channel electrode array plates (Axion Biosystems, Atlanta, GA) with DMEM (Gibco Invitrogen) containing 20% FBS. Four hours later, the medium was refreshed with neuronal culture medium (Neurobasal medium, 2% B-27, 2 mM Glutamax, Gibco Invitrogen). Media were changed by half every two days. All animal procedures followed the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Boston University Institutional Animal Care and Use Committee.
PKH26-labelled ADEV uptake in primary cultured neurons
Labelled CTL-ADEVs, IL-1β-ADEVs (4 µL, final concentration 10 μg/mL), or the equivalent amount of negative control (dye only) was added to each well of days in vitro (DIV) 10 primary cultured neurons in EV-depleted neuronal culture medium at a density of 50,000 cells/well in 24-well plates. For integrins inhibition experiment, final concentration of 0.1 or 0.1 μM RGD peptide was pre-treated with the same amount of IL-1β-ADEVs at 37°C for 30 min, thereafter adding to DIV 10 neurons in EV-depleted neuronal culture medium at a density of 50,000 cells/well in 24-well plates. Time-lapse images of ADEV uptake by neurons were obtained using a live cell imaging system (IncuCyte ZOOM system, Essen BioScience, Ann Arbor, MI) in a humidity chamber at 37°C and 5% CO2 as previously described [24]. Images were acquired from 16 fields per culture well every 6 h for total 72 h and further processed in Incucyte software (2019A) for analysis. Cellular PKH26 red fluorescent signal over time in each treatment was used as an indicator of EV uptake efficiency over time. Total integrated intensity of red fluorescence within cells per mm2 was calculated for each time point using Incucyte software with the analysis definition settings (Segmentation: Top-Hat; Radius (μm): 10; Threshold (OCU): 0.2; Filters: Area (μm2)-max 45 and integrated intensity-max 65), and the number of fluorescent cells were counted manually using Image J at 0, 24 h, 48 h and 72 h points and plotted as a time course across three independent experiments.
Neurite outgrowth analysis
Neurons (DIV 3) were cultured in PDL pre-coated 35 mm glass-coverslips at a density of 50,000 cells/well in 24-well plates. Prior to adding ADEVs, cells were washed with 1× HBSS for three times to remove exogenous vesicles. Purified ADEVs with gradient concentrations (1×, 1.25 μg/mL; 2×, 2.5 μg/mL; 4×, 5 μg/mL) were then added to each well in EV-depleted neuronal culture medium. After incubation with ADEVs for three days in vitro, neurons were fixed with 4% paraformaldehyde at 37°C for 15 min, and blocked in blocking buffer (4% normal goat serum, 4% BSA, 0.3% Triton X-100 in PBS) at room temperature for 1 h. Immunocytochemical staining was performed overnight using an anti-MAP2 antibody (1:1000, Millipore), followed by washing three times with PBS, and incubated with Alexa-488 secondary antibody (1:400, Life Technologies). Images were captured from 10 fields per culture well with a Nikon deconvolution wide-field Epifluorescence microscope (Nikon Instrument) using a 40× objective. Neurite outgrowth was manually traced and measured using Neurolucida imaging software (version 11.11.12; MBF Bioscience, Williston, VT) by a blinded experimenter. For each treatment condition, 15–20 neurons were measured per experiment across three independent experiments.
Multi-electrode array (MEA) recordings and analysis
Primary neurons (DIV 17) were cultured in PDL pre-coated 24-well MEA plate (Axion Biosystems) with 16 microelectrodes arranged over 4 × 4 square grid in each well at a density of 1 × 105 cells/well. Prior to recording, cells were washed with 1× HBSS, and 10 μg/mL ADEVs were added to each well in EV-depleted neuronal culture medium. The recordings started five min after the MEA plates were placed in a Maestro MEA system which was set to 37°C and 5% CO2, and lasted for 15 min. The electrical signals were collected using AxIS Navigator software (Axion Biosystems) with a filter for 200–3000 Hz cut-off frequencies. Spikes were detected using an adaptive threshold crossing set to six times the standard deviation of the estimated noise on each electrode. Bursts were identified in the data recorded from each individual electrode using an Inter-Spike Interval (ISI) threshold algorithm requiring a minimum of five spikes and a maximum ISI of 100 ms. Network bursts were calculated for each well using an Envelope algorithm with the threshold factor of 1.25–1.5, a minimum inter-beat interval (IBI) of 100 ms, and the minimum active electrodes of 35%. An active electrode was considered when there were five spikes over the period of one min (five spikes/min). MEA data were further analyzed and exported using the Axion Biosystems Neural Metrics Tool. For each experiment, neural activity was recorded for five consecutive days from DIV 17. We repeated total four independent experiments.
Statistical analysis
Graphical data were analyzed using GraphPad Prism versions 6.0. Data are presented as mean ± SEM. Figures S1(b–e) and 7(c, d) were analyzed by ordinary one-way ANOVA with Dunnett’s multiple corrections; Figures 2, 4(c) and S3(a, b) were analyzed by student’s t-test; Figures 5 and 6 were analyzed by repeated measure two-way ANOVA with Tukey’s multiple corrections; Figure 7(e–g) were analyzed by nonparametric test with Dunn’s multiple corrections; Figure 7(j, k) were analyzed by repeated measure two-way ANOVA with Sidak’s multiple corrections.
Figure 2.
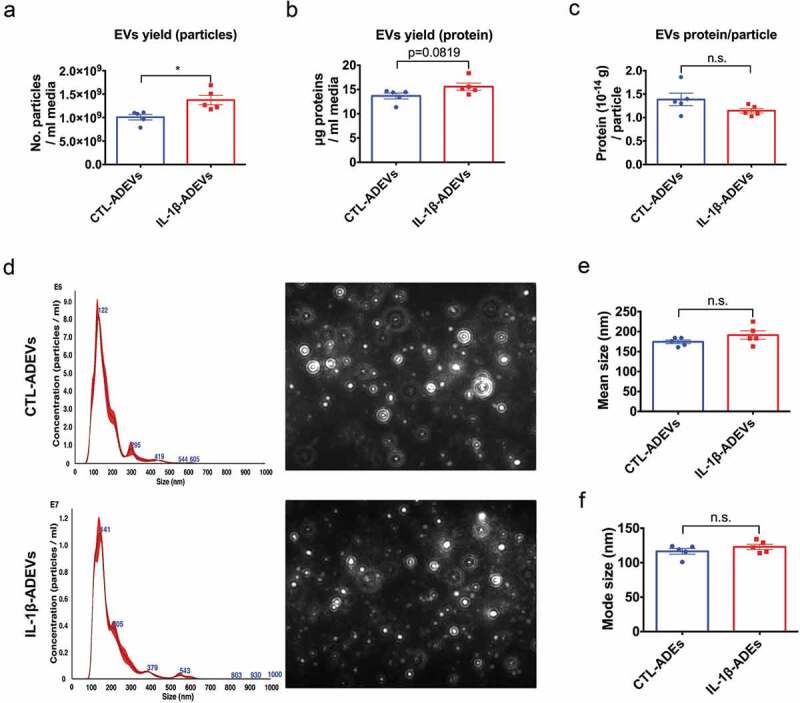
Isolation and characterization of extracellular vesicles from control astrocytes and IL-1β-treated astrocytes. (a) The yield of IL-1β-ADEVs, expressed as the number of isolated particles per mL media, was significantly higher than that of CTL-ADEVs (n = 5). (b) The nanogram protein of EVs per mL media (c) and the amount of protein per particle was not significantly different between the two groups (n = 5). (d) Representation of size distribution of CTL- and IL-1β-ADEVs by nanoparticle tracking analysis (n = 5). Data are expressed as mean ± SEM. n.s., no significance; *p < 0.05 compared to control group as determined by student’s t-test using Graphpad prism 6.
Figure 4.
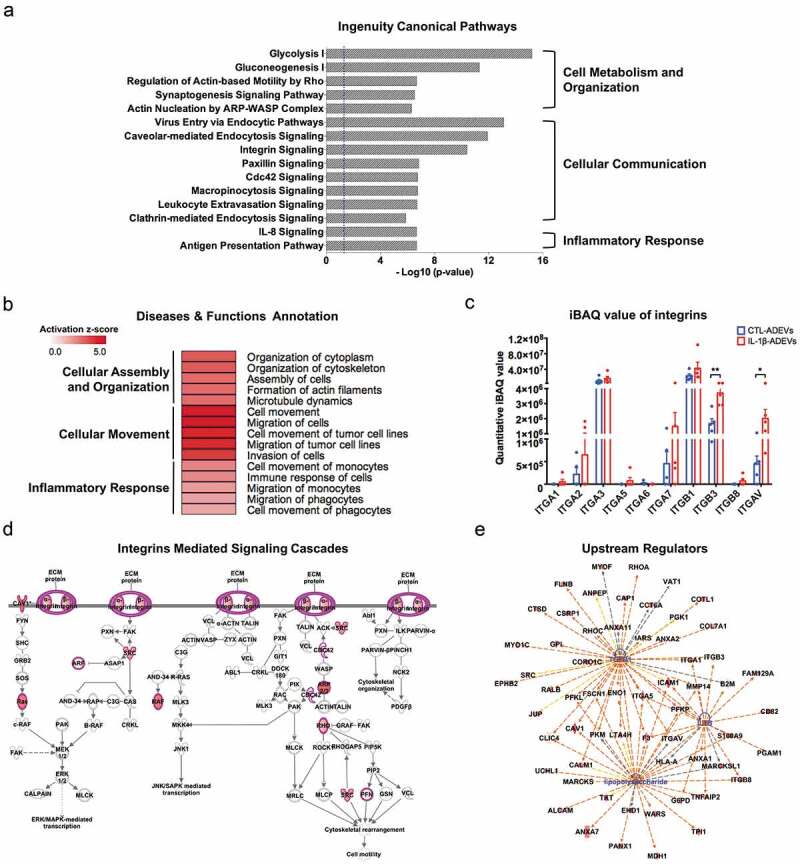
Ingenuity pathway analysis (IPA) of differentially expressed proteins (DEPs) in IL-1β- versus CTL-ADEVs. (a) Total 147 DEPs and their expression values were used as input for IPA. Curated list of the 15 most significantly enriched canonical pathways: cell metabolism and organization, cellular communication and inflammatory response. A statistically significant p-value of 0.05 is indicated on the plot as a vertical dashed line, at the x-value of 1.30. (b) Heatmap of result from diseases and functions annotation performed by IPA, which are associated with cellular assembly and organization, cellular movement and inflammatory response. (c) The iBAQ value of integrins between CTL- and IL-1β-ADEVs. Data are presented as mean ± SEM of n = 5 for both groups. *p < 0.05, **p < 0.01 compared to control group as determined by paired Student’s t-test. (d) DEPs in IL-1β-ADEVs mapping to the integrin-mediated signal cascades conducted by IPA. The highlighted proteins indicate the activated regulation. (e) Consistence of DEPs in IL-1β-ADEVs with the increased signalling of potential upstream regulators including TGF-β1, LPS and IL-1β. Blue colour indicates the upstream regulators; red colour scales the up-regulation of proteins in IL-1β-ADEVs; Red and orange lines indicate leading to activation by predicted relationships.
Figure 5.
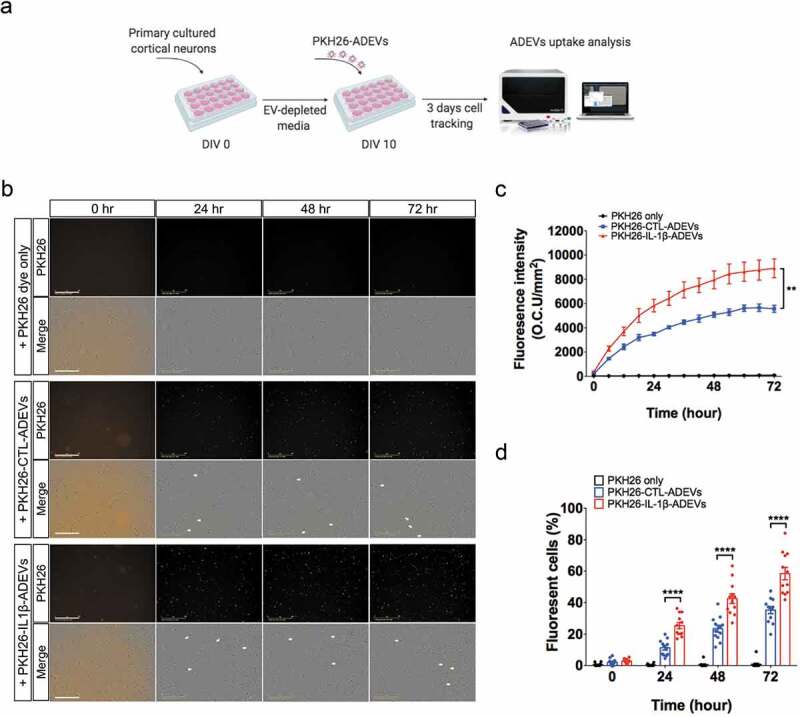
Increased cellular uptake of IL-1β-ADEVs compared to CTL-ADEVs by primary cultured neurons in vitro. (a) The scheme of in vitro ADEVs uptake. Both pooled CTL-ADEVs and IL-1β-ADEVs were labelled with PKH26 and equal amount of purified PKH26-labelled ADEVs were added to primary cultured cortical neurons at DIV 10 and imaged by in a live cell imaging system for three days. As a control, PKH26 dye was incubated and monitored in the same manner. (b) Representative images of the indicated time. White arrow shows the fluorescent signals within the cells. Scale bar, 200 μm. (c, d) Quantification of ADEVs uptake efficiency using Incucyte software to measure (c) total integrated intensity of red fluorescence per mm2 at each time point and Image J to count the number of PKH26+ cells at 0, 24, 48 and 72 h, across three independent experiments (d). Data are present as mean ± SEM. **p < 0.01, ****p < 0.0001 with PKH26-IL-1β-ADEVs compared to PKH26-CTL-ADEVs as determined by repeated measure two-way ANOVA with Tukey’s multiple comparisons.
Figure 6.
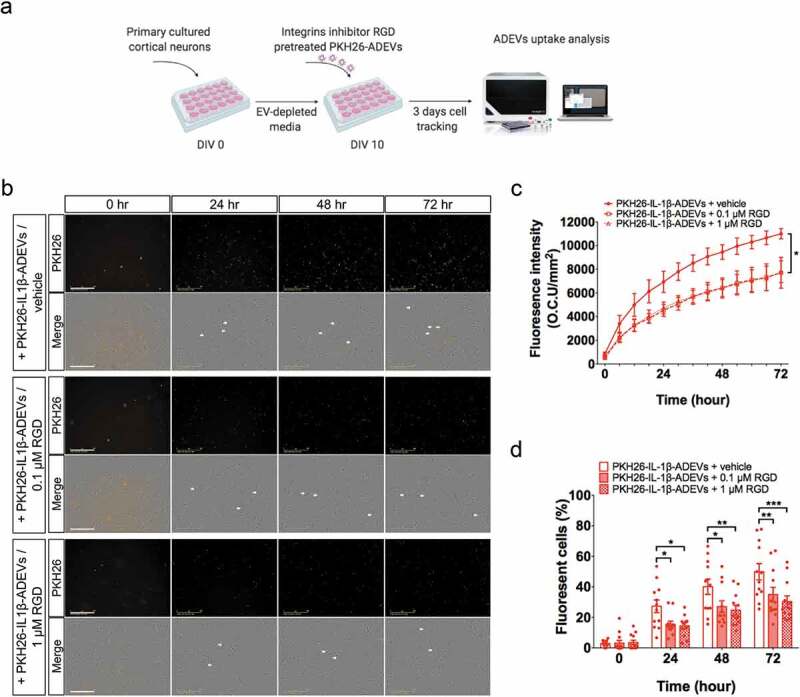
Inhibition of integrins in IL-1β-ADEVs reduced the EVs uptake by primary cultured neurons in vitro. (a) The scheme of in vitro IL-1β-ADEVs uptake with integrin inhibition. Purified PKH26-labelled IL-1β-ADEVs were pre-treated with integrin inhibitor RGD peptide (0.1μM or 1μM) at 37°C for 30 min, and equal amount of pre-treated PKH26-IL-1β-ADEVs were added to primary cultured mouse cortical neurons at DIV 10, which were imaged by a live cell imaging system for three days. (b) Representative images of the indicated time. White arrow shows the fluorescent signals within the cells. Scale bar, 200 μm. (c, d) Quantification of PKH26-IL-1β-ADEVs uptake using IncuCyte software to measure (c) total integrated intensity of red fluorescence per mm2 at each time point and Image J to count (d) the number of fluorescent cells at 0, 24, 48 and 72 h, across three independent experiments. Data are present as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 with PKH26-IL-1β-ADEVs + 0.1μM RGD peptide or PKH26-IL-1β-ADEVs + 1 μM RGD peptide compared to PKH26-IL-1β-ADEVs + vehicle as determined by repeated measure two-way ANOVA with Tukey’s multiple comparisons.
Figure 7.
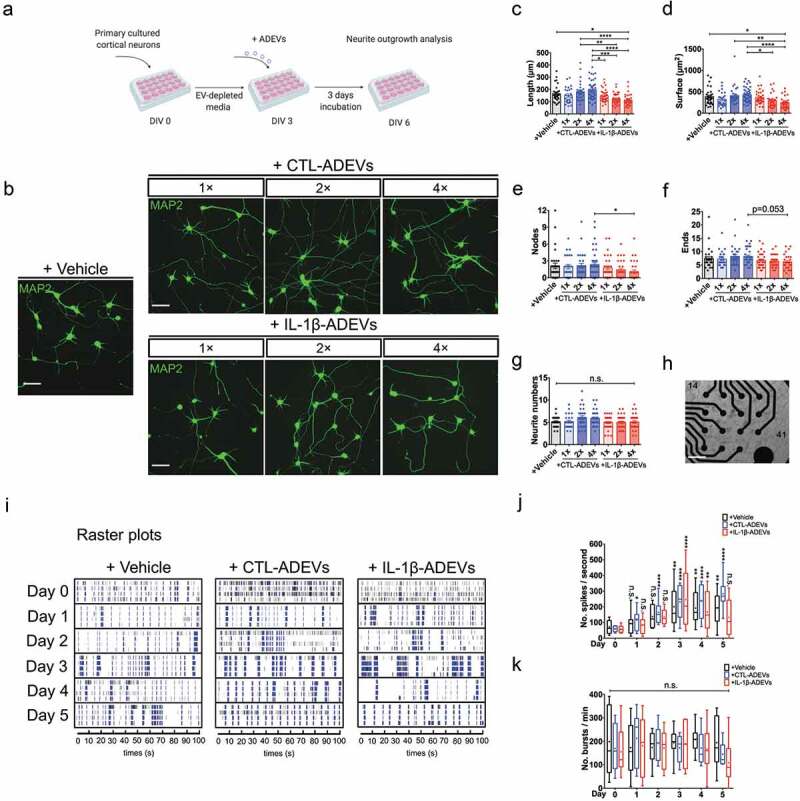
IL-1β-ADEVs exert suppressive effects on neurite outgrowth and neural spike firing. (a) The scheme of ADEV’s effects on neurite outgrowth. Different doses of purified CTL-ADEVs and IL-1β-ADEVs were added to primary cultured mouse cortical neurons at DIV 3 and incubated for three days. Neurite outgrowth analysis was performed at DIV 6 using Neurolucida imaging software. (b) Representative fluorescent images of MAP2+ cortical neurons after three-day exposure to the indicated concentrations of CTL- and IL-1β-ADEVs (1×, 1.25 μg/mL; 2×, 2.5 μg/mL; 4×, 5 μg/mL). Control group was treated with vehicle instead of ADEVs. Scale bar, 50 μm. Quantification of neurite outgrowth show (c) neurite length, (d) surface area, (e) number of nodes, (f) number of neurite ends, and (g) total neurite number for the indicated concentrations of ADEVs. Data are present as mean ± SEM from three independent experiments. n.s., no significance; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by ordinary one-way ANOVA with Tukey’s multiple corrections (c, d) and non-parametric test with Dunn’s multiple corrections (e–g). (h) Multi-electrode array (MEA) for monitoring neural spike firing after treating primary neurons with CTL- and IL-1β-ADEVs from DIV 17. Phase-contrast image of primary neurons cultured on an MEA plate at DIV 17. Scale bar, 500 μm. (i) Representative raster plots show the spontaneous firing activities recorded from primary neurons after incubation with ADEVs during a five-day time course. Quantitation of (j) the number of spikes per second (k) and the number of bursts per min from primary neurons incubated with vehicle, CTL- or IL-1β-ADEVs during day 0 to 5. Data were collected from 8 MEAs (2 replicates × 4 independent experiments). Box and whisker plots are used to show the median and the 5th to 95th percentiles; Plus sign indicates the means. n.s., no significance; *p < 0.05, **p < 0.01, ****p < 0.0001 compared to day 0, respectively, as determined by repeated measure two-way ANOVA with Sidak’s multiple corrections.
Results
Generation of human reactive astrocytes by IL-1β stimulation
Human astrocytes are highly sensitive to IL-1β stimulation [18]. Primary cultured human astrocytes were treated with 1 or 20 ng/mL IL-1β for 24 h in vitro for comparison. Astrocytes were activated by 1 ng/mL IL-1β and acquired a more polarized and shrunken morphology with intensive GFAP expression and elongated processes (Supplementary Figure 1(a)), which were considered as a cardinal feature of reactive astrocytes [25]. Stimulation with either 1 or 20 ng/mL IL-1β did not affect the astrocyte metabolic activity as determined by MTT assay (Supplementary Figure 1(b)), whereas their glutamate clearance activity was significantly reduced (Supplementary Figure 1(c)). This reflects the impaired glutamate transport capacity of reactive astrocytes [26].
In addition, the production of pro-inflammatory factors by IL-1β-treated astrocytes was examined by ELISA. CCL2 and CXCL8 secretion was significantly elevated in both 1 ng/mL and 20 ng/mL IL-1β stimulated astrocytes as compared to controls (Supplementary Figure 1(d, e). Given that the low concentration of IL-1β was sufficient to induce reactive-astrocyte phenotype, 1 ng/mL IL-1β was used to treat astrocytes for the remainder of the study (Figure 1).
Figure 1.
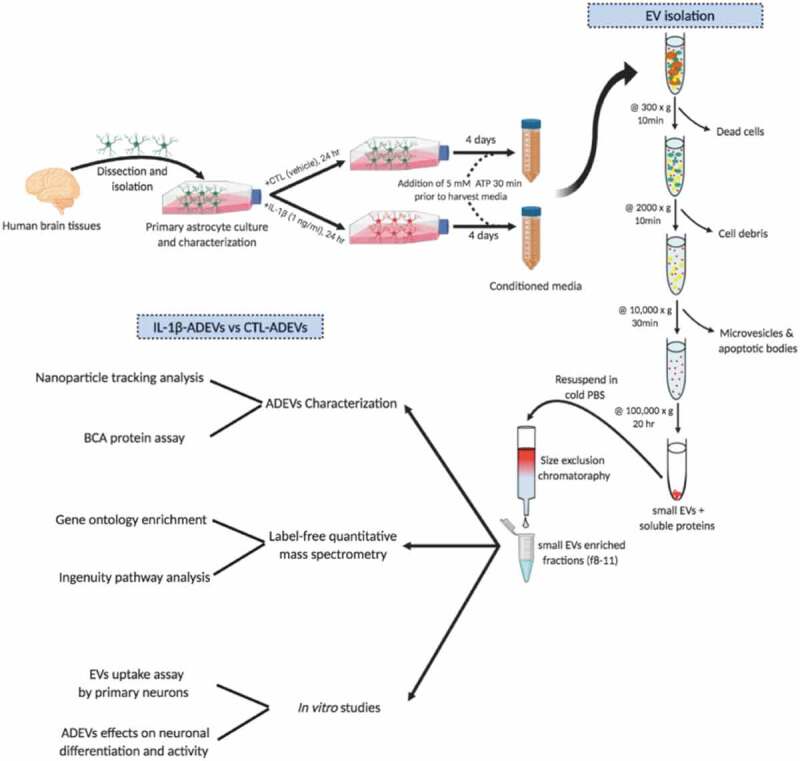
Study overview. Human primary astrocytes were isolated from conceptual brain tissues and cultured as adherent cells. IL-1β (1 ng/mL) was added in the culture to induce astrocyte reactivity. After 24 h, cultured media were removed and cells were washed with PBS three times, and then incubated with EV-depleted media for four days. EVs were isolated from the conditioned media by differential centrifugation combined with fractionation with qEV columns.
Isolation and characterization of EVS from control astrocytes and reactive astrocytes
Conditioned media of cultured astrocytes derived from five healthy human donors were collected (D1 to D5, Supplementary Table 1). CTL- and IL-1β-ADEVs were isolated using a combination of differential centrifugation and size-exclusion columns specifically formatted for EV isolation as described previously [20]. Subsequently, the EV size and protein distribution from different fractions were evaluated. EVs were enriched in fraction 8–11 (Supplementary Figure 2(a, b). Fractions 8–11 were pooled for further EV comparisons. The number of EV particles and protein concentrations per mL collected media were increased in IL-1β-ADEVs compared to CTL-ADEVs (Figure 2(a, b)), whereas the amount of protein per particle was similar (Figure 2(c)). Given the seeding density of astrocytes between two groups was comparable (Supplementary Table 1), these results also suggested that the number of EV particles secreted per cell was higher in IL-1β-ADEVs than that in CTL-ADEVs. Size distribution of isolated ADEVs in the two groups was determined by NTA (Figure 2(d–f) and Supplementary Figure 2(c, d)). A mean diameter of 174.4 ± 4.6 nm for CTL-ADEVs and 191.2 ± 10.5 nm for IL-1β-ADEVs was not statistically different (Figure 2(e)). The mode sizes of 116.6 ± 4.2 nm for CTL-ADEVs and 123.0 ± 3.8 nm for IL-1β-ADEVs were also similar (Figure 2(f)).
A distinct proteomic signature of EVS derived from reactive astrocytes compared to EVS from control astrocytes
To characterize the protein composition of CTL- and IL-1β-ADEVs, label-free quantitative mass spectrometry using equal amounts of proteins from both groups was performed. A total of 875 proteins were detected in the cargo of ADEVs from both groups, with 113 proteins uniquely present in IL-1β-ADEVs (Supplementary Table 2). Gene Ontology (GO) classification using Funrich functional enrichment analysis [22] indicated that most of the identified proteins were derived from exosomes (Figure 3(a)) and enriched for MFs of GTPase activity, MHC receptor activity and cell adhesion molecule activity (Figure 3(b)). These proteins were also enriched in the BP of cell growth and maintenance, transport, cell communication and immune response, which is consistent with the reported functions of ADEVs [27] (Figure 3(c)). Moreover, proteomic profiling identified a number of known exosomal proteins, including tetraspanins (such as CD9, CD81 and CD63) and ESCRT (endosomal sorting complexes required for transport) molecules (such as PDCD6IP/ALIX) [28], as well as astrocyte-specific proteins including GFAP, excitatory amino acid transporter 1 (SLC1A3/GLAST) and glucose transporter member 1 (SLC2A1/GLUT1) from both groups (Table 1). These data confirm significant enrichment of exosomal and astrocytic molecules in purified EVs.
Figure 3.
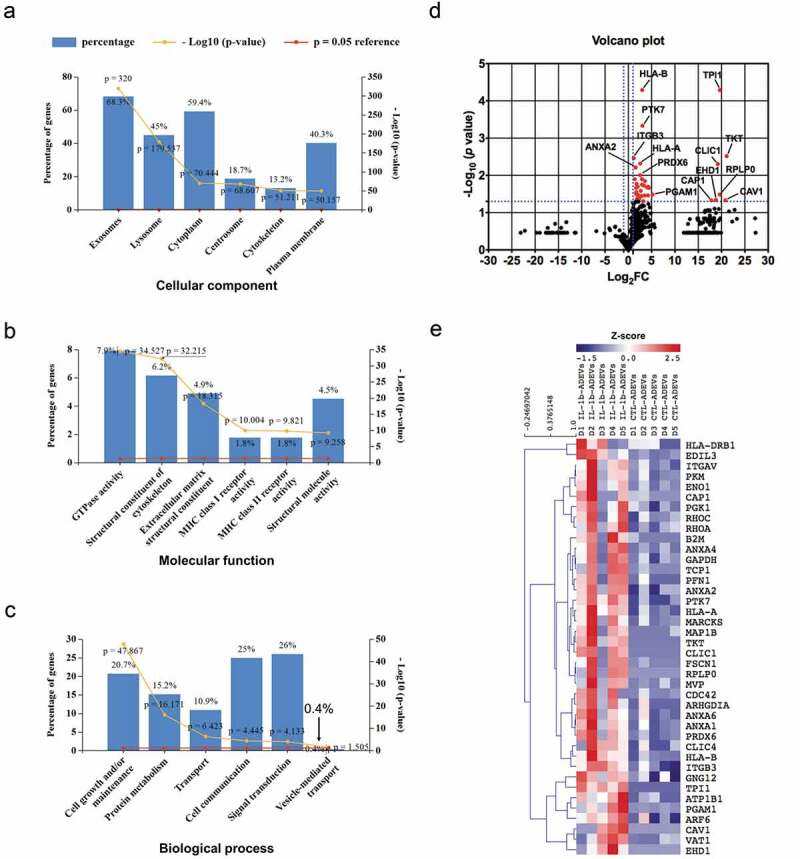
The proteome profile of CTL- and IL-1β-ADEVs. A total of 875 proteins were identified by mass spectrometry. All identified proteins were submitted to the GO classification system using Funrich analysis. (a–c) The percentage of proteins in the six most enriched categories and the enrichment significance (−log10 (p-value), p < 0.05) of identified proteins in (a) cellular components, (b) molecular functions, and (c) biological process categories were identified. The percentage of proteins identified in each category is indicated. (d) Volcano plot of the proteomic data between CTL- and IL-1β-ADEVs. Y axis of the plot represents significance (−log10 of p value) and the x axis shows the log2 of the fold-change (expression in IL-1β-ADEVs/expression in CTL-ADEVs). The red dots represent the proteins that are significantly upregulated in the IL-1β-ADEVs compared to CTL-ADEVs. The fold changes of proteins not statistically significant are represented as black dots. The dashed blue lines represent a criteria of p < 0.05 (−log10 (p value) >1.3) and fold change >2 (log2 FC >1 or <−1). The proteins with – log10 (p value) >2 or log2 FC> 5 are indicated. (e) Heatmap of 40 significantly upregulated proteins in IL-1β-ADEVs (n = 5) versus CTL-ADEVs (n = 5) using quantitative iBAQ value, with depletion depicted in blue and enrichment in red.
Table 1.
Presence of well-known EV markers and astrocyte-specific proteins in proteomic profiling of ADEVs.
| Identified proteins | Symbol | Molecular weight | p-value | Fold changea |
|---|---|---|---|---|
| sp|P21926|CD9_HUMAN CD9 antigen OS = Homo sapiens GN = CD9 PE = 1 SV = 4 | CD9 | 25 kDa | 9.99E−01 | 1.00E+00 |
| sp|P60033|CD81_HUMAN CD81 antigen OS = Homo sapiens GN = CD81 PE = 1 SV = 1 | CD81 | 26 kDa | 8.71E−02 | 1.72E+00 |
| sp|P08962-3|CD63_HUMAN Isoform 3 of CD63 antigen OS = Homo sapiens GN = CD63 | CD63 | 17 kDa | 1.99E−01 | 2.38E+00 |
| sp|Q8WUM4-2|PDC6I_HUMAN Isoform 2 of Programmed cell death 6-interacting protein OS = Homo sapiens GN = PDCD6IP | PDCD6IP | 97 kDa | 5.85E−02 | 2.27E+00 |
| sp|P14136-2|GFAP_HUMAN Isoform 2 of Glial fibrillary acidic protein OS = Homo sapiens GN = GFAP | GFAP | 50 kDa | 5.59E−01 | 2.19E+00 |
| sp|P43003-2|EAA1_HUMAN Isoform 2 of Excitatory amino acid transporter 1 OS = Homo sapiens GN = SLC1A3 | SLC1A3 | 55 kDa | 8.60E−01 | 8.05E−01 |
| sp|P11166|GTR1_HUMAN Solute carrier family 2, facilitated glucose transporter member 1 OS = Homo sapiens GN = SLC2A1 PE = 1 SV = 2 | SLC2A1 | 54 kDa | 9.35E−01 | 1.05E+00 |
aFold changes were calculated for all of the proteins by converting the 0 iBAQ values to 1 for calculation purposes.
To discover DEPs between IL-1β and CTL-ADEVs, we replaced the 0 iBAQ values with 1 in the proteomic data for the statistical significance calculation and a volcano plot was generated. A total of 40 proteins were significantly upregulated in IL-1β-ADEVs with a criteria of p < 0.05 and fold change > 2 (Figure 3(d)). Of note, most of the identified proteins are involved in cellular signalling and actin cytoskeleton regulation (Table 2). Furthermore, a heatmap of these significantly upregulated proteins was created to highlight the differences between CTL- and IL-1β-ADEVs (Figure 3(e)). Additionally, 113 proteins were uniquely present in IL-1β-ADEVs (Supplementary Table 3).
Table 2.
List of top 10 up-regulated proteins based on fold change in EVs from reactive astrocytes compare to control astrocyte.
| Uniprotein_ID | Symbol | Entrez gene name | Location | Type(s) | p-value | Fold change |
|---|---|---|---|---|---|---|
| P29401 | TKT | Transketolase | Cytoplasm | Enzyme | 3.05E−03 | 2.25E+06 |
| Q03135 | CAV1 | Caveolin 1 | Plasma Membrane | Transmembrane receptor | 4.67E−02 | 1.83E+06 |
| P05388 | RPLP0 | Ribosomal protein lateral stalk subunit P0 | Cytoplasm | Other | 3.27E−02 | 7.95E+05 |
| P60174 | TPI1 | Triosephosphate isomerase 1 | Cytoplasm | Enzyme | 5.12E−05 | 7.95E+05 |
| O00299 | CLIC1 | Chloride intracellular channel 1 | Nucleus | Ion channel | 4.96E−03 | 6.02E+05 |
| Q9H4M9 | EHD1 | EH domain containing 1 | Cytoplasm | Other | 4.99E−02 | 4.89E+05 |
| Q01518 | CAP1 | Cyclase associated actin cytoskeleton regulatory protein 1 | Plasma Membrane | Other | 4.69E−02 | 2.28E+05 |
| P18669 | PGAM1 | Phosphoglycerate mutase 1 | Cytoplasm | Phosphatase | 3.27E−02 | 3.63E+01 |
| Q16658 | FSCN1 | Fascin actin-bundling protein 1 | Cytoplasm | Other | 2.16E−02 | 2.13E+01 |
| P30447 | HLA-A | Major histocompatibility complex, class I, A | Plasma Membrane | Other | 3.50E−02 | 1.89E+01 |
Association of deps in EVS from reactive astrocytes with cellular metabolism, cellular movement and inflammatory response
Next, DEPs including either significantly expressed or uniquely present proteins in IL-1β-ADEVs were used as input for IPA. A list of 15 most highly significant canonical pathways associated with IL-1β-ADEVs was compiled (Figure 4(a) and Supplementary Table 4). Significant enrichment of both glycolysis and gluconeogenesis, the key processes of energy metabolism, represented by proteins including glucose-6-phosphate isomerase (GPI) and enolase 1 (ENO1) was observed. Some of actin regulation related pathways, essential for cellular organization, were also enriched, represented by a number of Rho GTPase family members, such as RhoA and cell division control protein 42 (Cdc42) in IL-1β-ADEVs (Supplementary Figure 3(a)). Interestingly, multiple significant pathways associated with endocytosis, cell interaction and communication, represented by integrins (e.g. ITGB1, ITGB3, ITGAV), caveolin 1 (CAV1) and clathrin light chain A (CLTA) were found. Furthermore, CXCL8/IL-8 signalling and antigen presentation pathway were identified with the increased expression of intercellular adhesion molecule 1 (ICAM1) and major histocompatibility complex (MHC) (e.g. HLA-A, HLA-B, HLA-DRB1; Supplementary Figure 3(b)), suggesting a potential role of IL-1β-ADEVs in cell adhesion and antigen presentation. Furthermore, diseases and function annotation in IPA of DEPs in IL-1β-ADEVs revealed the activation of similar pathways associated with cellular assembly and organization, cellular movement and inflammatory response (Figure 4(b)).
Integrins are known to control cell development, mediate cell interaction and modulate immune signalling in glial cells [29]. Quantitative proteome analysis demonstrated higher levels of integrin expression in IL-1β-ADEVs compared to CTL-EVs (Figure 4(c)), such as ITGB3 and ITGAV. Integrins and other IL-1β-altered proteins were mapped to integrins-mediated signal cascades, showing the involvement of IL-1β-ADEVs in cell motility and the protein kinases mediated gene transcription (Figure 4(d)).
Additionally, IPA revealed DEPs in IL-1β-ADEVs were consistent with the increased signalling of potential upstream regulators including transforming growth factor beta 1 (TGF-β1), lipopolysaccharide (LPS) and IL-1β (Figure 4(e)). They were also significantly associated with the increased signalling of pathogenic molecules such as amyloid precursor protein and tau (Supplementary Figure 3(c)). Overall, these findings demonstrated that EVs from reactive astrocytes may be capable of promoting cell communication, regulating cellular metabolism and organization, and transferring immune-reactive or pathological signals to the recipient cells.
Increased cellular uptake of EVS from reactive astrocytes compared to EVS from control astrocytes by neurons
A significant number of DEPs present in IL-1β-ADEVs are involved in endocytic pathways and actin cytoskeleton regulation, important for functioning cellular EV uptake [30]. On the other hand, EV uptake by recipient cells is a key step of trans-cellular EV communication. Therefore, we investigated the intercellular transfer of both IL-1β- and CTL-ADEVs to neurons in vitro. Murine primary cultured neurons (DIV 10) were treated with equal amount of PKH26-labelled CTL- or IL-1β-ADEVs pooled from five donors, and their uptake was imaged with a live-imaging system (Incucyte, Essen BioScience) every six hours for three days (Figure 5(a)). Both PKH26-labelled CTL-and IL-1β-ADEVs were taken up by neurons over this time period, whereas no signal was present in neurons incubated with dye alone (Figure 5(b)). Moreover, a significant increase in cellular uptake of IL-1β-ADEVs in neurons as compared to CTL-ADEVs was observed (Figure 5(b); Supplementary Movie 1–3). Two quantitative measures to evaluate EV uptake were assessed: the fluorescence intensity of cellular EVs per area and percentage of cells with PKH26-labelled EVs. The fluorescence intensity of PKH26 signal in neurons was higher after incubation with IL-1β-ADEVs than that with CTL-ADEVs over a three-day time period (Figure 5(c)). Similarly, there was a significantly higher percentage of fluorescent cells upon addition of IL-1β-ADEVs (58.47 ± 1.59% after 72 h incubation) as compared to CTL-ADEVs (35.23 ± 1.25% after 72 h incubation) (Figure 5(d)).
Accumulating evidence suggest that integrins are involved in EV uptake by recipient cells for internalization [30]. Blocking of ITGAV (CD51) and ITGB3 (CD61), which were significantly upregulated in IL-1β-ADEVs in our study, can reduce the uptake of EVs by dendritic cells [31]. To test whether integrins were involved in the elevated uptake of IL-1β-ADEVs by neurons, integrin binding was blocked using RGD peptide (Figure 6(a)). Pre-treatment of IL-1β-ADEVs with either 0.1 μM or 1 μM RGD peptide significantly prevented EV uptake by neurons (Figure 6(b, c); Supplementary Movie 4–6). Similar results were observed by assessing the percentage of PKH26+ cells over a three-day time course upon treatment of IL-1β-ADEVs with RGD (Figure 6(d)). Collectively, these data demonstrated an enhanced intercellular transfer of EVs from reactive astrocytes to neurons, and the significant contribution of integrins to the process.
Detrimental effects of EVS from reactive astrocytes on neurite development and neuronal activity compared to EVS from control astrocytes
To determine the biological effects of ADEVs on neuronal differentiation, primary cultured neurons were exposed to IL-1β-ADEVs or CTL-ADEVs on DIV 3 at three doses (1×, 1.25 μg/mL; 2×, 2.5 μg/mL; 4×, 5 μg/mL) for three days (Figure 7(a)). The neuron cultures were fixed on DIV 6 and immunostained with MAP2 to label neurons. No significant dose-dependent trophic effects were observed for neurite branching in CTL-ADEV-treated neurons compared to the vehicle control, whereas IL-1β-ADEV-treated neurons showed inhibition of on neurite outgrowth in a dose-response manner (Figure 7(b)). Quantitative analysis showed dose-dependent reductions of neurite length, total surface area and the number of nodes in IL-1β-ADEV-treated neurons compared to vehicle control and CTL-ADEVs groups (Figure 7(c–e)). In addition, there was a trend of reduction in number of neurite terminals between 4× CTL-ADEVs and 4× IL-1β-ADEVs (Figure 7(f)). The total number of neurites was similar among these groups (Figure 7(g)). These results showed a significant inhibitory effect of IL-1β-ADEVs on neurite extension and branching.
Finally, the effects of ADEVs on neuronal firing were assessed using multi-electrode arrays (MEA, Figure 7(h)) and DIV 17 neurons (marked as “day 0” in ADEVs, Figure 7(i)) for five consecutive days. Figure 7(i) shows raster plots of spike recordings from individual electrodes during a five-day time course (day 0–5, 4 out of 16 channels were shown for each day). In the vehicle controls, neurons gradually increased spike activity over the time and showed a significance after day 3 compared to day 0 (Figure 7(j), black box with whiskers). In particular, spike activity was significantly higher in CTL-ADEV group after day 1 than day 0 (Figure 7(j)), suggesting an acceleration of neuronal maturation after exposure to CTL-ADEVs. However, neurons treated with IL-1β-ADEVs did not show a significant increase in spike activity until day 3, which was decreased after day 3, resulting in no significant difference of spike activity between day 0 and day 5 (Figure 7(j)). Treatment of either CTL-ADEVs or IL-1β-ADEVs, on the other hand, had no effect on neuronal bursts over five days (Figure 7(k)). Together, these results indicated that CTL-ADEV treatment accelerated neuronal maturation for firing, which was delayed and regressed by IL-1β-ADEVs treatment.
Discussion
Astrocyte-shed EVs are considered as molecular regulators of the immunological response to inflammatory factors (e.g. IL-1β) in brain [10]. Here we presented the first study, to our knowledge, to characterize the proteomic profiles of both baseline human ADEVs and IL-1β-ADEVs, and their functional implications in primary cultured mouse cortical neurons. Using LC-MS/MS, we demonstrated that IL-1β-ADEVs showed a notable up-regulation of proteins compared to CTL-ADEVs, indicating a unique protein signature in EVs from reactive astrocytes. One protein cluster was surface proteins, such as integrins and CAV1, which regulate endocytic pathways. We further confirmed a partly integrins-dependent neuronal uptake for IL-1β-ADEVs in vitro. In addition, we found IL-1β-ADEVs exerted detrimental effects on neurite differentiation and neuronal firing, while CTL-ADEVs promoted neuronal maturation for firing.
As a downstream product of inflammasome activation, IL-1β is one of the most potent mediators of astrocyte activation upon brain injury and neuroinflammation. Our data presented typical reactive characteristics of astrocytes including morphological and functional transformations after IL-1β treatment. We also observed significantly increased release of EVs from reactive astrocytes, consistent with another report in rat primary astrocytes following TNF-α and IL-1β stimulation [9]. This might be explained by IL-1β-induced persistent activation of the neural sphingomyelinase-2, which functions in the ceramide and exosome biogenesis [16].
Increasing evidence suggests the cargo of EVs could be dynamically altered by the phenotype of the cells of origin. For example, oligodendrocyte-derived EVs contain myelin proteins, and proteins associated with protection against oxidative stress [32]. In our study, the astrocyte-specific proteins such as GFAP, SLC1A3 and SLC2A1 were found in both CTL-ADEVs and IL-1β-ADEVs. Further, elevated GFAP expression, a hallmark of astrocyte reactivity, was observed in IL-1β-ADEVs compared to CTL-ADEVs from our proteome data (fold change = 2.19, shown in Table 1), suggesting that EVs might reflect the activation status of astrocytes. Intriguingly, we found upregulated expression of some classic EV markers (CD81, CD63 and PDCD61IP/ALIX) from IL-1β-ADEVs, likely due to exosomes secreted by reactive astrocytes. It has been shown that different exosome types might have specific expression pattern of tetraspanins, which includes CD63+ CD81+ CD9+ EVs, CD63 – CD81 – CD9+ EVs, CD63 – CD81 – CD9 – EVs and EVs enriched in other factors [33]. However, their functional specificity still remains elusive. Of note, apart from TGFβ1, other known cytokines or growth factors present in astrocytes were not found in ADEVs. It is quite possible, however, that our methods may lack the sensitivity needed for identifying low-abundance proteins [34].
Previous study identified miRNA signatures in astrocyte-derived EVs response to inflammatory factors from a rodent model [9]. In this study, we identified 40 significantly upregulated proteins and a large number of uniquely expressed proteins in IL-1β-ADEVs while no proteins were significantly decreased, also revealing an activation of EVs biogenesis after treatment of astrocytes with IL-1β. These proteins are associated with pathways characteristic of reactive astrocytes including cell metabolism and organization, cellular communication and inflammatory response compared to control ADEVs. For example, functional changes in glutamate homoeostasis, energy metabolism and glucose metabolism occurred in reactive astrocytes, and we found enriched proteins in IL-1β-ADEVs were related to “glycolysis” and “gluconeogenesis” such as phosphoglycerate kinase 1 (PGK1), enolase 1 (ENO1) and phosphoglycerate mutase (PGAM) [25]. This might explain our observations of detrimental effects of IL-1β-ADEVs when exposed to primary cultured neurons as hyper-metabolism probably caused by astrocyte activation was found in neurodegenerative disease mouse models [35]. The immunomodulatory roles of astrocyte-derived EVs have also been reported in the brain injury and our proteome data showed increased expression of human MHC proteins in IL-1β-ADEVs, which might be involved in antigen presentation [16]. These HLA proteins were expressed by microglia and astrocytes in brain, and were dysregulated during neurodegenerative diseases [36]. In addition, the up-regulation of small GTPases in IL-1β-ADEVs also indicated a role of IL-1β-ADEVs in immune response, as they might contribute to increased CXCL8/IL-8 signalling, a well-known pathway in response to inflammatory signals of astrocytes [37]. Further studies on the detection of immune-responsive molecules after treatment of brain immune cells with IL-1β-ADEVs may help to better understand whether ADEVs play an immunosuppressive or activating role.
EVs can be targeted to recipient cells and deliver their cargo to mediate physiological processes and pathological progress [11]. The proteomic prevalence of surface proteins, particularly integrins in IL-1β-ADEVs, is intriguing as these molecules regulate endocytic pathways. They mediate targeted EV uptake by recipient cells by triggering membrane-associated signal cascades [30]. Here, we did observe a significantly increased cellular uptake of IL-1β-ADEVs compared to CTL-ADEVs by incubation of primary neurons with fluorescently labelled ADEVs. To further confirm whether it was due to the higher expression of integrins in IL-1β-ADEVs, we pre-incubated EVs with a specific integrin inhibitor RGD peptide and found neuronal uptake of IL-1β-ADEVs was partially suppressed. Some evidence in the literature showed an organ-specific metastasis of tumour cells was controlled by distinct exosomal integrins. Targeting the integrin α6β4 decreased exosome uptake by lung cells, while silencing integrin αvβ5 reduced exosome uptake by liver cells [38]. Additionally, neutralizing antibodies against ITGAV (CD51) and ITGB3 (CD61) can reduce dendritic cell uptake of EVs [31]. Thus, we interpret our results indicating that higher levels of integrins in IL-1β-ADEVs might increase their uptake by recipient cells, and may be involved in cellular specificity or targeting. Future studies using integrins antibodies (e.g. anti-CD51 or anti-CD61) to selectively block integrins activity in EVs may further elucidate the potential role of these proteins in regulating EV internalization and mediating EV functions. However, since uptake was not abrogated, other proteins likely also contribute to ADEV uptake by neurons. Collectively, our results suggest that proteins on the surface of the astrocytic EVs, which are essential for mediating EV uptake by target cells, can be dynamically regulated under the conditions of inflammation, thus providing a better understanding of the rules that underpin EV entry into cells.
Recent evidence shows ADEVs shed in response to acute cellular stressors (e.g. IL-1β, TNF-α, LPS) impaired neurite development and weakened neuronal activity [9]. Notably, our examination of ADEVs on neuronal differentiation also found that IL-1β-ADEVs exerted detrimental effects on neurite development and neural activity. The small GTPases cluster enriched in IL-1β-ADEVs, which play an important role in cytoskeletal reorganization, signal transduction, cell invasion, cellular interaction and inflammation, may explain how ADEVs could modulate neuritogenesis and synaptogenesis [39]. For example, profilin 1 (PFN1), fascin actin-bundling protein 1 (FSCN1) and destrin (DSTN), which were highly expressed in IL-1β-ADEVs, regulate actin dynamics; disruption of which might lead to abnormal neuronal development [40–42]. The Ras homolog family member A (RhoA), which showed significant upregulation in IL-1β-ADEVs, inhibited axonal outgrowth following CNS injury and counteracted regeneration [43]. A recent study reported an increased RhoA activity, which caused altered cell migration and impaired neuritogenesis in human induced pluripotent stem cell-derived neural cells with PARK2 mutation [44]. RhoA is also known to inhibit the formation, growth, and maintenance of synaptic spines [45]. Another explanation is the enrichment of MHC proteins in IL-1β-ADEVs compared to CTL-ADEVs. Indeed, emerging evidence suggests an important non-immune role of MHC proteins in brain. Soluble forms of MHC-I (HLA-A, B, C) negatively regulated neurite outgrowth in the embryonic mouse retina [46]. MHC-I also disrupted the establishment of neuronal connections and reduced synapse density [47]. Thus, our findings from functional validations were consistent with interpretations of proteomic analyses. Further studies to identify the critical proteins in IL-1β-ADEV-mediated effects are warranted to identify novel targets to control ADEV functions particularly under pathophysiological conditions.
In conclusion, this study presents the first proteomic profiling of EVs from primary cultured human astrocytes and IL-1β-induced reactive human astrocytes. Our data reveals a characteristic signature in the protein cargo of IL-1β-ADEVs. Proteomic analysis showed EV enrichment of proteins associated with cellular organization, cellular interaction and inflammatory response. Furthermore, we discovered the increased cellular transfer of IL-1β-ADEVs to neurons promoted by higher integrin compositions. IL-1β-ADEVs suppressed neurite development and neural maturation as determined by spike firing. These studies improve our understanding of the role of astrocyte EVs released during neurodegeneration or neuroinflammation, and are likely to promote development of novel diagnostic and therapeutic possibilities for neuroinflammatory disease.
Supplementary Material
Acknowledgments
We thank the Ikezu, Borgmann and Ghorpade lab members for experimental supports, and X. Li and S. Shaffer (Mass Spectrometry Facility, University of Massachusetts Medical School) for mass-spectroscopy services.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Author Contributions
Y.Y., A.G. and T.I. designed research; Y.Y., K.B., V.E. and S.S. performed research; Y.Y., K.B., V.E. and S.S. and T.I. analyzed data; Y.Y. and K.B. wrote the paper; and Y.Y., K.B., A.G. and T.I. edited the paper.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The Supplementary data for this article can be accessed here.
References
- [1].Colombo E, Farina C.. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37(9):608–18. [DOI] [PubMed] [Google Scholar]
- [2].Freeman MR, Rowitch DH.. Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron. 2013;80(3):613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Phatnani H, Maniatis T. Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol. 2015;7(6):a020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Teh DBL, Prasad A., Jiang W.,et al. Transcriptome analysis reveals neuroprotective aspects of human reactive astrocytes induced by interleukin 1beta. Sci Rep. 2017;7(1):13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang G, Li J., Purkayastha S. et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497(7448):211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kou W, Banerjee S., Eudy J. et al. CD38 regulation in activated astrocytes: implications for neuroinflammation and HIV-1 brain infection. J Neurosci Res. 2009;87(10):2326–2339. [DOI] [PubMed] [Google Scholar]
- [8].Ghorpade A, Holter S., Borgmann K. et al. HIV-1 and IL-1 beta regulate Fas ligand expression in human astrocytes through the NF-kappa B pathway. J Neuroimmunol. 2003;141(1–2):141–149. [DOI] [PubMed] [Google Scholar]
- [9].Chaudhuri AD, Dastgheyb RM, Yoo SW, et al. TNFalpha and IL-1beta modify the miRNA cargo of astrocyte shed extracellular vesicles to regulate neurotrophic signaling in neurons. Cell Death Dis. 2018;9(3):363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Delpech JC, Herron S., Botros M.B. et al. Neuroimmune crosstalk through extracellular vesicles in health and disease. Trends Neurosci. 2019;42(5):361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].You Y, Ikezu T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol Dis. 2019;130:104512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thompson AG, Gray E., Heman-Ackah S.M. et al. Extracellular vesicles in neurodegenerative disease - pathogenesis to biomarkers. Nat Rev Neurol. 2016;12(6):346–357. [DOI] [PubMed] [Google Scholar]
- [13].Ciregia F, Urbani A, Palmisano G. Extracellular vesicles in brain tumors and neurodegenerative diseases. Front Mol Neurosci. 2017;10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sami Saribas A, Cicalese S., Ahooyi T. M., et al. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017;8(1):e2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang S, Cesca F., Loers G.,et al. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J Neurosci. 2011;31(20):7275–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dickens AM, Tovar-y-Romo L.B., Yoo S.W. et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. 2017;10(473):eaai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shelke GV, Lässer C, Gho YS, et al. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles. 2014;3:24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tarassishin L, Suh HS, Lee SC. LPS and IL-1 differentially activate mouse and human astrocytes: role of CD14. Glia. 2014;62(6):999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Liu K., Liu Y. et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol. 2013;14(8):793–803. [DOI] [PubMed] [Google Scholar]
- [21].Benedikter BJ, Bouwman FG, Vajen T, et al. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Sci Rep. 2017;7(1):15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pathan M, Keerthikumar S., Chisanga D. et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6(1):1321455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saha B, Momen‐Heravi F., Furi I., et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology. 2018;67(5):1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zorina Y, Stricker J., Caggiano A. O., et al. Human IgM antibody rHIgM22 promotes phagocytic clearance of myelin debris by microglia. Sci Rep. 2018;8(1):9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ben Haim L, Carrillo-de Sauvage M. A., Ceyzériat K., et al. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Beurrier C, Faideau M, Bennouar KE, et al. Ciliary neurotrophic factor protects striatal neurons against excitotoxicity by enhancing glial glutamate uptake. PLoS One. 2010;5(1):e8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fruhbeis C, Fröhlich D, Kuo WP, et al. Extracellular vesicles as mediators of neuron-glia communication. Front Cell Neurosci. 2013;7:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thery C, Witwer K. W., Aikawa E., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu X, Reddy DS. Integrins as receptor targets for neurological disorders. Pharmacol Ther. 2012;134(1):68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3:24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Morelli AE, Larregina A. T., Shufesky W. J., et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104(10):3257–3266. [DOI] [PubMed] [Google Scholar]
- [32].Kramer-Albers EM, Bretz N, Tenzer S, et al. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: trophic support for axons? Proteomics Clin Appl. 2007;1(11):1446–1461. [DOI] [PubMed] [Google Scholar]
- [33].Kowal J, Arras G., Colombo M., et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nguyen DC, Lewis HC, Joyner C, et al. Extracellular vesicles from bone marrow-derived mesenchymal stromal cells support ex vivo survival of human antibody secreting cells. J Extracell Vesicles. 2018;7(1):1463778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sancheti H, Patil I., Kanamori K., et al. Hypermetabolic state in the 7-month-old triple transgenic mouse model of alzheimer’s disease and the effect of lipoic acid: a 13C-NMR study. J Cereb Blood Flow Metab. 2014;34(11):1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Song S, Miranda C. J., Braun L., et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat Med. 2016;22(4):397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hoffmann E, Dittrich‐Breiholz O., Holtmann H., et al. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72(5):847–855. [PubMed] [Google Scholar]
- [38].Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Johnson DS, Chen YH. Ras family of small GTPases in immunity and inflammation. Curr Opin Pharmacol. 2012;12(4):458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wu CH, Fallini C., Ticozzi N. et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kraft R, Escobar MM, Narro ML, et al. Phenotypes of drosophila brain neurons in primary culture reveal a role for fascin in neurite shape and trajectory. J Neurosci. 2006;26(34):8734–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frese CK, Mikhaylova M., Stucchi R., et al. Quantitative map of proteome dynamics during neuronal differentiation. Cell Rep. 2017;18(6):1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shao Z, Browning JL, Lee X, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45(3):353–359. [DOI] [PubMed] [Google Scholar]
- [44].Bogetofte H, Jensen P., Okarmus J. et al. Perturbations in RhoA signalling cause altered migration and impaired neuritogenesis in human iPSC-derived neural cells with PARK2 mutation. Neurobiol Dis. 2019;132:104581. [DOI] [PubMed] [Google Scholar]
- [45].Duman JG, Mulherkar S., Tu Y.K. et al. Mechanisms for spatiotemporal regulation of Rho-GTPase signaling at synapses. Neurosci Lett. 2015;601:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Washburn LR, Zekzer D., Eitan S. et al. A potential role for shed soluble major histocompatibility class I molecules as modulators of neurite outgrowth. PLoS One. 2011;6(3):e18439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Glynn MW, Elmer B.M., Garay P.A. et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14(4):442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.