

ABSTRACT
Aim of the study: Potential care implications of antifibrotic reimbursement restrictions were studied by forced vital capacity (FVC) decline, mortality and specialty care related healthcare resource utilization in patients with idiopathic pulmonary fibrosis (IPF).
Material and methods: IPF patients were identified from the electronic medical records of the Hospital District of Southwest Finland between 2005 and 2017. Text-mining was used for patient identification to exclude other interstitial lung diseases (ILD) from the cohort. FVC reimbursement restriction (FVC 50-90%) was used for stratification.
Results: Out of all patients with ILD, 27% (N = 266) were identified to have IPF. At baseline, 24% presented with FVC>90% and 63% with FVC 50-90% predicted. FVC at diagnosis did not improve during the study period. Median survival decreased by severity from 6.7 years in FVC>90% at baseline to 0.7 years in patient with FVC<50% predicted. In the FVC>90% group, 14% died before a change in FVC category could be noted. Overall, 4.7 million euro worth of specialty care resources were spent on IPF patients. The highest cost driver was inpatient days.
Conclusions: IPF is associated with a high burden of disease, and reimbursement restrictions are in conflict with early care. As there are antifibrotic treatment options for IPF patients, early diagnosis is important.
KEYWORDS: IPF, FVC decline, mortality, healthcare resource utilization
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrotic lung disease causing disrupted gas exchange, respiratory failure and death [1,2]. Disease mechanisms include genetic and environmental factors but are poorly understood [1,2]. Substantial international efforts have unified and improved IPF diagnostics and treatment recommendations [1,3–7]. The diagnosis is based on the histological and/or radiological appearance of a usual interstitial pneumonia (UIP) pattern, while excluding other interstitial lung diseases (ILD) [1,5].
IPF prevalence estimates vary from 2–29 cases/100 000 persons, with 8.6–19 cases/100 000 persons in Finland [1,8,9]. Differing study designs and historically ununiform definitions likely explain the variability in prevalence. In Finland, IPF and other ILD are diagnosed under ICD-10 code J84.1, leading to challenges in distinguishing between the diseases in retrospective studies, however, estimates are that 20-30% of these patients have IPF [9,10].
Due to strict inclusion and exclusion criteria, generalizing randomized clinical trial (RCT) data to real life remains challenging [11]. RCTs mainly investigated IPF patients with moderate physiological impairment, or even excluded those with forced vital capacity (FVC) >90% predicted [12–15]. Although post-hoc analyses show similar antifibrotic therapy responses between patients with preserved and significantly impaired lung function, the limited nature of the data has restricted reimbursement in many countries [16,17]. In Finland, the antifibrotic treatment reimbursement threshold is FVC 50-90% predicted [18].
IPF associates with a substantial humanistic and healthcare burden. Yet, information on disease burden and mortality by baseline FVC category is largely lacking [19–24]. Further, many studies assessing healthcare resource utilization (HCRU) have been claims based, leading to potential selection bias and unavailability of clinical data [19,23,24]. Because of reimbursement restrictions, FVC decline, mortality and HCRU are of high interest. Therefore, the aims of this study were to assess IPF progression measured by FVC decline, mortality, and healthcare resource utilization by disease severity in data reflecting real clinical practice and reimbursement restrictive categories.
Material and methods
Adult patients with ICD-10 codes for other interstitial pulmonary diseases with fibrosis or unspecified interstitial pulmonary disease (J84.1 or J84.9), at the Hospital District of Southwest Finland (HDSWF) during the years 2005–2017 were included in this retrospective registry based study (N = 993), and electronic medical records (EMR) were analysed. From the initial cohort, 266 IPF patients were identified based on high-resolution computed tomography (HRCT) and/or lung biopsy statements and clinical diagnosis. IPF patients were followed from the first J84.1 or J84.9 diagnosis (= index) until the end of 2017, the initiation of antifibrotic treatment (pirfenidone or nintedanib), death, or until lost to follow-up, defined as no specialty care contact in 18 months.
IPF cohort selection
For inclusion, fibrotic changes corresponding to IPF in HRCT or lung biopsy statements in addition to diagnosis of IPF symptoms in EMR were required. A clinical expert defined text patterns of affirmative and negative phrases for text-mining from the five most recent entries in the EMR (Table A1). Affirmative phrases yielded one point and negative phrases against IPF reduced one point. The IPF cohort was formed using a majority vote of the affirmative or negative entries of EMR texts and imaging/pathology statements. The inclusion criteria was met by 266 patients.
IPF cohort validation
Pirfenidone and nintedanib medication, available since 2013 and 2015 in Finland respectively, was utilized for cohort validation. Of the 993 patients with J84.1 or J84.9 diagnoses, 46 were prescribed pirfenidone or nintedanib. Of these, 43 patients were among those identified with IPF. Two patients with antifibrotic medication lacked HRCT or lung biopsy required for inclusion, and one false negative case was excluded based on text-mining results (3/46, specificity 93.5%).
The specificity of the text mining algorithm was verified by selecting 20 random positively identified patients. Based on manual text validation, IPF was verified in 19 persons. In the remaining patient, IPF was the most likely diagnosis, but interstitial fibrosis due to drug adverse event was not completely excluded (1/20, specificity 95.0%). Randomly selected texts of 10 excluded patients were further assessed for sensitivity. No evident IPF cases were identified, some were clearly not IPF (e.g. cryptogenic organizing pneumonia; myelofibrosis; or scleroderma with lung involvement). This proved that both the specificity and sensitivity, along with exclusion of non-IPF patients from the cohort, were good.
Lung function
Lung function was assessed by FVC at baseline (±6 months) and during follow-up. At baseline, 62% of patients had an original structured database spirometry recording available. Text-mining of EMR increased baseline FVC value coverage to 93%. Patients were divided by baseline FVC into groups, mirroring reimbursement restrictions in Finland: (1) FVC>90% predicted; (2) FVC 50-90% predicted; (3) FVC <50% predicted. Lung function was also assessed by diffusion capacity of carbon oxide (DLCO, mmol/min*kPa), available from structured data as an absolute value in 51% of IPF patients.
Time-to-event analyses were used to assess the change from one FVC category to the next or death. Censoring events were antifibrotic treatment initiation, end of study, or lost to follow-up. The association of FVC and DLCO was analysed with Spearman’s rank correlation.
Medication
The proportion of patients receiving N-acetylcysteine, oral corticosteroid, or azathioprine was assessed from hospital prescriptions (inpatient/outpatient care) and patient texts. Medication use was defined as the time between the first and the last recording. For medication combinations, overlapping timelines were required.
Survival
Survival was assessed by Kaplan-Meier fits (stratified by baseline FVC category) and Cox-proportional hazard models. Time to event was defined from index to death (event) with initiation of antifibrotic treatment, lost to, or end of follow-up as censoring events. Survival analyses included 260 patients (exclusions: one post-mortem diagnosis, five antifibrotic treatment initiations at index). Cox-proportional hazard models included univariable and multivariable models. FVC was treated as a time varying covariate using all available measures from the follow-up. Categorical FVC groups were treated as time varying covariates and considered ‘no return’. Other covariates included data ±6 months from index.
Oxygen supplementation
To investigate the need for oxygen supplementation, the lending of oxygen concentrators was assessed from the medical device lending database of the HDSWF. O2 supplementation period included the time from the first device lending day until the device was returned.
Healthcare resource utilization and cost
HCRU included specialty care contacts in the form of in-patient days, out-patient visits at the hospital, emergency room (ER) visits, procedures, surgical operations, imaging, neurophysiology, clinical physiology examinations, and laboratory tests. FVC categories were defined as no return. For each patient the time spent in each FVC category was used to map the corresponding HCRU until end of or lost to follow-up, antifibrotic medication initiation, or death.
Costs were computed utilizing standard HCRU unit costs from the HDSWF 2017 price listing. Each specialty care contact was evaluated by specialty and severity type (normal/demanding visit). Visits lacking these metadata were imputed as the mean costs of all corresponding visits. For prices not found at the HDSWF, other publicly available Finnish price listings were utilized. Operations, procedures, imaging and laboratory tests lacking data in any public price lists were imputed as the mean of all available corresponding prices. Costs were expressed in 2017 euros.
The Turku University Hospital administration approved the study (T76/2018) and the study was performed according to the EU General Data Protection Regulation (GDPR).
Results
Of the 993 patients with J84.1 or J84.9 diagnoses, 266 (27%) were identified as true IPF patients and included in further analyses. IPF patients were mainly identified through HRCT statements (77.1%, N = 205) or the combination of HRCT and biopsy (21.8%, N = 58), while biopsy alone was rare (1.1%, N = 3). The mean follow-up time was 3.03 (SD 2.97) years or 808 patient-years. Patients were most commonly treated with oral corticosteroids (68%) and azathioprine (11.7%), and rarely with N-acetylcysteine (2.6%). The combination of these three drugs was used by 1.1% of the patients.
The mean age at diagnosis was 74 years, with IPF more prevalent in men (64%), Table 1. Mean FVC at index was 77% predicted, with 24% of patients presenting with FVC >90% predicted and 63% with FVC 50-90% predicted. The median number of FVC measures in the cohort was 6 (IQR 3–12). No trend for improved FVC at diagnosis was observed during the study period (Figure 1). At the end of follow-up, 16% had initiated antifibrotic medication and 42% of patients had died (Table 1).
Table 1.
Baseline characteristics of IPF patients
| Trait | Value | MISSING, (%) | |
|---|---|---|---|
| N | 266 | ||
| MALE (%) | 64% | ||
| Age, years | 74.3 ± 8.45 | ||
| Follow-up time, years | 3.03 ± 2.97 | ||
| FVC % predicted | 77 ± 20 | 7% | |
| DLCO (mmol/min*kPa) | 4.02 ± 1.27 | 49% | |
| BMI kg/m2* | 27.8 ± 4.9 | 14% | |
| Charlson index | 2.1 ± 1.45 | ||
| Baseline 6 min walking test (m) | 335 ± 139 | 80% | |
| Any 6 min walking test (m)** | 349 ± 136 | 65% | |
| Baseline FVC | FVC>90% pred.; N (%) FVC % predicted |
64 (24%) 102 ± 10 |
|
| FVC 50-90% pred.; N (%) FVC % predicted |
167 (63%) 72 ± 11 |
||
| FVC <50% pred.; N (%) FVC % predicted |
17 (6%) 39 ± 8 |
||
| FVC unknown; N (%) FVC % predicted |
18 (7%) NA |
||
| Baseline smoking status * | Former, (%) | 31% | |
| Current smoker, (%) | 20% | ||
| Never smoked, (%) | 41% | ||
| Smoking status unknown, (%) | 9% | ||
| End of follow up type | Alive 31.12.2017, (%) | 41% | |
| Antifibrotic medication initiated, (%) | 16% | ||
| Dead, (%) | 42% | ||
*text-mined. Data presented as mean±SD if not otherwise indicated; **complementing baseline data with data from any time during follow-up; value closest to baseline used if multiple values were present
Figure 1.
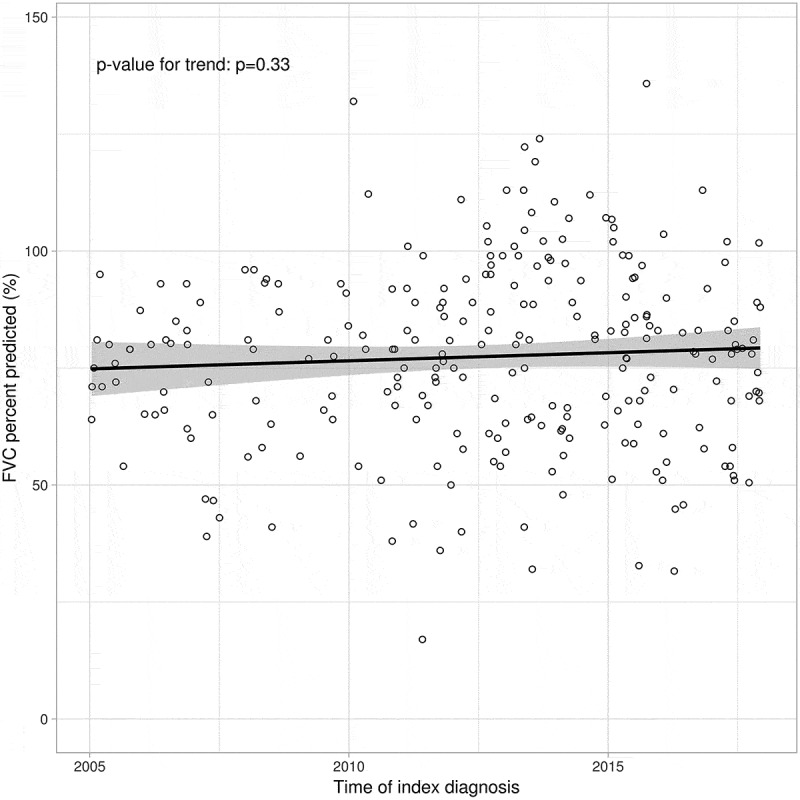
FVC (% predicted) at diagnosis by calendar year
At baseline vs. at end of follow-up, essential hypertension was diagnosed in 33% vs. 48% of patients; chronic ischemic heart disease in 19% vs. 27%. Pneumonia diagnoses increased from 16% to 41%, and chronic obstructive pulmonary disease (COPD) from 8% to 15% (Table A2).
FVC decline
IPF progression was described as the time from each baseline FVC category to the next. The median time from FVC>90% to below 90% or death was 2.4 years (Table 2). A change in category was observed in 44 (66%) of these patients and 14% died. Seventeen percent (N = 11) of patients remained in the FVC>90% category when their follow-up ended (Table 2). Only one patient with baseline FVC>90% reached FVC<50% predicted during follow-up, one third of patients died and follow-up ended in 58% of patient before this. Moreover, 6.3% of all patients initiated antifibrotic treatment and were censored.
Table 2.
Time to change of FVC category according to baseline status, Kaplan-Meier analysis
| Baseline value |
||||
|---|---|---|---|---|
| Event value | FVC>90% ↓ <90% |
FVC >90% ↓ <50% |
FVC 50-90% ↓ <50% |
|
| N total | 64 | 64 | 166 | |
| Time to event, years; median [95% CI] | 2.4 [1.1–3.1] | 4.9 [3.1 – NA] | 4.0 [2.8–5.4] | |
| Event | FVC below event value, N (%) | 42 (66%) | 1 (1.6%) | 30 (18%) |
| Dead, N (%) | 9 (14%) | 21 (33%) | 43 (26%) | |
| Transplant, N (%) | 0 | 0 | 0 | |
| Censoring | End of follow-up [FVC remained above event value], N (%) | 11 (17%) | 37 (58%) | 60 (36%) |
| Antifibrotic treatment initiated, N (%) | 1 (1.6%) | 4 (6.3%) | 32 (19%) | |
| Lost to follow-up, N (%) | 1 (1.6%) | 1 (1.6%) | 1 (0.6) | |
In patients with baseline FVC of 50-90% predicted, 18% reached an FVC<50% and 26% died. Censoring due to antifibrotic treatment was noted in 19% and end of follow-up in 36% before a category drop (Table 2).
DLCO was the best in those with baseline FVC >90% predicted and decreased with worsening FVC (median, IQR; FVC>90%: 4.4, 3.5–5.3; FVC 50-90%: 3.7, 3.1–4.7; FVC<50% 3.3, 3.3–3.6 mmol/min*kPa, p = 0.002). Long term continuous oxygen supplementation is provided to patients by clinical indication e.g. low oxygen at rest or exercise. Overall, 27% of patients utilized oxygen supplementation devices, 12.7% of patients with FVC >90% predicted; 15.7% of those with 50-90% predicted; and 47.9% of those with FVC<50% predicted.
Mortality
The median survival of the IPF cohort was 5.0 years (95% CI: 4.0–6.7), Figure 2. Median survival decreased by worsening FVC category from 6.7 years at baseline FVC>90% predicted to 5.3 years in FVC 50-90% predicted, and 0.7 years in patients with FVC<50% predicted (Figure 2). At the end of follow-up, 31% of patients with baseline FVC >90% predicted had died; 38% of FVC 50-90%, 82% of FVC<50%, and 88% of FVC unknown.
Figure 2.
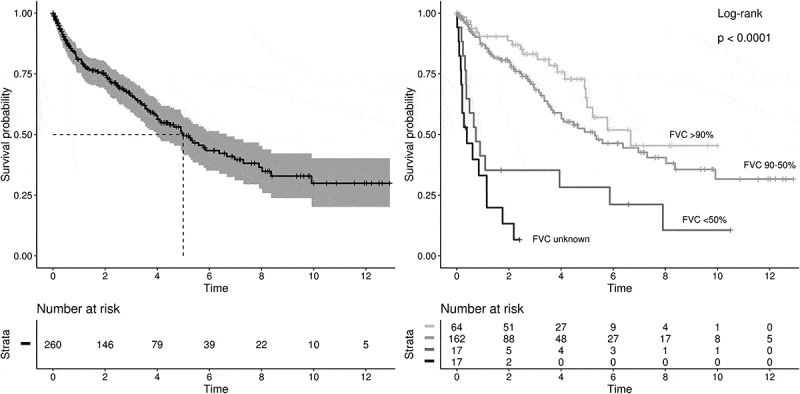
Overall survival of IPF patients (left) and stratified by baseline FVC (right) to FVC>90%; 50-90%; <50% predicted; and unknown FVC. Time is in years. Censoring events: antifibrotic treatment initiation, lost to follow-up, end of follow-up
In the multivariable Cox model, with FVC>90% as reference, FVC 50-90% was associated with a 2-fold increase in mortality [HR 1.98 (95% CI: 0.91–2.28), p = 0.084], but the association did not reach statistical significance. Patients with FVC <50% predicted, presented a significant 8.8-fold increased risk of death. Independent predictors of death were low BMI, and baseline oral corticosteroid (OCS) use (Figure 3, Table A3, model 1). Treating FVC as a continuous variable, each percent decline increased the mortality risk by 4%. Further, in this model higher age at diagnosis and low BMI (<20 kg/m2) were independently associated with mortality (Table A3, model 2).
Figure 3.
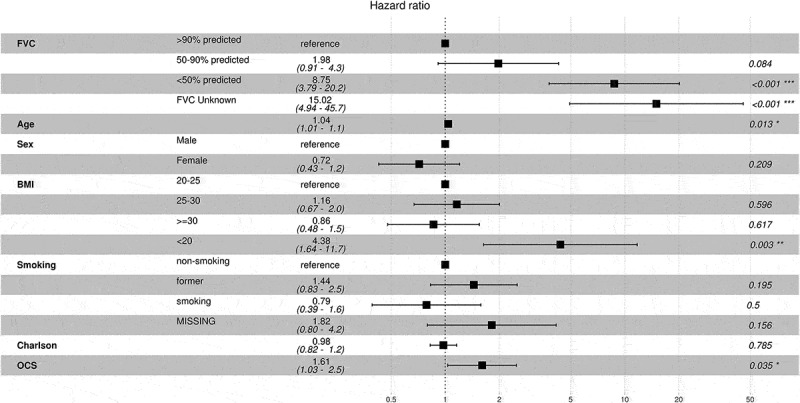
Cox regression analysis of overall mortality in IPF patients. FVC group was used as a time-varying covariable. BMI-body mass index, OCS-oral corticosteroid
Healthcare resource utilization
Healthcare resource utilization correlated with disease severity. IPF patients were on average hospitalized 5.5 days per patient-year. This increased by deteriorating FVC category and was 1.2-fold higher in patients with baseline FVC 50-90% predicted, and 2.8-fold higher in patients with FVC <50% predicted compared to FVC>90% predicted (Figure 4, Table A4). Unknown FVC presented an 8.1-fold increase in inpatient days per patient-year compared to FVC>90% predicted.
Figure 4.
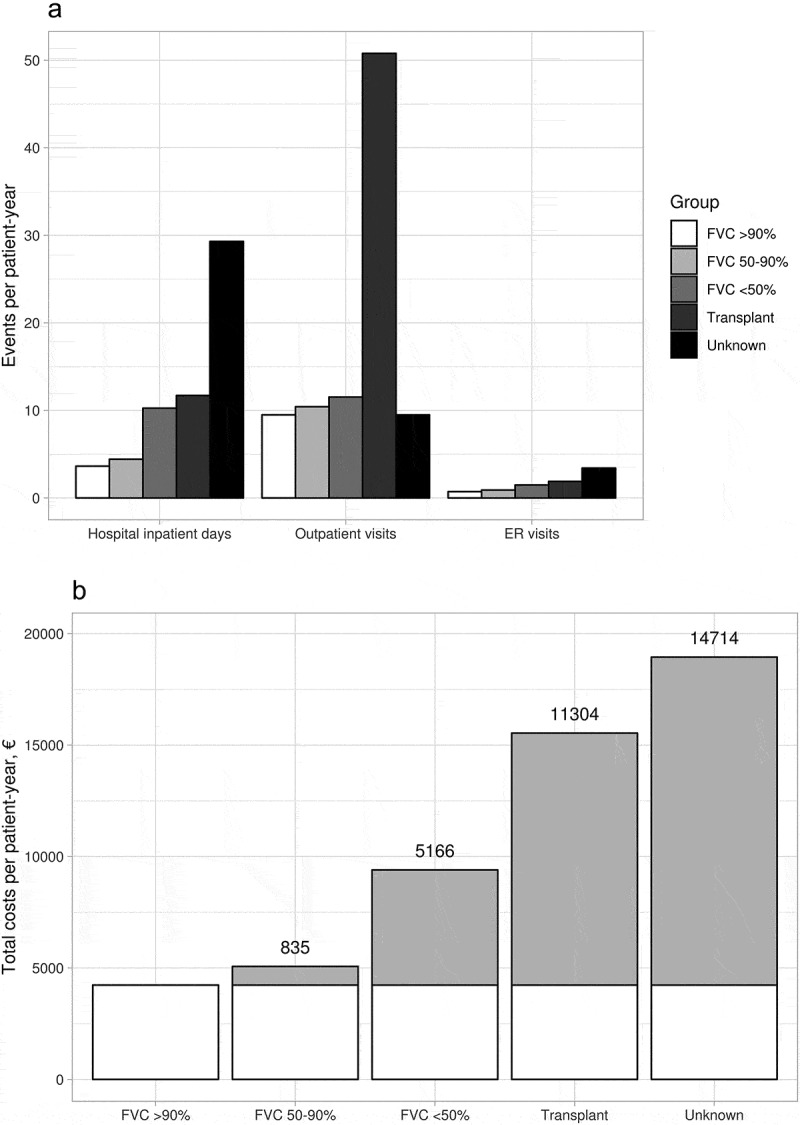
a) Hospital inpatient days, out-patient visits and emergency room visits (ER) per patient-year, stratified by FVC category. b) Overall healthcare resource utilization related costs (€) per patient-year, at specialty care of IPF patients, stratified by FVC category or lung transplant. The incremental costs per worsening category compared to FVC>90% predicated are presented on top of/in bars with grey
Overall, IPF patients utilized 4.7 million euros worth of specialty care resources between 2005 and 2017, calculated with 2017 prices. Incremental overall HCRU costs of patients with FVC 50-90% predicted compared to FVC>90% was 835€ per patient-year, increasing to 5,166€ per patient-year at FVC<50% predicted (Figure 4, Table A4). Lung transplanted patients and those lacking an FVC measure had the highest incremental costs (Figure 4, Table A4). Highest cost drivers were hospital inpatient days in all categories, with an average of 2,172€ per patient-year, followed by visits and procedures (1,290€ and 1,045€ per patient-year, Table A4).
Discussion
This retrospective registry study investigated 266 IPF patients during the years 2005–2017 for FVC decline, mortality and specialty care associated healthcare resource utilization. The Turku University Hospital is the sole provider of secondary care for severe respiratory diseases at the HDSWF, making data from the 480 000 catchment population representative. This study describes and defines an IPF population using data lakes for the first time. Investigating disease severity by FVC, over a period exceeding ten years, yielded insights on the disease course and healthcare resource utilization. Notably, one fourth of patients presented with mild disease and were unentitled to modern IPF treatment reimbursement.
Patient inclusion was based on positive HRCT and/or biopsy EMR results in combination with clinical diagnosis, whilst excluding other causes. This identified IPF in 27% of patients with diagnose codes J84.1 or J84.9. The proportion is consistent with previous studies showing that 20-30% of ILD patients have IPF [9,10].
Diagnosis over time
IPF guidelines and available treatment options have directed IPF care towards precise and earlier diagnosis [1,5,6]. Yet, no increase in FVC by year of diagnosis was observed in this study. This was surprising considering the evolution of diagnosis and the study period covering over 10 years of data, however, HRCT and biopsy criteria have been included in the diagnostic procedure in Finland since the beginning of the study period. A relatively preserved lung function at diagnosis, also seen in other Finnish studies, may partly explain this [9]. IPF diagnosis is often delayed up to 2 years from symptom onset to clinical diagnosis, by subtle progressive symptoms and non-specific clinical and physical presentation [25,26]. Diagnosis delays independently increase mortality risk, highlighting the importance of early diagnosis [25]. Additionally, clinical studies with pirfenidone and nintedanib show that early initiation of disease modifying treatment can postpone irreversible deteriorative changes in lung function [2,12,13,15].
Oxygen supplementation and quality of life
Impaired functional capacity, oxygen supplementation, and dyspnoea negatively affect the quality of life of IPF patients [27]. Unfortunately, quality of life data was not available in this study, as the 6 min walking test results were lacking for most of the patients, and the reported diffusion capacity results are more guiding in nature. Still, 13% of patients with FVC>90% received oxygen supplementation, supporting the notion that poor diffusion capacity may reduce quality of life already with mild disease. Whether they represent a sub-phenotype of IPF needs further research.
Mortality
A competing risk between FVC decline and mortality was observed. Notably, 14% of patients with baseline FVC>90% died before FVC<90% predicted was registered. Similarly, in the other baseline FVC categories one third of patients died before a change to the next category could be seen. Likely these patients represent rapid progressors [28]. Those lacking FVC data had the shortest survival, possibly explained by inability due to advanced disease, or incapacity due to morbidity, to perform spirometry. In addition to FVC, low BMI and OCS use were associated with poor survival. However, as exacerbations could not be evaluated, the context of OCS could not be assessed.
Median overall survival of IPF patients has been assessed in different studies with different methodological criteria to be 2–4 years [25,28,29] and 5-year survival to be 30% [30]. In this study the median overall survival for the whole cohort was 5.0 years, with 6.7 years for those with baseline FVC>90% predicted and decreasing by deteriorating lung function to 0.7 years for patients with FVC<50% predicted. In another study with differing FVC categories, mild and moderate-severe baseline FVC showed a similar trend in life-expectancy [20]. Importantly, we show a 9-fold increased mortality risk in patients with FVC <50% compared to FVC>90% predicted. Further, a 1% FVC drop was associated with a 4% increase in mortality, increasing mortality risk exponentially for each %-decline. The importance of these results is emphasized by the fact that prior survival data by baseline FVC categories are meagre.
Healthcare resource utilization
IPF progression, comorbidities and lung transplantations are key events affecting healthcare costs through hospital stays, visits and medication costs [23]. This is further supported by this study, where HCRU was mainly driven by hospital inpatient days. IPF patients presented an average of 5.5 hospital inpatients days per patient-year, in line with the previously reported 3.1–5.3 inpatient days per patient-year [19,24]. This study also showed, for the first time, an increase in hospital inpatient days by deteriorating FVC group compared to FVC>90% predicted. Correspondingly, IPF patients had on average 10.9 outpatient visits per patient-year, increasing 1.1-fold at FVC 50-90% predicted compared to FVC>90%, and 1.2-fold for those with FVC<50%. This is in line with other claims based studies reporting outpatient visits in the range of 5.7–28 per patient-year, however these studies did not assess HCRU at different disease stages [19,24,31].
Overall, HCRU costs were 5827€ per patient-year, which is a conservative estimate counting only direct specialty care related costs. Costs and mortality are substantially affected by exacerbations [21,23,32] but they could not be assessed in this study. Especially the prevalence of exacerbations at different disease severities, and their impact on HCRU and mortality will need further studies.
The strengths of the study include data collection in routine care that is not affected by strict inclusion or exclusion criteria. Further, text-mining tool utilization allowed for the exclusion of non-IPF patients from the cohort. The study period of 13 years led to a reliable assessment of mortality data.
Limitations are that as data was collected from a specific geographical region results are possibly affected by treatment praxis. However, big geographical differences are not expected, as the natural disease progression is described. Other limitations include those associated with real world data where all variables are not available for all patients. Further, incidence and prevalence were not investigated; however, this data could contribute to discussions on whether increasing IPF incidence is real or resulting from different methodologies and heterogenous patient populations [33].
Conclusions
This study raises several considerations for the care of IPF patients in the future. First, there is still a need to improve IPF diagnosis and enhance the collaboration between primary and specialty care to detect IPF at an early stage, as no improvement in FVC at diagnosis was observed during the study period. Furthermore, FVC alone may not always be an adequate measure of disease severity, rather efforts should be made to assess the quality of life in these patients utilizing other clinical indicators. As both the economic and the humanistic burdens of disease worsen with advancing disease, it is of outmost importance to postpone FVC decline in order to increase the IPF patients’ quality of life.
Acknowledgments
Juha Varjonen at Auria Clinical Informatics at the Turku University Hospital is acknowledged for his input on data extraction. Kirsi Tolonen at Boehringer Ingelheim is acknowledged for her comments during protocol development. Jaana Ahlamaa at Medaffcon is acknowledged for writing assistance.
Biographies
Mariann I Lassenius, PhD, is currently the Real World Evidence Lead and Senior Scientific Advisor at the healthcare consultancy company Medaffcon Oy. Before this she earned her doctoral degree within the field of diabetes research at the University of Helsinki, Department of Nephrology and at the Folkhälsan Research Institute, Helsinki, Finland.
Iiro Toppila, M.Sc. (Tech.), is currently Data Analysis Lead and biostatistician at the healthcare consultancy company Medaffcon Oy. He earned his Master’s degree from Aalto University, Finland, with Bioinformatics and Computational Systems Biology as his major.
Nora Pöntynen, PhD, is currently Medical Manager at Boehringer Ingelheim Finland Ky. Before that she held several positions in the academia, worked with IPR and in the pharmaceutical industry. She earned her MSc in cell biology from Åbo Akademi University, Finland, did a Maîtrise de Biologie Cellulaire et Physiologie at the Université de Reims Champagne-Ardennes, France and her doctoral degree focused on a rare monogenic autoimmune disease at the Department of Medical Genetics, University of Helsinki, Finland.
Laura Kasslin, M.Sc. (Pharm.), is currently Head of Market Access and External Affairs at Boehringer-Ingelheim Finland Ky. She earned her Master’s degree from the University of Helsinki, Finland, with Pharmacology as her major.
Jaana Kaunisto, MD is working as a pulmonologist at Turku University Hospital (Finland). Additionally, she is a PhD student and preparing a dissertation on the epidemiology and management of idiopathic pulmonary fibrosis.
Maritta Kilpeläinen, MD, PhD is currently the Head of the Department of Pulmonary Medicine at Turku University Hospital and special advisor in allergology. Earlier she has worked as a consultant in pulmonary diseases an allergology at the Turku University Hospital and as senior lecturer in at the University of Turku, Finland.
Tarja Laitinen, MD PhD is currently the Director of Research at the Tampere University Hospital. Before that she held several positions in the academia as well in the biotech industry: professorships in Respiratory Diseases, Biobank Research, and Clinical Informatics at the University of Turku and she has been the head of the Department of Pulmonary Diseases at the University Hospital of Turku. She has been the CSO and co-founder of the BioHealthCareInformatics Company Geneos Ltd. Her post-doctoral education was done in the Centre for Genome Research, MIT, USA.
Appendices.
Table A1.
Text patterns used to select the IPF patient cohort form the initial cohort of 993 patients with J84.1 or J84.9 diagnoses
| Image text statements | Pro IPF: honeycomb, UIP, IPF, idiopathic pulmonary fibrosis, traction bronchiectasis, reticular opacities Against IPF: NSIP-, COP-, LIP, alveolitis, asbestosis, stone dust lung, silicosis, eosinophilic pneumonia, hypersensitivity pneumonia, emphysema |
|---|---|
| Pathology statement text of lung biopsy |
Pro IPF: fibroblast focus, honeycomb, UIP, IPF, idiopathic pulmonary fibrosis Against IPF: unspecific/granulomatous inflammation, eosinophilia, NSIP, COP, LIP, DIP, bronchiolitis, hypersensitivity pneumonia |
| Clinical diagnosis from lung view of EMR |
Pro IPF: honeycomb, UIP, IPF, idiopathic pulmonary fibrosis, progressive restrictive lung disease/fibrosis, auscultation inspiratory rasping sound of small alveoli, no autoimmune antibodies, ANCA antibodies not elevated, ANA antibodies not elevated, ENA antibodies not elevated, rheumatoid factor not elevated, citrulline antibodies not elevated, DNA antibodies not elevated, ssDNA antibodies not elevated, SS-A antibodies not elevated, SS-B antibodies not elevated, anti SM not elevated, nucleus antibodies not elevated, MPO antibodies not elevated Against IPF: Rheumatoid arthritis, SLE, MCTD, Sjögren syndrome, scleroderma, myositis, autoimmune fibrosis, fibroelastosis, bronchiolitis, NSIP, COP, LIP, DIP, asbestosis, significant asbestos exposure, hypersensitivity pneumonia, radiation pneumonitis, condition after radiation, vasculitis, eosinophilic pneumonia, autoimmune antibodies elevated, ANCA antibodies elevated, ANA antibodies elevated, ENA antibodies elevated, rheumatoid factor elevated, citrulline antibodies elevated, DNA antibodies elevated, ssDNA antibodies elevated, SS-A antibodies elevated, SS-B antibodies elevated, anti SM elevated, nucleus antibodies elevated, MPO antibodies elevated |
All text patterns were searched in Finnish (as the patient texts are written in Finnish), including plausible different writing forms, most common typing errors and different ways of specifying negative phrases. The performance of text mining cohort formation was assessed by manual validation of randomly selected patients included and excluded from the cohort, and by assessing use of IPF specific antifibrotic medications (pirferidone or nintedanib), see material and methods.
ߓ.
Table A2.
Comorbidities in IPF patients at end of follow-up and their frequency in the patients at baseline
| ICD10 | End of follow-up | Baseline | Description |
|---|---|---|---|
| J84 | 100% | 100% | Other interstitial pulmonary diseases |
| R91 | 59% | 40% | Abnormal findings on diagnostic imaging of lung |
| I10 | 48% | 33% | Essential (primary) hypertension |
| J18 | 41% | 16% | Pneumonia, organism unspecified |
| R06 | 35% | 27% | Abnormalities of breathing |
| H25 | 33% | 17% | Senile cataract |
| Z01 | 29% | 21% | Other special examinations and investigations of persons without complaint or reported diagnosis |
| I25 | 27% | 19% | Chronic ischaemic heart disease |
| E11 | 25% | 18% | Non-insulin-dependent diabetes mellitus |
| Z03 | 23% | 11% | Medical observation and evaluation for suspected diseases and conditions |
| E78 | 22% | 14% | Disorders of lipoprotein metabolism and other lipidaemias |
| I48 | 21% | 12% | Atrial fibrillation and flutter |
| I50 | 20% | 6% | Heart failure |
| R05 | 18% | 15% | Cough |
| G47 | 17% | 11% | Sleep disorders |
| H90 | 15% | 11% | Conductive and sensorineural hearing loss |
| N40 | 15% | 11% | Hyperplasia of prostate |
| J44 | 15% | 8% | Other chronic obstructive pulmonary disease |
| J92 | 15% | 8% | Pleural plaque |
| M17 | 14% | 13% | Gonarthrosis [arthrosis of knee] |
| Z71 | 14% | 8% | Persons encountering health services for other counselling and medical advice, not elsewhere classified |
| R10 | 14% | 8% | Abdominal and pelvic pain |
| M16 | 12% | 10% | Coxarthrosis [arthrosis of hip] |
| R07 | 12% | 7% | Pain in throat and chest |
| I20 | 12% | 8% | Angina pectoris |
| J96 | 12% | 2 % | Respiratory failure, not elsewhere classified |
| K21 | 12% | 7% | Gastro-oesophageal reflux disease |
| Z95 | 11% | 7% | Presence of cardiac and vascular implants and grafts |
| I21 | 11% | 4 % | Acute myocardial infarction |
| C44 | 11% | 8% | Other malignant neoplasms of skin |
| E03 | 11% | 8% | Other hypothyroidism |
| J45 | 11% | 8% | Asthma |
| L57 | 10% | 6% | Skin changes due to chronic exposure to nonionizing radiation |
| M54 | 10% | 5 % | Dorsalgia |
| Z76 | 10% | 6% | Persons encountering health services in other circumstances |
ߓ.
Table A3.
Cox regression of overall mortality, with baseline FVC as time-varying class variable (multivariable 1) or continuous variable (multivariable 2)
| Univariable |
Multivariable (1) |
Multivariable (2) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-value | HR | 95% CI | p-value | HR | 95% CI | p-value | ||
| FVC (continuous, time-varying) | 0.96 | 0.95–0.98 | <0.001 | 0.96 | 0.95–0.97 | <0.001 | ||||
| FVC class (time-varying) | >90% | 1 | ref. | – | 1 | ref. | – | |||
| 50-90% | 1.77 | 0.87–3.61 | 0.114 | 1.98 | 0.91–4.28 | 0.084 | ||||
| <50% | 5.58 | 2.64–11.81 | <0.001 | 8.75 | 3.79–20.18 | <0.001 | ||||
| Transplanted | 2.03 | 0.25–16.81 | 0.511 | NA* | NA* | NA* | ||||
| Unknown | 17.36 | 7.47–40.32 | <0.001 | 15.02 | 4.94–45.71 | <0.001 | ||||
| Age | 1.04 | 1.02–1.07 | <0.001 | 1.04 | 1.01–1.07 | 0.013 | 1.04 | 1.01–1.07 | 0.018 | |
| Sex | Male | 1 | ref. | – | ref. | – | – | ref. | 1 | |
| Female | 0.59 | 0.39–0.88 | 0.010 | 0.72 | 0.43–1.20 | 0.209 | 0.82 | 0.48–1.40 | 0.466 | |
| BMI | <20 | 3.06 | 1.23–7.60 | 0.016 | 4.38 | 1.64–11.74 | 0.003 | 2.92 | 1.02–8.31 | 0.045 |
| 20-25 | 1 | ref. | – | 1 | ref. | – | ref. | 1 | ||
| 25-30 | 0.95 | 0.56–1.60 | 0.833 | 1.16 | 0.67–2.01 | 0.596 | 1.19 | 0.66–2.16 | 0.561 | |
| ≥30 | 0.84 | 0.48–1.46 | 0.529 | 0.86 | 0.48–1.55 | 0.617 | 0.95 | 0.51–1.77 | 0.868 | |
| Smoking | Non-smoker | 1 | ref. | – | 1 | ref. | – | ref. | 1 | |
| former | 1.41 | 0.92–2.16 | 0.114 | 1.44 | 0.83–2.52 | 0.195 | 1.24 | 0.70–2.20 | 0.463 | |
| Current | 0.58 | 0.33–1.03 | 0.063 | 0.79 | 0.39–1.58 | 0.500 | 0.75 | 0.38–1.49 | 0.412 | |
| Missing | 0.69 | 0.34–1.42 | 0.319 | 1.18 | 0.80–4.15 | 0.156 | 1.33 | 0.57–3.12 | 0.507 | |
| Charlson index | 1.11 | 0.97–1.28 | 0.127 | 0.98 | 0.82–1.16 | 0.785 | 1.07 | 0.9–1.28 | 0.452 | |
| BL OCS | No | 1 | ref. | – | 1 | ref. | – | ref. | 1 | |
| Yes | 2.03 | 1.38–3.00 | <0.001 | 1.61 | 1.03–2.49 | 0.035 | 1.55 | 0.98–2.45 | 0.062 | |
NA* patients with lung transplant dropped due to missing covariate values, BL- baseline, OCS-oral corticosteroids.
ߓ.
Table A4.
Overall healthcare resource utilization related costs per FVC or lung transplant categories
| HCRU TYPE | CLASS | EVENTS, N | COST, € | EVENTS PER PATIENT YEAR | COST PER PATIENT YEAR, € |
|---|---|---|---|---|---|
| OUTPATIENT VISITS | ALL | 8,817 | 885,219 | 10.91 | 1,095.53 |
| FVC >90% | 1,380 | 137,768 | 9.49 | 947.43 | |
| FVC 50-90% | 5,567 | 557,783 | 10.42 | 1,043.94 | |
| FVC <50% | 1,218 | 124,907 | 11.52 | 1,181.68 | |
| Transplant | 538 | 53,257 | 50.78 | 5,026.36 | |
| Unknown FVC | 114 | 11,504 | 9.49 | 957.57 | |
| ER VISITS | ALL | 804 | 156,780 | 1.00 | 194.03 |
| FVC >90% | 105 | 20,475 | 0.72 | 140.81 | |
| FVC 50-90% | 482 | 93,990 | 0.90 | 175.91 | |
| FVC <50% | 156 | 30,420 | 1.48 | 287.79 | |
| Transplant | 20 | 3,900 | 1.89 | 368.08 | |
| Unknown FVC | 41 | 7,995 | 3.41 | 665.49 | |
| HOSPITAL INPATIENT DAYS | ALL | 4,451 | 1,755,334 | 5.51 | 2,172.36 |
| FVC >90% | 528 | 198,155 | 3.63 | 1,362.71 | |
| FVC 50-90% | 2,363 | 887,994 | 4.42 | 1,661.96 | |
| FVC <50% | 1,084 | 469,430 | 10.26 | 4,441.03 | |
| Transplant | 124 | 48,637 | 11.70 | 4,590.34 | |
| Unknown FVC | 352 | 151,118 | 29.30 | 12,578.79 | |
| PROCEDURES | ALL | 2,638 | 844,370 | 3.26 | 1,044.97 |
| FVC >90% | 418 | 124,346 | 2.87 | 855.12 | |
| FVC 50-90% | 1,605 | 517,056 | 3.00 | 967.72 | |
| FVC <50% | 464 | 162,389 | 4.39 | 1,536.28 | |
| Transplant | 73 | 18,311 | 6.89 | 1,728.23 | |
| Unknown FVC | 78 | 22,267 | 6.49 | 1,853.50 | |
| OPERATIONS | ALL | 266 | 369,096 | 0.33 | 456.78 |
| FVC >90% | 38 | 49,381 | 0.26 | 339.59 | |
| FVC 50-90% | 169 | 245,081 | 0.32 | 458.69 | |
| FVC <50% | 50 | 64,719 | 0.47 | 612.27 | |
| Transplant | 3 | 4,150 | 0.28 | 391.68 | |
| Unknown FVC | 6 | 5,765 | 0.50 | 479.87 | |
| IMAGING ETC | ALL | 4,787 | 445,906 | 5.92 | 551.84 |
| FVC >90% | 620 | 58,040 | 4.26 | 399.14 | |
| FVC 50-90% | 3,036 | 275,254 | 5.68 | 515.16 | |
| FVC <50% | 866 | 80,164 | 8.19 | 758.39 | |
| Transplant | 145 | 18,875 | 13.69 | 1,781.42 | |
| Unknown FVC | 120 | 13,573 | 9.99 | 1,129.80 | |
| LABS | ALL | 117,216 | 251,783 | 145.06 | 311.60 |
| FVC >90% | 14,440 | 27,268 | 99.30 | 187.52 | |
| FVC 50-90% | 63,966 | 130,236 | 119.72 | 243.75 | |
| FVC <50% | 26,138 | 61,400 | 247.28 | 580.87 | |
| Transplant | 6,852 | 17,487 | 646.69 | 1,650.44 | |
| Unknown FVC | 5,820 | 15,392 | 484.45 | 1,281.19 | |
| TOTAL COSTS | ALL | – | 4,708,488 | – | 5,827.12 |
| FVC >90% | – | 615,433 | – | 4,232.32 | |
| FVC 50-90% | – | 2,707,395 | – | 5,067.13 | |
| FVC <50% | – | 993,429 | – | 9,398.31 | |
| Transplant | – | 164,617 | – | 15,536.55* | |
| Unknown FVC | – | 227,614 | – | 18,946.20 |
*including post-transplant care only.
Funding Statement
This study was funded by Boehringer Ingelheim.
Declaration of interest statement
MIL and IT are employees of Medaffcon Oy. NP and LK are employees of Boehringer Ingelheim Finland. JK has participated in congresses with support from Boehringer Ingelheim and Sanofi Genzyme. MK has nothing to declare. TL has served as a scientific advisory board member and participated in congresses with support from GSK, Boehringer Ingelheim, AstraZeneca, Teva, Roche and MSD.
References
- [1].Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Richeldi L, Collard HR, Jones MG.. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. [DOI] [PubMed] [Google Scholar]
- [3].Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment . International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161:19. [DOI] [PubMed] [Google Scholar]
- [4].Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a fleischner society white paper. Lancet Respir Med. 2018;6:138–153. [DOI] [PubMed] [Google Scholar]
- [5].Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. [DOI] [PubMed] [Google Scholar]
- [6].Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3–e19. [DOI] [PubMed] [Google Scholar]
- [7].Sköld CM, Bendstrup E, Myllärniemi M, et al. Treatment of idiopathic pulmonary fibrosis: a position paper from a nordic expert group. J Intern Med. 2017;281:149–166. [DOI] [PubMed] [Google Scholar]
- [8].Hodgson U. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 2002;57:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kaunisto J, Kelloniemi K, Sutinen E, et al. Re-evaluation of diagnostic parameters is crucial for obtaining accurate data on idiopathic pulmonary fibrosis. BMC Pulm Med. [Internet] 2015. [cited 2019 January21]; 15. Available from: http://bmcpulmmed.biomedcentral.com/articles/10.1186/s12890-015-0074-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history: IPF: diagnosis, epidemiology, course. Respirology. 2016;21:427–437. [DOI] [PubMed] [Google Scholar]
- [11].Kennedy-Martin T, Curtis S, Faries D, et al. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials. [Internet] 2015. [cited 2019 January15]; 16. Available from: http://trialsjournal.biomedcentral.com/articles/10.1186/s13063-015-1023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. [DOI] [PubMed] [Google Scholar]
- [13].Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Richeldi L, Cottin V, Du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med. 2016;113:74–79. [DOI] [PubMed] [Google Scholar]
- [15].Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. [DOI] [PubMed] [Google Scholar]
- [16].Albera C, Costabel U, Fagan EA, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48:843–851. [DOI] [PubMed] [Google Scholar]
- [17].Kolb M, Richeldi L, Behr J, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].284 Nintedanib och pirfenidon [Internet]. kela.sv [cited 2019. January 22].Available from: https://www.kela.fi/web/sv/lakemedel284.
- [19].Collard HR, Ward AJ, Lanes S, et al. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ. 2012;15:829–835. [DOI] [PubMed] [Google Scholar]
- [20].Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med. [Internet] 2018. [cited 2019 January21]; 18. Available from: https://bmcpulmmed.biomedcentral.com/articles/10.1186/s12890-018-0575-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Song JW, Hong S-B, Lim C-M, et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–363. [DOI] [PubMed] [Google Scholar]
- [22].Tarride J-E, Hopkins RB, Burke N, et al. Clinical and economic burden of idiopathic pulmonary fibrosis in Quebec, Canada. Clin Outcomes Res. 2018;10:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vaidya S, Hibbert CL, Kinter E, et al. Identification of key cost generating events for idiopathic pulmonary fibrosis: a systematic review. Lung. 2017;195:1–8. [DOI] [PubMed] [Google Scholar]
- [24].Wu N, Yu YF, Chuang -C-C, et al. Healthcare resource utilization among patients diagnosed with idiopathic pulmonary fibrosis in the USA. J Med Econ. 2015;18:249–257. [DOI] [PubMed] [Google Scholar]
- [25].Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Purokivi M, Hodgson U, Myllärniemi M, et al. Are physicians in primary health care able to recognize pulmonary fibrosis? Eur Clin Respir J. 2017;4:1290339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Graney BA, Lee JS. Impact of novel antifibrotic therapy on patient outcomes in idiopathic pulmonary fibrosis: patient selection and perspectives. Patient Relat Outcome Meas. 2018;9:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. [DOI] [PubMed] [Google Scholar]
- [29].Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vancheri C, Failla M, Crimi N, et al. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. [DOI] [PubMed] [Google Scholar]
- [31].Raimundo K, Chang E, Broder MS, et al. Clinical and economic burden of idiopathic pulmonary fibrosis: a retrospective cohort study. BMC Pulm Med. [Internet] 2016. [cited 2019 January21]; 16. Available from: http://www.biomedcentral.com/1471-2466/16/2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Teramachi R, Kondoh Y, Kataoka K, et al. Outcomes with newly proposed classification of acute respiratory deterioration in idiopathic pulmonary fibrosis. Respir Med. 2018;143:147–152. [DOI] [PubMed] [Google Scholar]
- [33].Hutchinson J, Fogarty A, Hubbard R, et al. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795–806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 284 Nintedanib och pirfenidon [Internet]. kela.sv [cited 2019. January 22].Available from: https://www.kela.fi/web/sv/lakemedel284.