

Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder caused due to loss of dopaminergic neurons in substantia nigra pars compacta, which occurs the presence of Lewy bodies made up of Alpha-synuclein (ASN) aggregation resulting in neuronal death. This study aims to identify potent 7,8-Dihydroxyflavone (DHF) derivatives to inhibit the ASN aggregation from in silico analysis. Molecular docking study reveals that carbamic ester derivatives of DHF [DHF-BAHPC (8q), DHF-BAHPEC (8s), DHF-BAHEC (8p), DHF-BDOPC (8c), DHF-BAPEC (8n) and DHF-BAMC (8h)] have good binding affinity towards ASN, when compared with DHF and L-DOPA; their docking score values are −16.3120, −16.1875, −15.2223, −14.3118, −14.2893, –14.2810, −14.0383, and −9.1560 kcal/mol respectively. The in silico pharmacological evaluation shows that these molecules exhibit the drug-likeness and ADMET properties. Molecular dynamics simulation confirms the stability of the molecules with ASN. The intermolecular interaction analyzed under the dynamic condition, allows to identify the candidate which potentially inhibits ASN aggregation. Hence, we propose that DHF derivatives are the potential lead drug molecules and preclinical studies are needed to confirm the promising therapeutic ability against PD.
Subject terms: Virtual drug screening, Molecular dynamics
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder characterized by impairment of motor functions due to complete loss of dopamine in the midbrain. Clinically, PD characterized by behavioral impairments such as rigidity, tremor, postural instability and bradykinesia1. Alpha-synuclein (ASN), a PD associated protein, misfolds and accumulates in the brain through protein aggregation. Post-mortem studies showed that these aggregated filamentous termed as a Lewy body, the neurotoxicity is a main factor involved in PD and other disorders such as Alzheimer and Prion disease2. Studies report that the loss of dopaminergic neurons in PD is partly due to the overexpression of ASN in the cytoplasm of neurons. ASN expression increases in the substantia nigra with the response of age in rhesus monkeys and humans3. The unfolded ASN protein does not have a secondary structure under the physiological condition. However, changes in various environmental factors (agitation, ion strength, pH) induces the formation of amyloid-like fibrils and ASN aggregates in vitro4. Comparatively, ASN has more cytotoxicity than the amyloid proteins, which generate amyloid-like fibrils5. To prevent the aggregation of protein and resultant proteotoxicity, therapeutic medicines could provide a significant neuroprotective effect against ASN aggregation6,7. However, these therapeutic medicines to inhibit the amyloid fibril formation, especially L-DOPA enhance the anti-cytotoxicity8. Bodner et al. reported that B2 (5-[4-(4-chlorobenzoyl)-1-piperazinyl]-8-nitro-quinoline) compound accelerates inclusion formation in PD and Huntington’s disease9. The plant extracts and phytoconstituents targeting ASN aggregation, oligomerization and fibrillation to reduce ASN toxicity in PD model10. The in vitro analysis states that the hydroxyl group moiety and nitrogen containing groups are important to inhibit the aggregation of ASN in different stages (oligomerization, fibrillation and aggregation). In addition, the binding of phytochemical molecules stabilizes the intrinsic structure of ASN. Similarly, in the present study, the DHF derivatives have hydroxyl and amine group; it may prevent the aggregation of ASN. Also, the monomer is the primary stage that leads to developing end stages of oligomeric and fibrils11,12, but the ASN aggregation is a major component of Lewy bodies. Furthermore, the folding of a monomer is very low compared with dimer and oligomeric forms. However, the inhibition of monomer prevents the aggregation of ASN. Numerous laboratory reports have been published in recent years concerning ASN related mechanisms that may be responsible for the observed neurodegeneration in PD. The binding region of ASN identified in the specific amino acid region (64–100), where the region is responsible for its self-aggregation13. The reports state that the hydrophobic cluster formed by NAC region (85–95) and C terminus (110–130) residues. These regions are also crucial for the aggregation mechanism of ASN14. Currently, there are no therapeutic agents to prevent the formation of protein aggregation thus it is a main concern in this PD research field.
Xiao-Huan Li et al. reported a biological evaluation of DHF molecule that suppressed ASN expression and oxidative stress against MPTP induced Parkinson mice model15. The 7,8-Dihydroxyflavone (DHF) is a member of the flavonoid family, and highly present in vegetables and fruits. The metabolite forms of flavonoids are promoted to enhance memory and knowledge through their interfaces with neuronal signal pathways, which is essential for controlling long-term potentiation and memory in human subjects16. Thus reducing chances of RNS and ROS protect from neurodegenerative disorder including PD’ as oxidative stress plays a role in degeneration of neurons in PD17–19. In recent studies, DHF is thought to be a promising therapeutic agent for various neurodegenerative diseases20–23. However, only a few of the synthesized molecules have shown potent biological activity24,25. The 9-Fluorenylmethoxycarbonyl (Fmoc) is protecting group used in experimental peptide synthesis to keep the amino group for the further chemical reaction26,27. The fluorenyl ring of Fmoc forms the hydrophobic and π-π stacking interactions with reactive molecules due to its hydrophobicity and aromaticity. Moreover, the physicochemical properties of Fmoc-modified or conjugated amino acids and short peptides is significantly varied and it is used in the various applications including antibiotics, catalysis, therapeutic, drug delivery, cell cultivation, optical devices and templating28–31. Considering the potential pharmaceutical applications, the DHF derivatives would produce value added product and inhibit the ASN aggregation. Therefore, the present study aims to design novel potent DHF derivatives, which are linked with Fmoc-amino acids and find out the potential neuroprotective agents against human ASN using molecular docking and molecular dynamics simulations.
Results and Discussion
Designed DHF derivatives (Ligands)
The DHF has been linked with Fmoc-amino acid to produce amino acid ester of DHF [1 to 4 (4a-4t)] and carbamate esters of DHF [1, 2, 5–8 (8a-8t)] (Fig. 1 and Supplementary Table S1). Both the phenolic OH groups of DHF reacted with an in-situ generated Fmoc-amino acids and isocyanate followed by the cleavage of Fmoc-unit to afford the target conformed. The DHF hydrogen atoms of hydroxyl groups substituted with 20 different Fmoc-amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine) to produce DHF derivatives.
Figure 1.

The DHF derivatives designed (A) Amino acid esters and (B) Carbamate esters.
Molecular docking
Molecular docking (MD) plays a vital role in the field of computer-aided drug design. This study helps to identify the small molecules by docking towards the binding site of the protein. The ProToss analysis (build in the program) helps to predict the hydrogen bond network in the active site also could increase the number of hydrogen bonds, which hikes the scoring function32. Here, LeadIT suite was used for FlexX scoring function to identify the first best 200 poses based on the scoring function. To evaluate the final poses, the scoring function used HYDE function33. The primary purpose of the study is to find out the inhibition of a specific protein based on their score value34. For ASN protein, the newly designed ligands were virtually screened along with DHF and L-DOPA; and their binding energies were calculated. The designed structures and their score values are given in Supplementary Fig. S1. The molecules were chosen for further investigation based on the binding energy values. The intermolecular interactions between the ASN protein and ligands (8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA) are shown in Fig. 2. Moreover, the conventional hydrogen bonds, van der Waals and carbon-hydrogen bonds played a significant role to maintain the stability of the complexes. The Lys97 formed Pi-cation and Pi-lone pair interactions with all molecules rather than DHF; the DHF forms two hydrogen bonds with the Lys97. The L-DOPA forms one hydrogen bond and one salt bridge with Lys97. The 8c and L-DOPA compounds form a salt bridge bond with Asp98. Similarly, the amino acid Leu100 forms Pi-alkyl interaction with the ligands rather than 8n and L-DOPA; these two compounds altered their interactions resulting in a strong hydrogen bond. Generally, the Val95, Gly93 and Gln99 residues form hydrogen bonding interactions with all the above said ligands, but DHF does not have hydrogen bonding interaction with Val95 and Gly93 residues. The 8c form unfavorable donor-acceptor interaction with Val95 and Gly93; these hydrogen bonds were diminished in L-DOPA; apart from this, each molecule solidly have two hydrogen bonds with the same residue. The 8q showed more binding capacity than other molecules. The best pose for each molecule was taken to analyze the intermolecular interactions. In the present study, 8q showed high binding energy (−16.312 kcal/mol) and also interacts with Gln99, Ala90, Gly93, Ser87, Val95, Gly93, Lys96, Leu100 and Lys97. The 8s (−16.188 kcal/mol) molecule interacts with Lys97, Lys96, Leu100, Gln99, Gly93, Ser87, Val95, Ala90 and Ala91. Similarly, the 8p (−15.222 kcal/mol) interacts with Gln99, Ala90, Gly93, Ser87, Val95, Lys97, Lys96 and Leu100. The 8c (−14.312 kcal/mol) molecule interacts with the residues Gln99, Asp98, Val95, Phe94, Ala90, Ala91, Gly93, Lys97, Phe94 and Leu100. Further, the 8n (−14.289 kcal/mol) interacts with Lys97, Gln99, Leu100, Val95, Gly93, Lys96, Ala90 and Ala91. The 8h (−14.281 kcal/mol) interacts with Ser87, Gln99, Ala90, Val95, Gly93, Lys97 and Leu100. DHF (−14.038 kcal/mol) interacts with Gln99, Lys97, Phe94 and Leu100. The L-DOPA (−9.156 kcal/mol) interacts with Asp98, Phe94, Gln99, Leu100, Val95, Phe94 and Lys97 of ASN. All these molecules form more than 5 hydrogen bonding interactions with the active site amino acid residues. All the molecules showed the highest negative LeadIT score, which reveals that the compounds are capable of binding with ASN to inhibit the fibrillation. Previous reports state that NAC-region (65–100) of ASN is the primary target to inhibit the aggregation process6. Moreover, our previous study demonstrated that CNB-001 (Docking score: −13.6158 kcal/mol) showed potent inhibitory effect against ASN followed by, DHF (−13.0499 kcal/mol), Curcumin (−12.0386 kcal/mol), Naringenin (−11.1311 kcal/mol) and emodin (−8.8539 kcal/mol). In silico and animal studies showed that CNB-001 diminished the expression of ASN against MPTP induced Parkinson model35. Interestingly, our reports revealed that DHF derivative molecules (8q, 8s, 8p, 8c, 8n and 8h) showed better results when compared with DHF and L-DOPA; their docking scores values are listed in Supplementary Table S3 (−16.3120, −16.1875, −15.2223, −14.3118, −14.2893, −14.2810, −14.0383 and −9.1560 kcal/mol). The interaction distances of each molecule with their respective active site amino acid residues are shown in Supplementary Table S2.
Figure 2.

Intermolecular interactions between DHF derivatives with ASN protein, (a) 3D and (b) 2D view of 8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA intermolecular interaction with ASN.
ADMET investigation
The ADMET profile was found to play a vital role to understand the pharmacokinetic properties of the ligand molecules for therapeutic intervention. In this analysis, we revealed that the drug likenesses of selected compounds studied by calculating the ADMET properties using “TOPKAT” module of DS 3.5. The compounds (8q, 8s, 8p, 8c, 8n, and 8h) were chosen based on Veber’s rule36, which satisfies the molecular weight (~500 g/mol) of all drugs. The compounds were investigated using Lipinski’s rule of five37; in which, the H-bond donors and acceptors were counted. The counting of H-bond is based on the electronegative atoms present in the drug molecules. All the compounds have ~6 donors and ~8 acceptors, and the ratio of compound concentration in a mixture of two immiscible phases of partition-coefficient (logP). The logP values of the ligand molecules were an acceptable threshold (<5) level (Supplementary Table S3). The AQ SOL LEV prediction tool was used to investigate the biological transportation functions of the drug molecules through a water-based solvent, the values are lies between 2 to 3 except L-DOPA (5).
Further, BBB LEV analysis reveals the information about drugs passing through the blood-brain barrier and their values were calculated based on logP, AQ SOL LEV and lipophilic characters. The BBB LEV value of the molecules are high (4) on compared with DHF (2). The critical parameters, such as Plasma Protein Binding levels (PPB LEV) and Hepatotoxicity (HEPATOX) with CYP2D6 scores were calculated from ADMET properties. The values of CYP2D6 scores were found to be less than 0, which shows that the molecules are a non-inhibitory function to the Cytochrome P450 2D6 enzyme; whereas CYP2D6 ≥ 1 shows that they are inhibitory. Calculated PPB LEV provides the detail of drug concentration and an insight into the plasma protein. PPB LEV with high values paves the way for the adsorption of the drug in the renal passage and prevents PD. All the compounds exhibited an excellent binding capacity and diffuse through the membrane, which also confirms the high levels of PPB. The hepatotoxicity levels were predicted, to study the molecular toxicity of the organ. When the HEPATOX value is 1, then the molecules could be highly toxic; on the other hand, if the value is 0, then they are non-toxic. The HEPATOX value of the molecule is < 0 shows that they are non-toxic. In the present study, NTP carcinogenicity, mutagenicity and developmental toxicity were performed to predict the toxicity profiles of the molecules. The skin irritation studies include a topical application for skin or mucous membranes. TOPKAT features is a patented algorithm (US Patent 6,036,349, issued March 14, 2000), which decides whether the compound lies within the optimum prediction space (OPS) for toxicity analysis and skin irritation studies. If the range of OPS is lies between 0 to 0.29, the compound belongs to the non-toxic group. Consequently, the range is between 0.3 to 0.69 they are indeterminate and the score lies between 0.7 to 1, the molecule is considered to be highly toxic. The OPS score (1.0) of 8c is high; which indicates that it is highly toxic for the skin. On the other hand, the molecules 8q, 8s, 8p, 8n, 8h, DHF and L-DOPA exhibits low OPS score (0.0, 0.0, 0.247, 0.0, 0.0, 0.0 and 0.303). Further, the AMES mutagenicity prediction analysis has been carried out, it shows that all the molecules are non-mutagenic except 8 h.
Molecular dynamics
The molecular dynamics (MD) simulation was performed to find out the stability, confirmation and intermolecular interaction of the ligand molecules with ASN protein. The time-dependent modification of the complexes was calculated over 50 ns using Desmond package. The MD simulation was performed under the thermodynamical conditions (applied volume, density, pressure and temperature). The complete system was annealed and equilibrates using ensembles. Moreover, the final production step performed to investigates the structural modification of the complex. Further, the trajectories of each complex subjected to specific parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein secondary structure element (SSE), conformational modification of ligands and intermolecular interactions to analyze the level of structural changes.
RMSD and RMSF
The backbone deviation (N, Cα, C) of protein was calculated from the RMSD value during the MD simulation. The complex structures were highly fluctuated (1 to 27 Å) up to equilibration. After equilibration (5 ns), the system gets stabilized, and this trend continued up to 50 ns. After 5 ns, the RMSD of the complexes varied ~1 to 4 Å. Notably, the RMSD of DHF and its derivatives are low on compared with L-DOPA. In which, the RMSD of 8q, 8p, 8c, 8n, 8h and DHF is not much varied except 8s. It indicates that the molecules were highly stable during the MD simulation (Fig. 3). The RMSD analyzes confirm that the DHF derived molecules 8q, 8p, 8c, 8n, 8h and DHF showed structural stability during the MD simulation.
Figure 3.
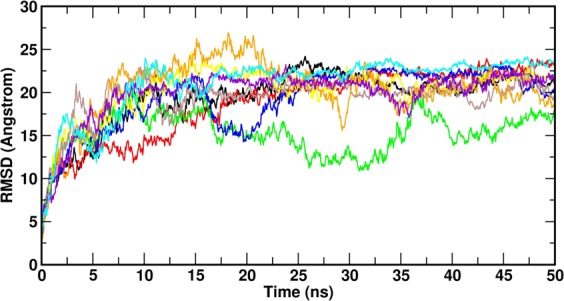
The RMSD plot for all complexes during the MD simulation. (8q–red, 8s–green, 8p–blue, 8c–yellow, 8n–brown, 8h–orange, DHF–meganta, L-DOPA–cyan and ASN alone–black).
Further, the flexibility of the complexes was analyzed, while the ligands present in the active site of the protein. The RMSF was used to investigate the fluctuation of the complexes in the function of time. The N-terminal (~25 Å) has high fluctuation compared with C terminal (~22 Å). The DHF derivative molecules were highly stable in the catalytic region except for 8n, due to the weak intermolecular interaction with the protein (Fig. 4). The complexes exhibit low fluctuation in NAC region (64–100); it is due to the intermolecular interactions of the ligand molecules with ASN. Moreover, the intermolecular interactions and secondary structure elements (alpha helices and beta strands) make the protein molecule is slightly rigid.
Figure 4.
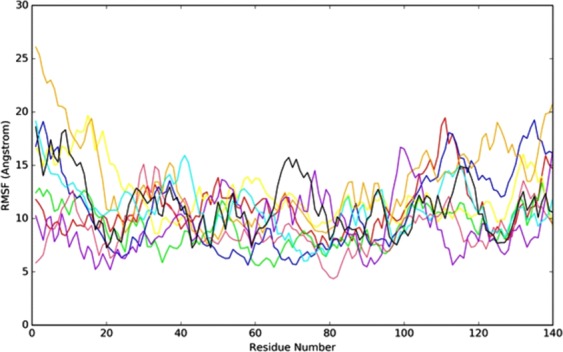
The RMSF plot for all complexes during the MD simulation. (8q–red, 8s–green, 8p–blue, 8c–yellow, 8n–brown, 8h–orange, DHF–meganta, L-DOPA–cyan and ASN alone–black).
Protein secondary structure elements (SSE)
The SSE (alpha-helices and beta-strands) monitored throughout the simulation process. Supplementary Figs. S2 and S3 describe the SSE distribution and SSE composition analysis of the respective residue index of the ASN. Except 8s, 8h, and L-DOPA complexes, other complexes have beta-strands; those complexes were highly helical nature. SSE analyzes of ASN confirms that, the secondary structure is not presented in the C-terminal region (100–140 residues). The torsion angle potential plot exhibits the relation between the torsion angle present in the ligands and their corresponding potential energy. The information of torsion angle is necessary to predict the rotatable bonds of ligand molecule (Supplementary Fig. S4). Here, 8s and 8c each contain 15 rotatable bonds; whereas, 8q, 8p, 8n, 8h, DHF and L-DOPA contain 13, 13, 13, 9, 3 and 5 rotatable bonds respectively. The histogram and torsion potential relationships give an insight into the conformational strain of the ligands, which is used to understand the protein-bound ligand conformation. The ligand modification showed in Supplementary Fig. S5 includes RMSD, radius of gyration, intra-molecular hydrogen bonds, molecular surface area (MolSA), solvent accessible surface area (SASA) and polar surface area (PSA).
Intermolecular interaction
The atomic level information is essential to predict the binding mode of 8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA in the binding site of ASN protein. For binding mode analysis, the intermolecular interactions such as hydrogen bond, hydrophobic contact, ionic interaction and salt bridge were analyzed over 50 ns MD simulation studies. The report states that the hydrophobic cluster formed by NAC region (85–95) and C terminus (110–130) residues. These regions are also crucial for the aggregation mechanism of ASN14. The present study also confirms that the DHF derivatives form strong intermolecular interactions with both NAC (65–100) and C-terminal (101–140) residues. The 8q forms strong ionic interactions with binding site residues of Glu105. Along with this, the 8q forms hydrophobic interactions with Leu100 and Val118 by making Pi- Pi stacking wall, also one polar interaction with Asn103. The 8s form one hydrophobic interaction with Phe94. The 8p forms two ionic contacts (Glu28, Glu104) and one water mediated bridge bond (Glu105). Similarly, the 8c forms ionic contact (Glu104, Glu105), polar (Gly93) and charged interaction (Lys97) with the active site residues. Likewise, the 8n forms hydrophobic (Leu100) and three charged interactions (Glu83, Lys97 and Glu104). Further 8h and DHF compounds display very less accountable interactions. Here, 8h has only one charged interaction (Glu61) and DHF forms two hydrophobic interactions (Phe94 and Ile112). Whereas, L-DOPA loss the intermolecular interactions with active site residues and slightly moved away from the active site on compared with all other complexes (Fig. 5 and Supplementary Fig. S6). The reports state that the ligand molecules induce the α-helix formation of ASN38,39. Similarly, in the present study, the 8q and 8s form the possible interactions with NAC and C-terminal of ASN. In which, the 8q molecule induces the folded state of ASN (Supplementary Fig S6), it leads to α-helix formation. This mechanism inhibits the aggregation of ASN. The protein-ligand intermolecular interactions confirm the binding strength and stability of DHF derivatives with the active site amino acid throughout the MD simulations (Fig. 6 and Supplementary Fig. S7).
Figure 5.

2D Intermolecular interactions of 8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA ligands with ASN protein.
Figure 6.

Protein-ligand interactions between ASN with respective compounds 8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA.
Materials and Methods
Computational feature
The in silico analyses were performed using HP workstation Z220 with Next-generation 22 nm processor. The DHF derivative molecules were drawn using ChemDraw Ultra 12 (ChemDraw)40. Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties of DHF derivative molecules were analyzed using Accelery Discovery Studio (DS) 3.541,42. Further, the docking studies were carried out using Biosolve IT (Lead IT) software package43 and Desmond v3.6 Package was used to run the MD simulation. The intermolecular interactions were analyzed using Pymol and Chimera44,45.
Designing of DHF derivatives
The chemical structure of DHF shown in Fig. 7. DHF derivatives designed and their structures drawn using ChemDraw software. The DHF derivatives depicted in two different schemes, each scheme of DHF reacts with 20 different Fmoc amino acids to produce 40 DHF derivatives and it is used in in silico analysis.
Figure 7.
Chemical structure of 7,8-Dihydroxyflavone.
Docking study
Molecular docking studies used to find out the binding affinity of the ligand molecule with protein. The experimental structure of ASN protein retrieved from PDB (PDB ID: 1XQ8) database13. In which, the water molecules were removed, followed by hydrogen atoms, charges, standard bond orders and the missing residues were added using protein preparation wizard in DS 3.5 software46. Minimization was carried out up to 500 steps using smart minimizer methods and the standard dynamic cascade was performed using DS 3.5 simulation protocol47. The ligands and protein were prepared using Ligprep and protein preparation wizard available in the DS 3.5. The refined molecules docked using LeadIT software package, which is based on the FlexX docking approach. The FlexX algorithm was used to generate up to 200 poses for each ligand; the best conformer will be scored in high. The interaction modes between the ligands (DHF derivatives) and protein were studied using Biosolve IT FlexX48.
ADMET properties
The molecules were subjected to ADMET analysis using DS 3.5 protocol to predict the pharmacokinetics and toxicity properties49,50. The ADMET studies provide insight into the pharmacokinetics properties such as Plasma Protein Binding level (PPB LEV), Hepatotoxicity (HEPATOX), CYP 2D6, Blood Brain Barrier Level (BBB LEV) and Aqueous Solubility level (AQ SOL LEV). The toxicity profile of the compounds was predicted using TOPKAT 6.1, which uses a range of robust, cross-validated and Quantitative Structure-Toxicity Relationship (QSTR) models for identifying specific toxicological activity51. Toxicity profiles were tested, including NTP Carcinogenicity Call (Male Mouse) (v3. 2), NTP Carcinogenicity Call (Female Mouse) (v3. 2), Developmental Toxicity Potential (DTP) (v3. 1), Skin Irritation (v6. 1) and Ames Mutagenicity (v3. 1).
Molecular dynamics
The MD simulations were performed using Desmond v3.6 Package to elucidate the fact behind the effectiveness of these compounds against ASN inhibition52,53. The lead compounds such as DHF, L-DOPA and the DHF derivatives with ASN protein were prepared using the OPLS2005 force field54. Further, the pre-defined TIP3P water model was used to build the system, which could act as water molecules and these are constructed in the orthorhombic periodic boundary conditions at the distances of 10 Å units55. Moreover, the charge of the complexes electrically neutralized with balancing Na+/Cl− ions and also the system minimized their energies by heating and equilibrium processes before the MD simulations. The complexes were subjected to the minimization protocol based on the steepest descent method, then heated at 0–300 K with the annealing steps of 2000 and the time steps of 0.001 ps. Further, the system normalized in an equilibrium state at 1000 steps with the time step of 0.001 ps. The final production step of the system continued up to 50 ns, at the time steps of 0.001 ps; 300 K temperature and 1 Atm pressure, applied using Nose-Hoover method56 with NPT ensemble57,58. The best conformations were selected based on the interactions and dynamical properties of the complexes59.
Conclusion
In this study, 40 different DHF derivatives were designed through the amino acid esters and carbamate esters. Among the 40 DHF derived complexes, the molecule 8q exhibited the highest molecular docking score (−16.3120 kcal/mol), indicates that it has high binding towards ASN. The ADMET properties reveal that 8q, 8s, 8p, 8c, 8n, 8h, DHF and L-DOPA compounds are non-toxic. The stability of the ligand-protein complexes were evaluated from the molecular dynamics simulation showed that the ligands 8q, 8s, 8p, 8c, 8n, 8h and DHF with ASN complexes have subtle structural modification throughout the MD simulations. From the binding mode analysis, it is confirmed that the DHF and its derivatives are forming stable interactions with the ASN protein except L-DOPA. The DHF derivatives 8q, 8s, 8p, 8c, 8n and 8h potentially inhibit the ASN when compared with DHF and L-DOPA; among these, presumably, 8q molecule may be the potential candidate to inhibit the ASN aggregation. On the whole, these compounds of novel scaffolds provide valuable leads for further optimization in both in vitro and in vivo as potent inhibitors against ASN to treat PD. In conclusion, these results suggest that the carbamate ester of DHF showed better efficiency than the L-DOPA.
Supplementary information
Acknowledgements
We thank Prof. A.K. Mohanakrishnan, Head, Department of Organic Chemistry, University of Madras, for discussions on synthetic studies.
Author contributions
M.T., V.C., L.H.S. and E.N. designed the research, M.T., V.C., K.P., S.M., H.G. and E.N. performed the in silico study and analyzed the results, M.T., V.C., R.L.J., R.V., B.R., M.D. and E.N. prepared the manuscript. All the authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-57417-9.
References
- 1.Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. PNAS. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu YJ, Kordower H. Age-associated increases of α-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol. Dis. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto M, et al. Human recombinant NACP/α-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain. Res. 1998;799:301–306. doi: 10.1016/S0006-8993(98)00514-9. [DOI] [PubMed] [Google Scholar]
- 5.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 6.El-Agnaf OM, et al. A strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB. J. 2004;18:1315–1317. doi: 10.1096/fj.03-1346fje. [DOI] [PubMed] [Google Scholar]
- 7.Nagai Y, et al. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J. Biol. Chem. 2000;275:10437–10442. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 8.Conway KA, Rochet JC, Bieganski RM, Lansbury PT. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 9.Bodner RA, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. PNAS. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Javed H, et al. Plant Extracts and Phytochemicals Targeting α-Synuclein Aggregation in Parkinson’s Disease Models. Front. Pharmacol. 2018;19:1555. doi: 10.3389/fphar.2018.01555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng, X., Munishkina, L. A., Fink, A. L. & Uversky, V. N. Effects of various flavonoids on the-synuclein fibrillation process. Parkinson’s Disease. 2010 (2010). [DOI] [PMC free article] [PubMed]
- 12.Li X, et al. Early stages of aggregation of engineered α-synuclein monomers and oligomers in solution. Sci. Rep. 2019;9:1734. doi: 10.1038/s41598-018-37584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 14.Bertoncini CW, et al. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. PNAS. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XH, et al. 7, 8‐dihydroxyflavone Ameliorates Motor Deficits Via Suppressing α‐synuclein Expression and Oxidative Stress in the MPTP‐induced Mouse Model of Parkinson’s Disease. CNS. Neurosci. Ther. 2016;22:617–624. doi: 10.1111/cns.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spencer JP. Food for thought: the role of dietary flavonoids in enhancing human memory, learning and neuro-cognitive performance: Symposium on ‘Diet and mental health’. Proc. Nutr. Soc. 2008;67:238–252. doi: 10.1017/S0029665108007088. [DOI] [PubMed] [Google Scholar]
- 17.Pandey RP, Parajuli P, Koffas MA, Sohng JK. Microbial production of natural and non-natural flavonoids: pathway engineering, directed evolution and systems/synthetic biology. Biotechnol. Adv. 2016;34:634–662. doi: 10.1016/j.biotechadv.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Jäger A, Saaby L. Flavonoids and the CNS. Molecules. 2011;16:1471–1485. doi: 10.3390/molecules16021471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertog MG, Feskens EJ, Kromhout D, Hollman PCH, Katan MB. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. 1993;342:1007–1011. doi: 10.1016/0140-6736(93)92876-U. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, et al. 7, 8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2014;39:638. doi: 10.1038/npp.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian M, et al. 7, 8-Dihydroxyflavone induces synapse expression of AMPA GluA1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology. 2015;89:43–53. doi: 10.1016/j.neuropharm.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhao S, Gao X, Dong W, Chen J. The role of 7,8-dihydroxyflavone in preventing dendrite degeneration in cortex after moderate traumatic brain injury. Mol. Neurobiol. 2015;53:1884–1895. doi: 10.1007/s12035-015-9128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.García-Díaz Barriga G, et al. 7, 8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCγ1 pathway. Hum. Mol. Genet. 2017;26:3144–3160. doi: 10.1093/hmg/ddx198. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, et al. A synthetic 7, 8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 2010;3:8274–8286. doi: 10.1021/jm101206p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chitranshi N, Gupta V, Kumar S, Graham SL. Exploring the molecular interactions of 7,8-dihydroxyflavone and its derivatives with TrkB and VEGFR2 proteins. Int. J. Mol. Sci. 2015;16:21087–21108. doi: 10.3390/ijms160921087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972;37:3404–3409. doi: 10.1021/jo00795a005. [DOI] [Google Scholar]
- 27.Carpino LA, Sadat-Aalaee D, Chao HG, DeSelms RH. [(9-Fluorenylmethyl) oxy] carbonyl (FMOC) amino acid fluorides. Convienient new peptide coupling reagents applicable to the FMOC/tert-butyl strategy for solution and solid-phase syntheses. J. Am. Chem. Soc. 1990;112:9651–9652. doi: 10.1021/ja00182a041. [DOI] [Google Scholar]
- 28.Mahler A, Reches M, Rechter M, Cohen S, Gazit E. Rigid, self‐assembled hydrogel composed of a modified aromatic dipeptide. Adv. Mater. 2006;18:1365–1370. doi: 10.1002/adma.200501765. [DOI] [Google Scholar]
- 29.Kuang Y, Gao Y, Shi J, Lin HC, Xu B. Supramolecular hydrogels based on the epitope of potassium ion channels. Chem. Commun. 2011;47:8772–8774. doi: 10.1039/c1cc13115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming S, Ulijn RV. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014;43:8150–8177. doi: 10.1039/C4CS00247D. [DOI] [PubMed] [Google Scholar]
- 31.Tao K, Levin A, Adler-Abramovich L, Gazit E. Fmoc-modified amino acids and short peptides: simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev. 2016;45:3935–3953. doi: 10.1039/C5CS00889A. [DOI] [PubMed] [Google Scholar]
- 32.Lippert T, Rarey M. Fast automated placement of polar hydrogen atoms in protein-ligand complexes. J. Cheminform. 2009;1:13. doi: 10.1186/1758-2946-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reulecke I, Lange G, Albrecht J, Klein R, Rarey M. Towards an integrated description of hydrogen bonding and dehydration: decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem. 2008;3:885–897. doi: 10.1002/cmdc.200700319. [DOI] [PubMed] [Google Scholar]
- 34.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002;6:439–446. doi: 10.1016/S1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 35.Jayaraj RL, Elangovan N. In silico identification of potent inhibitors of alpha-synuclein aggregation and its in vivo evaluation using MPTP induced Parkinson mice model. Biomed. Aging. Pathology. 2014;4:147–152. doi: 10.1016/j.biomag.2014.01.002. [DOI] [Google Scholar]
- 36.Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 37.Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem. Rev. 1971;71:525–616. doi: 10.1021/cr60274a001. [DOI] [Google Scholar]
- 38.Abedini A, Raleigh DP. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 2009;6:015005. doi: 10.1088/1478-3975/6/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharya S, Xu L, Thompson D. Molecular simulations reveal terminal group mediated stabilization of helical conformers in both amyloid-β42 and α-synuclein. ACS Chem. Neurosci. 2019;10:2830–2842. doi: 10.1021/acschemneuro.9b00053. [DOI] [PubMed] [Google Scholar]
- 40.Cousins KR. Computer review of ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011;133:8388–8388. doi: 10.1021/ja204075s. [DOI] [PubMed] [Google Scholar]
- 41.Nikolic K, Agababa D. Prediction of hepatic microsomal intrinsic clearance and human clearance values for drugs. J. Mol. Graph. Model. 2009;28:245–252. doi: 10.1016/j.jmgm.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Mercader, A. G., Duchowicz, P. R. & Sivakumar, P. M. Chemometrics Applications and Research: QSAR in Medicinal Chemistry. CRC Press. (Eds) (2016).
- 43.Kalhotra P, Chittepu V, Osorio-Revilla G, Gallardo-Velázquez T. Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study. Molecules. 2018;23:1368. doi: 10.3390/molecules23061368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsletter On Protein Crystallography. 2002;40:82–92. [Google Scholar]
- 45.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 47.Sahoo BR, et al. Activation of nucleotide-binding oligomerization domain 1 (NOD1) receptor signaling in Labeo rohita by iE-DAP and identification of ligand-binding key motifs in NOD1 by molecular modeling and docking. Appl. Biochem. Biotechnol. 2013;170:1282–1309. doi: 10.1007/s12010-013-0263-6. [DOI] [PubMed] [Google Scholar]
- 48.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 49.Cook D, et al. Pangalos, Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat. Rev. Drug. Discov. 2014;13:419. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 50.Lin J, et al. The role of absorption, distribution, metabolism, excretion and toxicity in drug discovery. Curr. Top. Med. Chem. 2003;3:1125–1154. doi: 10.2174/1568026033452096. [DOI] [PubMed] [Google Scholar]
- 51.Venkatapathy R, Moudgal CJ, Bruce RM. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Model. 2004;44:1623–1629. doi: 10.1021/ci049903s. [DOI] [PubMed] [Google Scholar]
- 52.Desmond Molecular Dynamics System, version 3.6, D. E. Shaw Research, New York, NY, 2013. Maestro-Desmond Interoperability Tools, version 3.6, Schrödinger, New York, NY (2013).
- 53.Shivakumar D, et al. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory. Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 54.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 55.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 56.Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984;81:511–519. doi: 10.1063/1.447334. [DOI] [Google Scholar]
- 57.Reddy SVG, Reddy KT, Kumari VV, Basha SH. Molecular docking and dynamic simulation studies evidenced plausible immunotherapeutic anticancer property by Withaferin A targeting indoleamine 2, 3-dioxygenase. J. Biomol. Struct. Dyn. 2015;33:2695–2709. doi: 10.1080/07391102.2015.1004834. [DOI] [PubMed] [Google Scholar]
- 58.Basha SH, Bethapudi P, Majji Rambabu F. Anti-angiogenesis property by Quercetin compound targeting VEGFR2 elucidated in a computational approach. European J. Biotechnol. Biosc. 2014;2:30–46. [Google Scholar]
- 59.Raja Naika H, et al. Suresh, Molecular docking and dynamic studies of bioactive compounds from Naravelia zeylanica (L.) DC against glycogen synthase kinase-3β protein. JTUSCI. 2015;9:41–49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.