

Abstract
Mycobacterium tuberculosis (Mtb) possesses a two-component regulatory system, DosRST, that enables Mtb to sense host immune cues and establish a state of nonreplicating persistence (NRP). NRP bacteria are tolerant to several antimycobacterial drugs in vitro and are thought to play a role in the long course of tuberculosis therapy. Previously, we reported the discovery of six novel chemical inhibitors of DosRST, named HC101A–106A, from a whole cell, reporter-based phenotypic high throughput screen. Here, we report functional and mechanism of action studies of HC104A and HC106A. RNaseq transcriptional profiling shows that the compounds downregulate genes of the DosRST regulon. Both compounds reduce hypoxia-induced triacylglycerol synthesis by ∼50%. HC106A inhibits Mtb survival during hypoxia-induced NRP; however, HC104A did not inhibit survival during NRP. An electrophoretic mobility assay shows that HC104A inhibits DosR DNA binding in a dose-dependent manner, indicating that HC104A may function by directly targeting DosR. In contrast, UV–visible spectroscopy studies suggest HC106A directly targets the sensor kinase heme, via a mechanism that is distinct from the oxidation and alkylation of heme previously observed with artemisinin (HC101A). Synergistic interactions were observed when DosRST inhibitors were examined in pairwise combinations with the strongest potentiation observed between artemisinin paired with HC102A, HC103A, or HC106A. Our data collectively show that the DosRST pathway can be inhibited by multiple distinct mechanisms.
Introduction
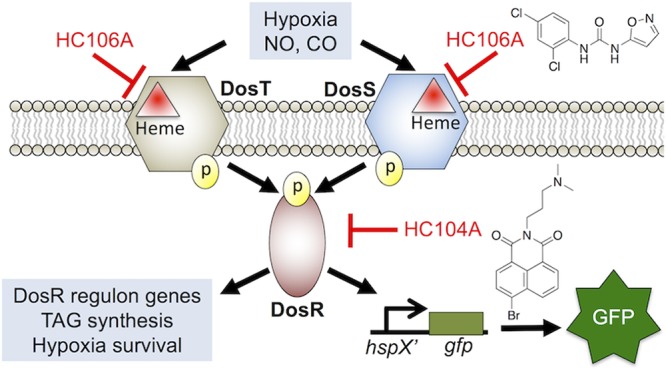
Mycobacterium tuberculosis (Mtb) can establish a dormant state known as nonreplicating persistence (NRP) where the bacterium modulates its metabolism in response to environmental and host immune cues, such as hypoxia, acidic pH, and nutrient starvation.1,2 DosRST is a two-component regulatory system that regulates Mtb persistence.3−5 It consists of two sensor histidine kinases, DosS and DosT, and the cognate response regulator DosR, which regulates expression of about 50 genes in the DosRST regulon.5−7 The pathway can be induced by host intracellular stimuli, such as nitric oxide (NO), carbon monoxide (CO), and hypoxia, through DosS and DosT.8−10 DosS is an oxygen and redox sensor, whereas DosT acts as an oxygen sensor.11−13 Both kinases sense ligands via the heme group and are inactive when the heme exists as either the Met (Fe3+) form (DosS) or the oxy (Fe2+-O2) form (DosT) in the presence of O2.12 However, hypoxic conditions activate the kinases by inducing the conversion of DosS to the ferrous form and DosT to the deoxy form. Therefore, DosS/T play overlapping and distinct roles in sensing the redox status and oxygen level of the environment to turn on the DosR pathway.10,14
NRP Mtb is tolerant to several antimycobacterial drugs in vitro.15−17 The NRP population of bacteria is thought to be responsible, in part, for the 6-month long course of TB treatment. dosRST mutants are attenuated in in vitro models of hypoxia-driven NRP18 and in animal models that generate hypoxic granulomas, including nonhuman primates, guinea pigs, and C3HeB/FeJ mouse models of TB infection.3,19−21 Furthermore, deletion of DosR-regulated gene tgs1, which is involved in triacylglycerol (TAG) synthesis, causes reduced antibiotic tolerance.22,23 Therefore, antivirulence strategies to inhibit the DosRST pathway may function to reduce virulence and deplete the reservoir of drug-tolerant NRP Mtb.24
In an effort to discover new chemical probes that inhibit Mtb persistence, we previously performed a whole cell phenotypic high throughput screening (HTS) of a >540,000 compound library using the DosRST regulon reporter strain CDC1551 (hspX’::GFP).25 We discovered six compounds that inhibit the DosR-dependent, hypoxia-induced GFP fluorescence. In the previous report, we showed that the HC101, HC102, and HC103 series functioned to inhibit NRP associated physiologies, including TAG accumulation, survival during hypoxia, and isoniazid tolerance. Mechanism of action studies showed that the HC101 series, composed of artemisinin and related analogs, functioned by oxidizing and alkylating the DosS and DosT heme. HC102 and HC103 did not modulate the DosS/T heme and were instead found to inhibit sensor kinase autophosphorylation. The purpose of this study was to characterize the activity and mechanism of action of two additional compounds, HC104A and HC106A.
Results
HC104A and HC106A Inhibit DosR Regulated Genes
Half-maximal effective concentration (EC50) studies using the CDC1551 (hspX’::GFP) DosRST-dependent fluorescent reporter strain show that HC104A and HC106A (Figure 1a) inhibit DosRST-dependent GFP florescence with EC50 values of 9.8 μM and 2.5 μM, respectively (Figure 1b and 1c). RNA-seq-based transcriptional profiling was undertaken to determine if the DosRST regulon was inhibited by the compounds. Mtb was treated with 40 μM HC104A, HC106A, or dimethyl sulfoxide (DMSO) control for 6 d in a standing flask, and following incubation RNA was extracted, sequenced, and analyzed for differential gene expression relative to the DMSO control. As a control for the DosR regulon, transcriptional profiling was also previously conducted on a DMSO treated CDC1551(ΔdosR) mutant strain.25 The transcriptional profiles showed that the genes strongly repressed following exposure to HC104A and HC106A (>2-fold; q < 0.05) are from the dosR regulon (Figure 2a–c, Supporting Information (SI) Data Sets 1 and 3).
Figure 1.

HC104A and HC106A inhibit DosRST reporter fluorescence. (a) Chemical structures of HC104A and HC106A. HC104A (b) and HC106A (c) inhibited DosR-driven GFP fluorescence signal in a dose-dependent manner, while having minimal impact of Mtb growth (OD, optical density). The EC50 values of fluorescence inhibition for HC104A and HC106A are 9.8 μM and 2.5 μM, respectively.
Figure 2.

HC104A and HC106A inhibit DosR regulated genes during hypoxia. Differential gene expression scatter plots of Mtb cells treated with 40 μM HC104A (a) or HC106A (b). The labeled genes represent selected genes that belong to the DosRST regulon. The red dots represent genes with significant differential expression, q < 0.05. (c) A Venn diagram for the downregulated genes (>2-fold; q < 0.05) of WT CDC1551 treated with HC104A or HC106A compared to that of CDC1551 (ΔdosR). (d) Venn diagram for downregulated genes (>2-fold; q < 0.05) of CDC1551 (ΔdosR) treated with HC104A or HC106A.
HC106A treatment caused remarkably strong reduction of gene expression, with transcripts for tgs1 and hspX being almost undetectable by RNA-seq following HC106A treatment. Interestingly, while HC106A broadly inhibited genes of the DosRST regulon, HC104A only strongly inhibited part of the DosR regulon, with the strongest inhibition reserved for hspX, the promoter used to drive reporter fluorescence in the screen. These RNA-seq results were validated by semiquantitative RT-PCR, with HC104A causing downregulation of dosR, hspX, and tgs1 in vitro by 6-, 570-, and 13-fold, respectively, whereas HC106A downregulated these three genes by 49-, 1360-, and 1424-fold, respectively (SI Figure 1), with hspX and tgs1 transcripts being below the level of detection by qRT-PCR.
Comparisons of transcriptional profiles from the inhibitor treated wild type (WT) Mtb strain to a CDC1551(ΔdosR) mutant strain showed that there are a total of 26 genes and 53 genes from dosR regulon inhibited by HC104A and HC106A, respectively. Notably, HC104A and HC106A caused an additional 119 genes and 35 genes to be repressed that were not repressed in the CDC1551(ΔdosR) mutant strain (Figure 2c). This observation suggested that these two compounds exhibit some DosR-independent activities. To confirm the specificity of the compounds, RNA-seq was also performed on CDC1551(ΔdosR) mutant background (SI Data Sets 2 and 3) treated with HC104A or HC106A. This analysis identified 171 genes and 51 genes that are downregulated (>2-fold; q < 0.05) by HC104A and HC106A, respectively (Figure 2d). This finding indicates that HC104A and HC106A impact other targets besides DosR regulon, with HC106A showing greater on-target specificity than HC104A. Based on these findings, we conclude that (1) HC106A strongly and specifically inhibits the DosRST regulon and (2) HC104A strongly inhibits a portion of the DosRST regulon, with several notable off-target activities.
To assess the impact of the inhibitors on the DosRST pathway of intracellular Mtb, murine bone marrow-derived macrophages were infected with Mtb and treated with 40 μM HC104A and HC106A for 48 h. Total bacterial RNA was isolated and analyzed by RT-PCR for hspX and tgs1 gene differential expression. The results demonstrate that the induction of hspX and tgs1 was inhibited 185- and 10-fold by HC104A and 6- and 4-fold by HC106A, respectively (Figure 3a). These findings confirm that HC104A and HC106A can access Mtb inside the macrophage; however, the reduced repression of the pathway by HC106A as compared to broth culture suggests that the molecule may not be able to efficiently target intracellular Mtb.
Figure 3.

Inhibition of DosR regulon and persistence-associated physiologies by HC104A and HC106A. (a) Inhibition of DosR regulon in Mtb in murine macrophages infected with Mtb and treated with HC104A and HC106A for 48 h. Differential gene expression of hspX and tgs1 were quantified by qRT-PCR. The error bars represent the standard derivation of three biological replicates. (b) HC104A and HC106A inhibit DosR regulon induction by vitamin C and NO. Transcripts of DosR-regulated genes, hspX and tgs1, were quantified by qRT-PCR. The differences in the drug treated samples compared to DMSO treated samples in response to vitamin C or NO are significant with a p-value <0.001 based on a t test, except the one marked as nonsignificant (n.s.). The error bars represent the standard deviation of three replicates. The experiment was repeated twice with similar results. (c) Inhibition of TAG accumulation of Mtb treated with HC104A or HC106A for 6 d.. The error bars represent the standard deviation of two biological replicates. (d) Mtb survival during NRP when treated with HC104A or HC106A during NRP. The error bars represent the standard deviation of three biological replicates. The experiment was repeated twice with similar results.
To examine whether HC104A and HC106A can repress the induction of the DosRST pathway by vitamin C or NO, Mtb cells, cultured under aerobic conditions to reduce expression of hypoxia-induced genes, were pretreated with HC104A or HC106A for 24 h followed by vitamin C or NO induction for 2 h. The expression of DosR-regulated genes (hspX and tgs1) was examined by real time-PCR. Vitamin C and DETA-NONOate (NO donor) strongly induced hspX and tgs1 as previously reported25 (Figure 3b). For instance, vitamin C induced hspX and tgs1 by 2162- and 58-fold, respectively, whereas DETA-NONOate upregulated hspX and tgs1 3024- and 113-fold, respectively (Figure 3b). Mtb cells pretreated with HC106A showed strong inhibition of hspX and tgs1 induction by vitamin C and DETA-NONOate. For example, HC106A inhibited the hspX and tgs1 transcripts by 78- and 14-fold following vitamin C treatment, respectively, and 362- and 151-fold following DETA-NONOate treatment. Following vitamin C treatment, HC104 showed inhibition of hspX by 3.4-fold and no effect on tgs1, or 302- and 6.6-fold inhibition of hspX and tgs1 following DETA-NONOate treatment. These findings show that HC104A and HC106A act as inhibitors of the DosRST pathway in response to redox signals.
Inhibition of Mtb Persistence Physiology
DosR directly regulates tgs1, which encodes a TAG synthase that is involved in the last step of TAG biosynthesis and is required for TAG accumulation during hypoxia.26 Transcriptional profiling showed that HC104A and HC106A repress expression of tgs1. Therefore, we hypothesized these compounds may inhibit TAG biosynthesis during NRP. To test this hypothesis, Mtb cells were radiolabeled with 14C-acetate and treated with HC104A or HC106A for 6 d. Lipids were isolated and analyzed by thin layer chromatography (TLC). As previously observed, DMSO treated CDC1551(ΔdosR) mutant displayed a strong (87%) reduction of TAG accumulation as compared to DMSO treated WT (Figure 3c and SI Figure 2). Mtb cells treated with HC104A or HC106A showed a ∼50% reduction of TAG accumulation, supporting our hypothesis that the compounds can inhibit TAG biosynthesis.
DosRST has been previously reported to be required for survival during NRP, where deletion of dosR causes greatly reduced survival during prolonged hypoxic stress.18 The impact of HC104A and HC106A on Mtb survival during NRP was examined using the hypoxic shift-down model.27 Mtb survival was examined following 10 d of treatment with the compounds at 40 μM. The ΔdosR mutant control had 15% survival relative to DMSO and was partially complemented, supporting the proposal that survival during hypoxia is DosR dependent. Mtb cells treated with HC106A displayed 50% survival relative to DMSO control (Figure 3d), whereas HC104A had no impact on Mtb survival during NRP, an observation that suggests the portion of the DosR regulon inhibited by HC104A is not essential for survival during NRP.
To determine that HC106 kills NRP Mtb in a dosR-dependent manner, we examined killing of the ΔdosR mutant by HC106, reasoning that if the compound is specific, there should be no killing of the mutant by HC106 treatment. Using the compound MSU-39446 (Table 2), an analog of HC106A that is ∼4.5-fold more potent, we observed treated cells had 32% survival relative to the DMSO treated control during NRP (SI Figure 3). The DMSO treated ΔdosR mutant exhibited 12% survival, not significantly different from the 15% survival of the mutant treated with MSU-39446. These data are consistent with HC106 killing Mtb in a dosR-dependent manner.
Table 2. SAR Studies of “A-ring” Analogs of HC106a.

The HC106 analogs with different R-groups were synthesized. The reporter strain CDC1551 (hspX’::GFP) was treated with doses of each analog from 200 μM to 0.328 μM. The EC50 values of fluorescence inhibition were calculated for each analog to determine their potency.
HC104A Inhibits DosR DNA Binding
To investigate the biochemical mechanisms of action of HC104A and HC106A, inhibition of DosS autophosphorylation was initially evaluated. The DosS protein was treated with different concentrations of HC104A and HC106A from 10 μM to 40 μM, or with 40 μM HC103A as a positive control that was previously discovered to be a DosS/T autophosphorylation inhibitor.25 As previously observed, HC103A strongly inhibited DosS autophosphorylation, but HC104A and HC106A had no inhibitory activity (SI Figure 4). This suggests HC104A and HC106A are not directly inhibiting DosS/T autophosphorylation activity.
Next, a UV–visible spectroscopy assay was employed to investigate if HC104A targets to the heme of the sensor kinase DosS. Treatment of DosS protein with the reducing agent dithionite (DTN) caused a shift of the Soret peak from 403 to 430 nm as previously described.12,25 Addition of HC104A to reduced DosS did not shift the peak to the oxidized position, suggesting that HC104A does not modulate DosS heme redox (SI Figure 5).
Inspection of the HC104A structure revealed it had similarity to the compound virstatin (SI Figure 6A).28 Virstatin inhibits ToxT protein dimerization and subsequently interferes with DNA binding, thereby inhibiting the transcription of downstream genes involved in toxin production.28 Therefore, we hypothesized that HC104A may be targeting DosR and interfering with DNA binding. An electrophoretic mobility shift assay (EMSA) was employed to investigate the impact of HC104A on DosR DNA binding. Recombinant DosR protein, ranging from 0.5 μM to 4 μM, was treated with 40 μM HC104A or a DMSO control and tested for binding to fluorescently labeled hspX promoter DNA. In the DMSO treated control, DosR bound promoter DNA beginning at a concentration of 2 μM DosR protein (Figure 4a). Treating the reaction containing 2 μM DosR protein with HC104A significantly inhibited DNA binding by ∼22-fold compared to DMSO control (Figure 4b). To further characterize the impact of HC104A on DosR binding of DNA, a dose–response study was performed. Reactions containing 2 μM recombinant DosR proteins were treated with different concentrations of HC104A or virstatin ranging from 1–80 μM (Figure 4c and SI Figure 6c). HC104A inhibited DosR binding of DNA beginning at 10 μM HC104A. The fraction of free DNA increased as HC104A concentration increased (Figure 4c and d). Thus, HC104A significantly inhibits DosR-DNA binding in a dose-dependent manner. Virstatin did not have any impact on DosRST signaling in the whole cell Mtb fluorescence reporter assay (SI Figure 6b) or on DosR binding of DNA (SI Figure 6c).
Figure 4.

Inhibition of DosR DNA-binding by HC104A. (a) DosR was treated with DMSO or 40 μM HC104A, and binding to the hspX promoter was examined by EMSA. HC104A inhibits DosR DNA binding at 2 μM concentration. (b) Quantification of free DNA (**P value <0.005 based on a t-test). The error bars represent the standard deviation of two biological replicates. (c) Dose-dependent impact of HC104A on DosR DNA binding. DosR protein at 2 μM was treated with HC104A at concentrations from 1 μM to 80 μM. (d) Quantification of free DNA (*P value <0.05 and **P value <0.005 based on a t-test). The error bars represent the standard deviation of two biological replicates.
HC106A Modulates DosS Heme
DosS and DosT have a channel that exposes the heme to the environment and enables interactions with gases.29,30 Previously, it was shown that artemisinin modulates DosS/T by oxidizing and alkylating heme carried by the kinases.25 UV–visible spectroscopy studies were conducted to examine if HC106A modulated DosS heme. Recombinant DosS was purified from E. coli and degassed, and the change of the DosS heme spectrum was monitored under anaerobic conditions by UV–visible spectroscopy. Treating DosS with the reducing agent dithionite (DTN) caused the Soret peak to shift to 430 nm.12,25 HC106A was added to the reaction following DTN treatment to observe the impact on the DosS heme UV–visible spectrum. HC106A caused the DosS Soret peak to immediately shift to 422 nm, where the peak was stably maintained for 2 h (Figure 5a). A similar shift was observed in DosT treated with HC106A (SI Figure 7). This spectrum shift is different from artemisinin, where under identical conditions, artemisinin causes the DosS Soret peak to gradually shift back to the oxidized state at 403 nm25 or the DosT Soret peak to decrease in amplitude (consistent with heme alkylation25). These findings show that HC106A may also interact with sensor kinase heme, but via a mechanism that is distinct from artemisinin–heme interactions.
Figure 5.
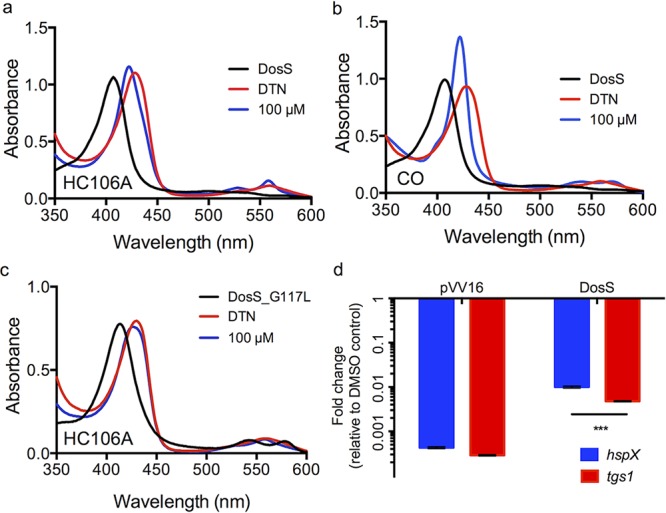
Interactions between HC106A and DosS heme. WT DosS protein was treated with dithionite (DTN) and then 100 μM HC106A (a) or 100 μM pf CORM-2 (a CO donor) (b). The UV–visible spectra of the two treatments exhibited a shift of the Soret peak to a common position of 422 nm. (c) DosS with a G117L amino acid substitution provides resistance to HC106A. (d) Overexpression of DosS protein promotes resistance to 20 μM HC106A treatment in Mtb. hspX and tgs1 transcripts were analyzed by qRT-PCR (***P value <0.0001 based on a t-test). The error bar represents the standard derivation of the mean for three technical replicates.
The Soret peak at 422 nm is consistent with previously described spectra that are observed when DosS heme interacts with NO or CO.12 To confirm this observation, DosS was treated with 100 μM CORM-2 (a CO donor) which caused a shift of the Soret peak to 422 nm, similar to what was observed for HC106A (Figure 5b). This finding supports a hypothesis that HC106A may also be directly binding to the heme. Amino acid substitutions in the channel exposing the DosS heme to the environment, such as DosS E87L or G117L, can limit access of artemisinin to modulate heme.25 Therefore, we tested the impact of these amino acid substitutions on HC106A/DosS heme interactions. Treating DosS(E87L) with HC106A exhibited a profile similar to wild type DosS with the Soret peak shifting to 422 nm (SI Figure 8). However, DosS(G117L) had no change to the overall spectrum after HC106A treatment (Figure 5c). This finding indicates that DosS(G117L) is resistant to HC106A and confirms that HC106A accesses the heme via a similar mechanism as artemisinin.
To confirm DosS is a target of HC106A in Mtb, we examined the impact of overexpressing DosS protein in Mtb. WT DosS protein was constitutively expressed from the hsp60 promoter in Mtb and Mtb was grown under conditions of mild hypoxia. The vector control showed that both hspX and tgs1 genes were strongly downregulated by HC106A (Figure 5d). Overexpressing DosS provided significant resistance to HC106A, with hspX and tgs1 showing 23- and 16.5-fold less inhibition, respectively, relative to the empty vector control. This observation of resistance in Mtb is consistent with the biochemical data supporting the view that DosS is a direct target of HC106A. We also examined if overexpression of DosS(G117L) or DosT (G115L) provides resistance to treatment with the HC106 analog MSU-39446 in the hypoxic shift-down assay. Unexpectedly, we observed overexpression of the control DosS or DosT proteins caused a significant 80% and 60% reduction of Mtb survival, respectively (SI Figure 3). Treatment of the DosS overexpressor with MSU-39446 caused a significant reduction of survival relative to the DMSO control, whereas no significant difference in survival was observed in the DosS(G117L) overexpressor. These data are suggestive of the DosS (G117L) mutant providing HC106 resistance but are not conclusive given the confounding survival defect in the DosS overexpressor. The DosT (G115L) overexpressor had significantly reduced survival when treated with MSU-39446, supporting limited resistance provided by this mutation during NRP.
Synergistic Interactions of Inhibitors
To define interactions between DosRST regulon inhibitors, checkerboard assays were performed with pairwise comparisons of artemisinin (HC101A), HC102A, HC103A, HC104A, and HC106A. CDC1551 (hspX’::GFP) was treated with combinations of two compounds ranging from 50 μM to 0.08 μM in 96-well plates. The combination index (CI) was calculated for each drug pair based on the Chou-Talalay method in the CompuSyn software package,31,32 where CI values of <1, =1, and >1 indicate synergistic, additive, or antagonistic interactions, respectively. Among all 64 compound pairs, artemisinin combined with HC102A, HC103A, HC104A, and HC106 showed 46, 49, 41, and 50 combinations that have CI < 1, respectively (Figure 6). Notably, some CI values are below 0.1 when artemisinin was paired with HC102A, HC103A, or HC106A combinations. Example dose response curves illustrate these synergistic interactions (Figure 6). Several other pairwise comparisons also demonstrated synergy (SI Figure 9). Overall, these studies provide the evidence that the inhibitors function by distinct mechanisms and may be combined to improve potency.
Figure 6.

Synergistic interactions between DosRST inhibitors. CDC1551 (hspX’::GFP) was treated with pairwise combinations of two compounds at concentrations of 50 μM to 0.08 μM. GFP fluorescence was measured and used to calculate percentage inhibition. The combination index (CI) for the panel of each drug combination is presented, including (a) artemisinin and HC102A; (b) artemisinin and HC103A; (c) artemisinin and HC104A; and (d) artemisinin and HC106A. Example EC50 curves are presented with individual compounds or a selected synergistic combination to illustrate the potentiating interactions.
Structure–Activity Relationship Studies
We conducted a catalog search for HC104 and HC106 analogs and obtained commercial analogs for each series to define initial structure activity relationships (SAR). For HC104A we observed that a bromine in the 5-position is required for activity and that the R2 dimethylamine group is not required for full activity (SI Table 1). For HC106A (Table 1), catalog SAR work led to new understandings of the nature of the series. We first found that the simple removal of an ortho chloro on the “A” ring of HC106A leads to ∼2-fold enhanced activity, with an EC50 in the whole cell Mtb assay for DosRST inhibition of 1.33 μM (HC106F). It was also found that the use of an alternative isomer of the isoxazole had no detectable activity (HC106C).
Table 1. Initial SAR Studies of the HC106 Seriesa.

The HC106 analogs with different R-groups were synthesized or purchased. The reporter strain CDC1551 (hspX’::GFP) was treated with doses of each analog from 200 μM to 0.328 μM. The EC50 values of fluorescence inhibition were calculated for each analog to determine their potency.
To further understand the SAR of the HC106 series, additional analogs were synthesized to examine the need of the central urea functionality and whether modifications can be tolerated (Table 1). A pyridyl analog (MSU-41425), designed to replace the isoxazole, also demonstrated no activity as was the case for the symmetrical 4-chloroaniline derived urea (MSU-41324). However, the bis-isoxazole urea (MSU-39444) provides an EC50 of 1.7 μM, indicating that the isoxazole is important for function. Isoxazoles are unique among heterocycles in that they exist in multiple tautomeric forms as supported by initial NMR studies.33 We next explored the need of one of the -NHs of the urea, capping it with a methyl (MSU-39451), integrating it into a ring for conformational restriction (MSU-39453), and replacing with a methylene unit (MSU-39449). In all cases, reduced activity (0.5–1 log) was observed but not all activity was lost.
To further test the SAR, we conducted a Topliss Tree evaluation of the “A-ring” aniline (Table 2).34 To reliably prepare the derivatives, we explored and established a general preparation (SI Figure 10). Using HC106F and HC106A as starting points, we prepared the 3,4-diclorochloro and 3-chloro derivatives (MSU-39452 and MSU-39445, respectively). Both the 3- and 4-chloro derivatives demonstrated greater activity than 3,4-dichloro (MSU-39452). We found that replacing the 4-chlorophenyl ring with pyridyl analogs (MSU-39448 and MSU-39450) lead to similar activity. Focusing on 4-position derivatives, we found that fluoro (MSU-39446), bromo (MSU-41464), and methoxy (MSU-39447), as electron p-orbital donating substituents, also lead toward incrementally increased activity. p-tert-Butyl phenyl (MSU-41442) provided slightly diminished activity. Electron withdrawing substituents, such as 4-CO2Me (MSU-4165), 4-trifluoromethyl (MSU-41463), and biphenyl (MSU-41443) saw activity similar to the 4-chlorophenyl derivative (HC106F). Overall, several analogs were discovered with significantly ∼4-fold enhanced potency, with several inhibitors having whole cell DosRST inhibitory EC50 below 1 μM.
There appears to be little sensitivity, positive or negative, for electron-drawing substituents other than the biphenyl derivative (MSU-41443), which is likely the result of negative steric interactions in the binding domain. Additional derivatives (replacement of dichlorophenyl) were also prepared to further probe the size and nature of the binding domain. We began by replacing the chlorophenyl ring of HC106F with benzyl- (MSU-41462), isobutyl- (MSU-41542), cyclopentyl- (MSU-41546) and cyclohexyl- (MSU-42002) analogs. All analogs demonstrated similar activities, relative to HC106F and the simple phenyl analog (MSU-33189), suggesting flexibility of fragments that could bind in this domain.
Kinetic solubility assays were conducted for selected analogs, and all exhibited excellent aqueous solubility greater than >100 μM, except for MSU-41443 (Table 2). This finding shows that the urea group present in the HC106A does not have a detrimental impact on HC106 aqueous solubility. All of the tested derivatives also demonstrated favorable mouse microsomal stability. Overall, the nanomolar whole cell potency, flexible SAR, good microsomal stability, and excellent solubility confirm that the HC106 series is a suitable series for continued optimization to identify a drug-like lead.
Discussion
The UV–visible spectrum of HC106A-treated DosS is similar to those of CO- or NO-treated DosS. The overlap between the CO and HC106A spectra supports that HC106A may also directly bind to the heme of DosS. Interestingly, CO activates sensor kinases, whereas HC106A inhibits them. This could be due to the difference in conformational changes induced by CO and HC106A, or binding of HC106A may lock the sensor kinases into an inactive state. Furthermore, the DosS G117L substitution in recombinant DosS blocks the heme exposing channel and provides resistance to HC106A. This means that, similar to artemisinin, this channel is also important for the activity of HC106A. These findings provide additional evidence that the heme-exposing channel in DosS/T can be exploited by small molecules to inhibit the heme from sensing signals and to disrupt signal transduction of a two-component regulatory system.
Mechanistic studies via EMSA indicate that HC104A may function by targeting DosR and inhibiting DosR DNA binding. Virstatin had no effect on DosR DNA binding and no impact on the DosRST signaling in whole cells, showing that although both compounds share a similar structure, virstatin does not disrupt DosR signaling or the DosR/DNA complex. Transcriptional profiling shows that the most repressed genes by HC104A are from the DosR regulon, providing additional evidence that HC104A is somewhat selective for specific DosR regulated genes. Interestingly, the genes most downregulated by HC104A, including hspX, Rv2030c, pfkB, and Rv2028c, are from the same operon under control of hspX promoter. This result suggests that HC104A is more specific to target hspX operon genes as compared to other DosR regulated genes. This finding leads to the speculation that HC104A may be more efficient to prevent DosR binding to the hspX promoter than the other DosR promoters. This observation also supports that HC104A may not have an impact on DosR protein dimerization, which would lead to universal downregulation of DosR-regulated genes.
The transcriptional profiling data showed off-target impacts in the treated dosR mutant strain. These effects may be due to inhibition of DosS/T signaling that functions independent of DosR. Notably, DosT can interact with the other noncognate response regulators, including NarL and PrrA.35 Our transcriptional profiling of the compound treated dosR mutant strain suggests that some genes downregulated by HC101A, HC103A, and HC106A may be DosS/T-dependent but DosR-independent (SI Figure 11). In the prior study, six genes are similarly regulated between artemisinin and HC103A in the treated dosR mutant strain, including Rv0260c that encodes a putative response regulator.25 HC106A and HC103A share four differentially regulated genes in the dosR mutant, including argC, argJ, argB, and argF, which are genes involved in arginine biosynthesis. These data suggest that DosST may modulate gene expression independently of DosR. Notably, de novo arginine biosynthesis has been shown to be required for Mtb survival,36 supporting that DosR-independent, but DosS/T-dependent pathways may be associated with HC106 killing of Mtb during NRP.
Synergistic interactions were observed between artemisinin, HC102A, HC103A, HC104, or HC106A. Moreover, artemisinin exhibited the greatest synergistic activities with HC102A, HC103A, or HC106A, indicating that inhibition of histidine kinases by a second inhibitor can lead to synergistic inhibition of the DosRST pathway. This interaction could be due to both sensor kinases being required for full induction of the DosR regulon, where DosT responds early during hypoxia and DosS further induces the regulon at later times,37 and it is possible that the inhibitors have different affinities for DosS or DosT. Thus, multiple inhibitors could inhibit both DosS and DosT better than an inhibitor alone. Interestingly, artemisinin shows the greatest synergism with HC106A. Both compounds are proposed to target the heme of DosS/T, but through different mechanisms. This finding suggests that both inhibitors can enter the channel of DosST to interact with the heme and do so without antagonizing interactions.
Methods
Bacterial Strains and Growth Conditions
Mtb CDC1551 and CDC1551 (ΔdosR) strains were used in this study. All cultures were grown at 37 °C and 5% CO2 in 7H9Middlebrook medium supplemented with 10% OADC (oleic acid albumin dextrose catalase) and 0.05% Tween-80 in standing, vented tissue culture flasks, unless stated otherwise.
EC50 Assays
The assay was performed as previously described.25 Briefly, the (hspX’::GFP) reporter strain culture was diluted to an OD600 of 0.05 in fresh 7H9 media, pH 7.0, and 200 μL of diluted culture was aliquoted in clear-bottom, black, 96-well plates (Corning). Cells were treated with an 8-point (2.5-fold) dilution series ranging from 200 μM to 0.32 μM. For the structure relationship studies for the HC106 series, a 12-point (2.5-fold) dilution series of HC106 analogs ranging from 200 μM to 8.4 nM was used. GFP fluorescence and optical density were measured following 6 d incubation. Percentage fluorescence and growth inhibitions were normalized to a rifampin-positive control (100% inhibition) and DMSO-negative control (0% inhibition). Each experiment was performed with two technical replicates per plate and two biological replicates, and the error bar represents the s.d. of the biological replicates. Experiments were performed twice with similar results.
Transcriptional Profiling and Data Analysis
Transcriptional profiling studies were conducted as previously described in Zheng et al.25 Briefly, CDC1551 or CDC1551 (ΔdosR) cultures were treated with 40 μM HC104A, HC106A, or DMSO control for 6 d. The starting OD600 was 0.1 in 8 mL of 7H9 medium in standing T25 vented tissue culture flasks. Bacterial growth consumes oxygen and stimulates the DosRST pathway. The total bacterial RNA from two biological replicates was isolated and prepared for sequencing as previously described.38 The RNA-seq data were processed and analyzed using the SPARTA software package.39 Sequencing data are available at the GEO Database (Accession GSE115892).
Acknowledgments
We acknowledge ChemAxon for academic use of Marvin Sketch and MarvinSpace. This project was supported by start-up funding and a Molecular Discovery Grant from Michigan State University, AgBioResearch, grants from the NIH-NIAID (R21AI105687 and R01AI116605), and Grand Challenges Explorations grants from the Bill and Melinda Gates Foundation (OPP1059227 and OPP1119065).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00849.
Methods; SI Table 1 showing the structure and properties of the HC104 series; and SI Figures 1-11 demonstrating: inhibition of the DosR regulon by HC104 and HC106A during hypoxia, TLC of TAG reduction in Mtb treated with HC104A and HC106A, inhibition of Mtb survival by HC106 analog MSU-39446 during NRP in the hypoxic shift-down model, autoradiograph, investigation of interaction between HC104A and DosS, impact of virstatin on DosR DNA-binding and DosRST signaling in Mtb, investigating the interaction between HC106A and DosT heme, investigating the interaction between HC106A and DosS heme, checkerboard assays examining paired interactions of DosRST inhibitors, synthetic scheme for the HC106 analogs, and comparison between artemisinin, HC103 and HC106 for interactions (PDF)
Differential gene expression tables of WT treated with HC104A or HC106A compared to DMSO (XLSX)
Differential gene expression tables of dosR mutant treated with HC104A or HC106A compared to DMSO (XLSX)
Complete gene expression tables of WT or dosR mutant treated with HC104A or HC106A compared to DMSO (XLSX)
Author Contributions
H.Z., E.E., and R.B.A. conceived the experiments. H.Z. and J.T.W. performed the Mtb physiology and biochemistry experiments. B.A. synthesized the analogs. E.E. directed the medicinal chemistry optimizations. H.Z., E.E., and R.B.A. wrote the manuscript.
The authors declare the following competing financial interest(s): RBA is the founder and owner of Tarn Biosciences, Inc., a company that is working to develop new TB drugs.
Supplementary Material
References
- Rustad T. R.; Sherrid A. M.; Minch K. J.; Sherman D. R. (2009) Hypoxia: a window into Mycobacterium tuberculosis latency. Cell. Microbiol. 11, 1151–1159. 10.1111/j.1462-5822.2009.01325.x. [DOI] [PubMed] [Google Scholar]
- Wayne L. G.; Sohaskey C. D. (2001) Nonreplicating persistence of mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163. 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- Mehra S.; Foreman T. W.; Didier P. J.; Ahsan M. H.; Hudock T. A.; Kissee R.; Golden N. A.; Gautam U. S.; Johnson A. M.; Alvarez X.; Russell-Lodrigue K. E.; Doyle L. A.; Roy C. J.; Niu T.; Blanchard J. L.; Khader S. A.; Lackner A. A.; Sherman D. R.; Kaushal D. (2015) The DosR Regulon Modulates Adaptive Immunity and Is Essential for Mycobacterium tuberculosis Persistence. Am. J. Respir. Crit. Care Med. 191, 1185–1196. 10.1164/rccm.201408-1502OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudock T. A.; Foreman T. W.; Bandyopadhyay N.; Gautam U. S.; Veatch A. V.; LoBato D. N.; Gentry K. M.; Golden N. A.; Cavigli A.; Mueller M.; Hwang S. A.; Hunter R. L.; Alvarez X.; Lackner A. A.; Bader J. S.; Mehra S.; Kaushal D. (2017) Hypoxia Sensing and Persistence Genes Are Expressed during the Intragranulomatous Survival of Mycobacterium tuberculosis. Am. J. Respir. Cell Mol. Biol. 56, 637–647. 10.1165/rcmb.2016-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N.; Kapur V.; Singh K. K.; Das T. K.; Sachdeva S.; Jyothisri K.; Tyagi J. S. (2000) Characterization of a two-component system, devR-devS, of Mycobacterium tuberculosis. Tubercle and lung disease: the official journal of the International Union against Tuber Lung Dis 80, 141–159. 10.1054/tuld.2000.0240. [DOI] [PubMed] [Google Scholar]
- Park H. D.; Guinn K. M.; Harrell M. I.; Liao R.; Voskuil M. I.; Tompa M.; Schoolnik G. K.; Sherman D. R. (2003) Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843. 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini D. K.; Malhotra V.; Dey D.; Pant N.; Das T. K.; Tyagi J. S. (2004) DevR-DevS is a bona fide two-component system of Mycobacterium tuberculosis that is hypoxia-responsive in the absence of the DNA-binding domain of DevR. Microbiology (London, U. K.) 150, 865–875. 10.1099/mic.0.26218-0. [DOI] [PubMed] [Google Scholar]
- Voskuil M. I.; Schnappinger D.; Visconti K. C.; Harrell M. I.; Dolganov G. M.; Sherman D. R.; Schoolnik G. K. (2003) Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713. 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Deshane J. S.; Crossman D. K.; Bolisetty S.; Yan B. S.; Kramnik I.; Agarwal A.; Steyn A. J. (2008) Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283, 18032–18039. 10.1074/jbc.M802274200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. M.; Cho H. Y.; Cho H. J.; Ko I. J.; Park S. W.; Baik H. S.; Oh J. H.; Eom C. Y.; Kim Y. M.; Kang B. S.; Oh J. I. (2008) O2- and NO-sensing mechanism through the DevSR two-component system in Mycobacterium smegmatis. Journal of bacteriology 190, 6795–6804. 10.1128/JB.00401-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa E. H.; Tuckerman J. R.; Gonzalez G.; Gilles-Gonzalez M. A. (2007) DosT and DevS are oxygen-switched kinases in Mycobacterium tuberculosis. Protein Sci. 16, 1708–1719. 10.1110/ps.072897707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Toledo J. C.; Patel R. P.; Lancaster J. R. Jr.; Steyn A. J. (2007) Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. U. S. A. 104, 11568–11573. 10.1073/pnas.0705054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioanoviciu A.; Yukl E. T.; Moenne-Loccoz P.; de Montellano P. R. (2007) DevS, a heme-containing two-component oxygen sensor of Mycobacterium tuberculosis. Biochemistry 46, 4250–4260. 10.1021/bi602422p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. J.; Park K. J.; Ko I. J.; Kim Y. M.; Oh J. I. (2010) Different roles of DosS and DosT in the hypoxic adaptation of Mycobacteria. J. Bacteriol. 192, 4868–4875. 10.1128/JB.00550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. P.; Alonso S.; Rand L.; Dick T.; Pethe K. (2008) The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105, 11945–11950. 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams K. N.; Takaki K.; Connolly L. E.; Wiedenhoft H.; Winglee K.; Humbert O.; Edelstein P. H.; Cosma C. L.; Ramakrishnan L. (2011) Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53. 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. S.; Kaufmann B. B.; Chand N. S.; Haseley N.; Hung D. T. (2012) Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc. Natl. Acad. Sci. U. S. A. 109, 12147–12152. 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leistikow R. L.; Morton R. A.; Bartek I. L.; Frimpong I.; Wagner K.; Voskuil M. I. (2010) The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 192, 1662–1670. 10.1128/JB.00926-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Converse P. J.; Karakousis P. C.; Klinkenberg L. G.; Kesavan A. K.; Ly L. H.; Allen S. S.; Grosset J. H.; Jain S. K.; Lamichhane G.; Manabe Y. C.; McMurray D. N.; Nuermberger E. L.; Bishai W. R. (2009) Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77, 1230–1237. 10.1128/IAI.01117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam U. S.; Mehra S.; Kaushal D. (2015) in-vivo gene signatures of Mycobacterium tuberculosis in C3HeB/FeJ Mice. PLoS One 10, e0135208 10.1371/journal.pone.0135208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam U. S.; McGillivray A.; Mehra S.; Didier P. J.; Midkiff C. C.; Kissee R. S.; Golden N. A.; Alvarez X.; Niu T.; Rengarajan J.; Sherman D. R.; Kaushal D. (2015) DosS is required for the complete virulence of Mycobacterium tuberculosis in mice with classical granulomatous lesions. Am. J. Respir. Cell Mol. Biol. 52, 708–716. 10.1165/rcmb.2014-0230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C.; Lee C. M.; Dubey V. S.; Daniel J.; Abomoelak B.; Sirakova T. D.; Pawar S.; Rogers L.; Kolattukudy P. E. (2009) A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4, e6077 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A.; Abouelhassan Y.; Zuo R.; Yousaf H.; Ding Y.; Huigens R. W. (2017) Antimicrobial peptide-inspired NH125 analogues: bacterial and fungal biofilm-eradicating agents and rapid killers of MRSA persisters. Org. Biomol. Chem. 15, 5503–5512. 10.1039/C7OB01028A. [DOI] [PubMed] [Google Scholar]
- Johnson B. K.; Abramovitch R. B. (2017) Small Molecules That Sabotage Bacterial Virulence. Trends Pharmacol. Sci. 38, 339–362. 10.1016/j.tips.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Colvin C. J.; Johnson B. K.; Kirchhoff P. D.; Wilson M.; Jorgensen-Muga K.; Larsen S. D.; Abramovitch R. B. (2017) Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat. Chem. Biol. 13, 218–225. 10.1038/nchembio.2259. [DOI] [PubMed] [Google Scholar]
- Baek S. H.; Li A. H.; Sassetti C. M. (2011) Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 9, e1001065 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P. A.; Rao S. P.; Ping Tan M.; Lin X.; Chyba J.; Tay J.; Ng S. H.; Tan B. H.; Cherian J.; Duraiswamy J.; Bifani P.; Lim V.; Lee B. H.; Ling Ma N.; Beer D.; Thayalan P.; Kuhen K.; Chatterjee A.; Supek F.; Glynne R.; Zheng J.; Boshoff H. I.; Barry C. E. 3rd; Dick T.; Pethe K.; Camacho L. R. (2012) A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 7, 1190–1197. 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung D. T.; Shakhnovich E. A.; Pierson E.; Mekalanos J. J. (2005) Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674. 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- Podust L. M.; Ioanoviciu A.; Ortiz de Montellano P. R. (2008) 2.3 A X-ray structure of the heme-bound GAF domain of sensory histidine kinase DosT of Mycobacterium tuberculosis. Biochemistry 47, 12523–12531. 10.1021/bi8012356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. Y.; Cho H. J.; Kim Y. M.; Oh J. I.; Kang B. S. (2009) Structural insight into the heme-based redox sensing by DosS from Mycobacterium tuberculosis. J. Biol. Chem. 284, 13057–13067. 10.1074/jbc.M808905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T., and Martin N. (2005) CompuSyn for drug combinations: PC software and user’s guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values, ComboSyn, Paramus, NJ.
- Chou T. C.; Talalay P. (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Boulton A. J.; Katritzky A. R. (1961) The tautomerism of heteroaromatic compounds with five-membered rings—I: 5-hydroxyisoxazoles-isoxazol-5-ones. Tetrahedron 12, 41–50. 10.1016/0040-4020(61)80097-5. [DOI] [Google Scholar]
- Topliss J. G. (1972) Utilization of operational schemes for analog synthesis in drug design. J. Med. Chem. 15, 1006–1011. 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- Lee H. N.; Jung K. E.; Ko I. J.; Baik H. S.; Oh J. I. (2012) Protein-protein interactions between histidine kinases and response regulators of Mycobacterium tuberculosis H37Rv. J. Microbiol. (Seoul, Repub. Korea) 50, 270–277. 10.1007/s12275-012-2050-4. [DOI] [PubMed] [Google Scholar]
- Tiwari S.; van Tonder A. J.; Vilcheze C.; Mendes V.; Thomas S. E.; Malek A.; Chen B.; Chen M.; Kim J.; Blundell T. L.; Parkhill J.; Weinrick B.; Berney M.; Jacobs W. R. Jr. (2018) Arginine-deprivation-induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 115, 9779–9784. 10.1073/pnas.1808874115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honaker R. W.; Leistikow R. L.; Bartek I. L.; Voskuil M. I. (2009) Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect. Immun. 77, 3258–3263. 10.1128/IAI.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. J.; Johnson B. K.; Abramovitch R. B. (2014) Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 94, 56–69. 10.1111/mmi.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. K.; Scholz M. B.; Teal T. K.; Abramovitch R. B. (2016) SPARTA: Simple Program for Automated reference-based bacterial RNA-seq Transcriptome Analysis. BMC Bioinf. 17, 66. 10.1186/s12859-016-0923-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.