

Abstract
Background
Principal component analysis (PCA) is an essential method for analyzing single-cell RNA-seq (scRNA-seq) datasets, but for large-scale scRNA-seq datasets, computation time is long and consumes large amounts of memory.
Results
In this work, we review the existing fast and memory-efficient PCA algorithms and implementations and evaluate their practical application to large-scale scRNA-seq datasets. Our benchmark shows that some PCA algorithms based on Krylov subspace and randomized singular value decomposition are fast, memory-efficient, and more accurate than the other algorithms.
Conclusion
We develop a guideline to select an appropriate PCA implementation based on the differences in the computational environment of users and developers.
Keywords: Single-cell RNA-seq, Cellular heterogeneity, Dimension reduction, Principal component analysis, Online/incremental algorithm, Randomized algorithm, Out-of-core, Sparse data format, R, Python, Julia
Background
The emergence of single-cell RNA sequencing (scRNA-seq) technologies [1] has enabled the examination of many types of cellular heterogeneity. For example, cellular subpopulations consisting of various tissues [2–6], rare cells and stem cell niches [7], continuous gene expression changes related to cell cycle progression [8], spatial coordinates [9–11], and differences in differentiation maturity [12, 13] have been captured by many scRNA-seq studies. As the measurement of cellular heterogeneity is highly dependent on the number of cells measured simultaneously, a wide variety of large-scale scRNA-seq technologies have been developed [14], including those using cell sorting devices [15–17], Fludigm C1 [18–21], droplet-based technologies (Drop-Seq [2–4], inDrop RNA-Seq [5, 6], the 10X Genomics Chromium system [22]), and single-cell combinatorial-indexing RNA-sequencing (sci-RNA-seq [23]). Such technologies have encouraged the establishment of several large-scale genomics consortiums, such as the Human Cell Atlas [24–26], Mouse Cell Atlas [27], and Tabula Muris [28]. These projects are analyzing a tremendous number of cells by scRNA-seq and tackling basic life science problems such as the number of cell types comprising an individual, cell-type-specific marker gene expression and gene functions, and molecular mechanisms of diseases at a single-cell resolution.
Nevertheless, the analysis of scRNA-seq datasets poses a potentially difficult problem; the cell type corresponding to each data point is unknown a priori [1, 29–35]. Accordingly, researchers perform unsupervised machine learning (UML) methods, such as dimensionality reduction and clustering, to reveal the cell type corresponding to each individual data point. In particular, principal component analysis (PCA [36–38]) is a commonly used UML algorithm applied across many situations.
Despite its wide use, there are several reasons why it is unclear how PCA should be conducted for large-scale scRNA-seq. First, because the widely used PCA algorithms and implementations load all elements of a data matrix into memory space, for large-scale datasets such as the 1.3 million cells measured by 10X Genomics Chromium [39] or the 2 million cells measured by sci-RNA-seq [23], the calculation is difficult unless the memory size of the user’s machine is very large. Furthermore, the same data analysis workflow is performed repeatedly, with deletions or additions to the data or parameter changes for the workflow, and under such trial-and-error cycles, PCA can become a bottleneck for the workflow. Therefore, some fast and memory-efficient PCA algorithms are required.
Second, there are indeed some PCA algorithms that are fast and memory-efficient, but their practicality for use with large-scale scRNA-seq datasets is not fully understood. Generally, there are trade-offs between the acceleration of algorithms by some approximation methods and the accuracy of biological data analysis. Fast PCA algorithms might overlook some important differential gene expression patterns. In the case of large-scale scRNA-seq studies aiming to find novel cell types, this property may cause a loss of clustering accuracy and not be acceptable.
Finally, actual computational time and memory efficiency are highly dependent on the specific implementation, including the programming language, the method for loading input files, and the data format. However, there is no benchmarking to evaluate these properties. Such information is directly related to the practicality of the software and is useful as a guideline for users and developers.
For the above reasons, in this research, we examine the practicality of fast and memory-efficient PCA algorithms for use with large-scale scRNA-seq datasets. This work provides four key contributions. First, we review the existing PCA algorithms and their implementations (Fig. 1). Second, we present a benchmark test with selected PCA algorithms and implementations. To our knowledge, this is the first comprehensive benchmarking of PCA algorithms and implementations with large-scale scRNA-seq datasets. Third, we provide some original implementations of some PCA algorithms and utility functions for quality control (QC), filtering, and feature selection. All commands are implemented in a fast and memory-efficient Julia package. Finally, we propose guidelines for end-users and software developers.
Fig. 1.

Overview of benchmarking in this work. a Schematic overview of this work. b Evaluation metrics of the benchmarking with real-world datasets. c Evaluation metrics of the benchmarking with synthetic datasets
Results
Review of PCA algorithms and implementations
PCA is widely used for data visualization [39–41], data QC [42], feature selection [13, 43–49], de-noising [50, 51], imputation [52–54], confirmation and removal of batch effects [55–57], confirmation and estimation of cell-cycle effects [58], rare cell type detection [59, 60], cell type and cell state similarity search [61], pseudotime inference [13, 62–66], and spatial reconstruction [9].
Additionally, principal component (PC) scores are also used as the input of other non-linear dimensionality reduction [67–73] and clustering methods [74–77] in order to preserve the global structure, avoid the “curse of dimensionality” [78–81], and save memory space. A wide variety of scRNA-seq data analysis tools actually include PCA as an internal function or utilize PC scores as input for downstream analyses [22, 82–89].
We reviewed the existing PCA algorithms and implementations and classified the algorithms into six categories, namely similarity transformation-based (SimT), downsampling-based (DS), singular value decomposition (SVD) update-based (SU), Krylov subspace-based (Krylov), gradient descent-based (GD), and random projection-based (Rand) (Additional file 1 [22, 42–44, 49–52, 55–61, 63, 65, 69, 74–77, 82, 85, 89–113]). We have listed 21 PCA implementations (comprising 10 algorithms) that are freely available and easy to download, install, and use for analyses. The correspondence of the reviewed PCA implementations and scRNA-seq studies is summarized in Table 1.
Table 1.
Use cases of PCA implementations in scRNA-seq studies
| scRNA-seq studies | PCA algorithms | Commands or functions used in the studies |
|---|---|---|
| In most cases [13, 42, 43, 51, 52, 55, 56, 58, 60, 63, 65, 74, 77, 82, 85, 91, 93] | Golub-Kahan method | prcomp/svd (R) PCA (Python, sklearn) |
| Bhaduri et al. [94] | Downsampling | Unknown |
| Loompy [93] | SKL | IncrementalPCA (Python, sklearn) |
| Scanpy [93] | IRLBA | PCA (Python, sklearn) |
| SKL | IncrementalPCA (Python, sklearn) | |
| Halko’s method | TruncatedSVD (Python, sklearn) | |
| Cell Ranger [22] | IRLBA | irlb (Python, from scratch) |
| Seurat2 [49] | IRLBA | irlba (R, irlba) |
| Scran [50] | Golub-Kahan method | svd (R) |
| IRLBA | irlba (R, irlba) | |
| SAFE [76] | IRLBA | irlba (R, irlba) |
| MAGIC [52] | Golub-Kahan method | svds (MATLAB) |
| Halko’s method | randPCA (MATLAB, from scratch) | |
| Halko’s method | PCA (Python, sklearn) | |
| Harmony [57] | IRLBA | irlba (R, irlba) |
| Scater [82] | Golub-Kahan method | prcomp (R) |
| IRLBA | irlba (R, irlba) | |
| GiniClust2 [59] | IRLBA | propack.svd (R, svd) |
| SIMLR[75] | Halko’s method | fast.rsvd (R, from scratch) |
| SEQC[89] | Golub-Kahan method | PCA (Python, sklearn) |
| Halko’s method | PCA (Python, sklearn) | |
| CellFishing.jl [61] | Li’s method | rsvd (Julia, from scratch) |
To extend the scope of the algorithms used in the benchmarking, we originally implemented some PCA algorithms in an out-of-core manner (Additional file 1). The pseudocode and source code of all the algorithms benchmarked in this study are summarized in Additional files 2 and 3, respectively.
Benchmarking of PCA algorithms and implementations
Next, we performed the benchmarking tests of the PCA algorithms and implementations. The results of the benchmarking are summarized in Fig. 2 [69, 90, 92, 94–99, 107–109, 114, 115].
Fig. 2.

Summary of results. a Theoretical properties summarized by our literature review. b Properties related to each implementation. c Performance evaluated by benchmarking with real-world and synthetic datasets. d User-friendliness evaluated by some metrics
Real-world datasets
In consideration of the trade-offs among the large number of methods evaluated with our limited time, computational resources, and manpower, we carefully selected real-world datasets for the benchmarking. The latest scRNA-seq methods are divided into two categories, namely full-length scRNA-seq methods and high-throughput scRNA-seq methods with specific cell dissociation and cellular/molecular barcoding technologies such as droplet-based and split-and-pool experiments [34, 35]. Because the number of cells measured by scRNA-seq has been increased by the latter technology, we selected the following four datasets generated by such technologies: human peripheral blood mononuclear cells (PBMCs), human pancreatic cells (Pancreas), mouse brain and spinal cord (BrainSpinalCord), and mouse cells from the cortex, hippocampus, and ventricular zone (Brain) (Table 2). These datasets have been used in many previous scRNA-seq studies [61, 76, 94, 116–122].
Table 2.
Real-world datasets for benchmarking
| Dataset | No. of genes | No. of cells | No. of cell types | PCs used | File size (LogCPMED, CSV) | File size (count, CSV) | File size (count, binary) |
|---|---|---|---|---|---|---|---|
| PBMCs | 17,484 | 713 | 6 | PC1–3 | 45 MB | 24 MB | 2.1 MB |
| Pancreas | 17,499 | 3605 | 14 | PC1–12 | 530 MB | 287 MB | 22 MB |
| BrainSpinalCord | 25,893 | 156,049 | 73 | PC1–16 | 9.3 MB | 7.5 GB | 197 MB |
| Brain | 18,782 | 130,6127 | 60 | PC1–20 | 290 GB | 58 GB | 3.2 GB |
The accuracy of PCA algorithms
Here, we evaluate the accuracy of the various PCA algorithms by using the four real-world datasets. For the analyses of the PBMCs and Pancreas datasets, we set the result of prcomp as the gold standard, which is a wrapper function for performing SVD with LAPACK subroutines (Additional file 1). The other implementations are compared with this result (Figs. 1b and 2). For the BrainSpinalCord and Brain dataset analyses, full-rank SVD by LAPACK is computationally difficult. According to the benchmarking guidelines developed by Mark D. Robinson’s group [123], comparing the methods against each other is recommended when the ground truth cannot be defined. Therefore, we just compared the results of the methods against each other using several different criteria, such as the magnitude of the eigenvalues and the clustering accuracy.
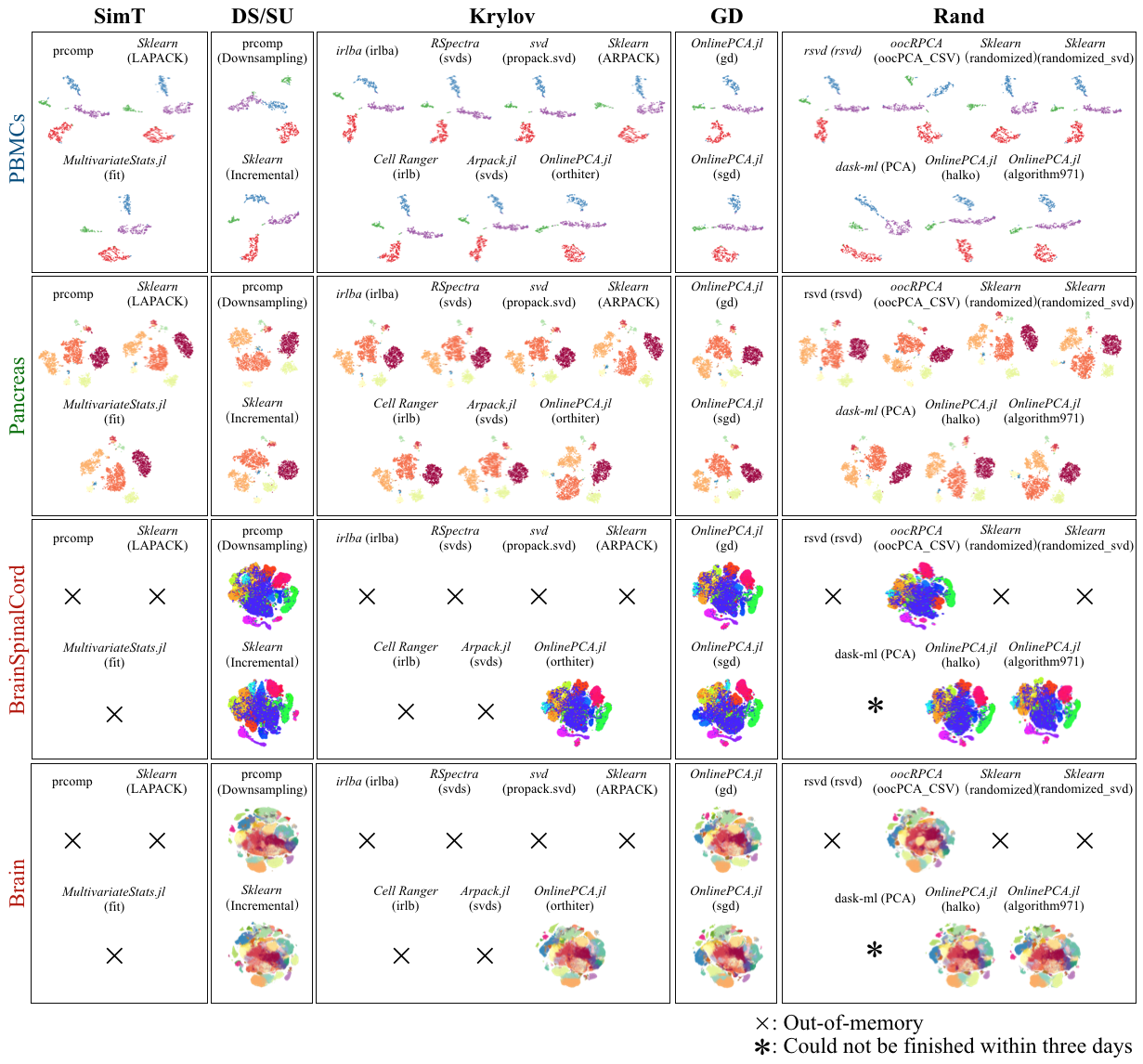
First, we performed t-stochastic neighbor embedding (t-SNE [67, 68]) and uniform manifold approximation and projection (UMAP [71, 72]) for the results of each PCA algorithm and compared the clarity of the cluster structures detected by the original studies (Figs. 1b and 3, Additional files 4, and 5). For the BrainSpinalCord and Brain datasets, only downsampling, IncrementalPCA (sklearn), orthiter/gd/sgd/halko/algorithm971 (OnlinePCA.jl), and oocPCA_CSV (oocRPCA) could be performed, while the other implementations were terminated by out-of-memory errors on 96 and 128 GB RAM machines. For the PBMCS and Pancreas datasets, compared with the gold standard cluster structures, the structures detected by downsampling were unclear, and some distinct clusters determined by the original studies were incorrectly combined into single clusters (red circled cluster in Fig. 3). In the realistic situation when the cellular labels were unavailable a priori, the labels were exploratorily estimated by confirming differentially expressed genes, known marker genes, or related gene functions of clusters. In such a situation, downsampling may overlook subgroups hiding in a cluster.
Fig. 3.

The comparison of t-stochastic neighbor embedding (t-SNE) plots. Comparison of multiple principal component analysis (PCA) implementations performed with empirical datasets: PBMCs (102 cells), Pancreas (103 cells), BrainSpinalCord (105 cells), and Brain datasets (106 cells). t-SNE was performed with the result of each PCA implementation. Some distinct clusters determined by the original studies were incorrectly combined into single clusters (red circled cluster)
We also performed four clustering algorithms on all the results of the PCA implementations and calculated the adjusted Rand index (ARI [124]) to evaluate clustering accuracy (Additional file 6). Here, we only show the result of Louvain clustering [125] (Figs. 1b and 4). The ARI values show that the results of downsampling and sgd (OnlinePCA.jl) were worse compared with the gold standard or other implementations.
Fig. 4.

Clustering accuracy comparison. Clustering accuracy was evaluated by the adjusted Rand index (ARI) of the result of Louvain clustering. Multiple principal component analysis (PCA) implementations were performed for PBMCs (102 cells), Pancreas (103 cells), BrainSpinalCord (105 cells), and Brain datasets (106 cells); Louvain clustering was performed for the PCA results. For each PCA result, Louvain clustering calculations were performed ten times and the average values were calculated. The cluster labels are the same as those of the respective original papers
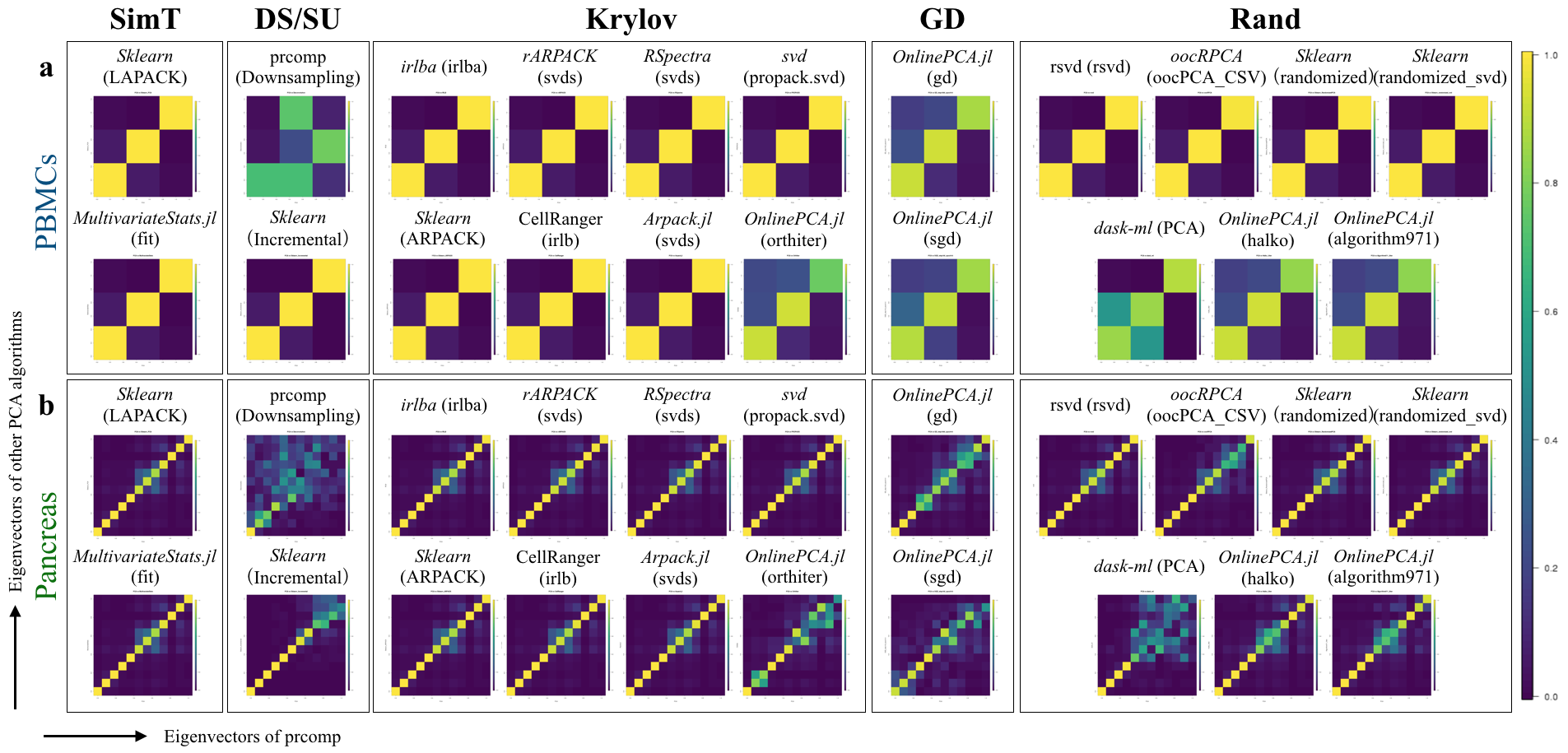
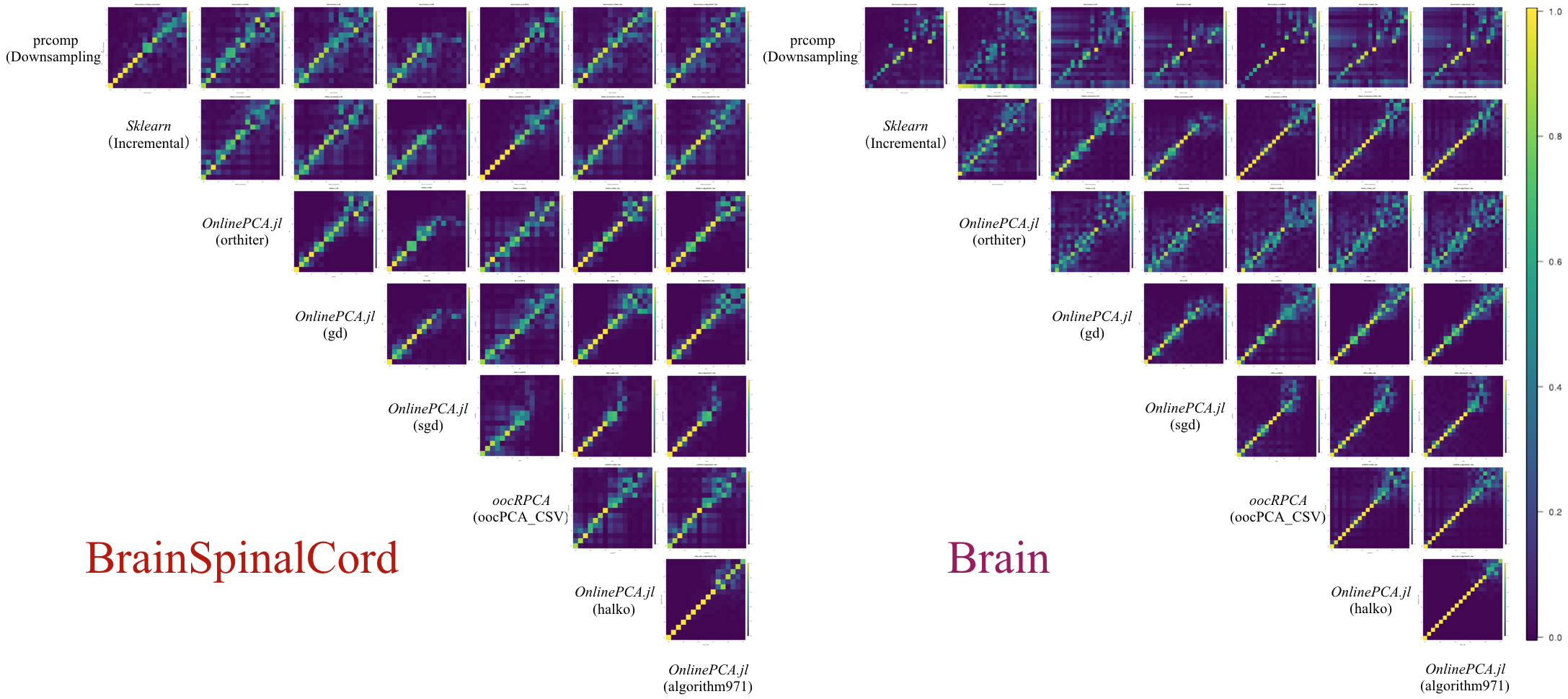
Next, we performed an all-to-all comparison between PCs from the gold standard and the other PCA implementations (Figs. 1b and 5a, and Additional file 7). Because the PCs are unit vectors, when two PCs are directed in the same or opposite direction, their cross product becomes 1 or − 1, respectively. Both the same and opposite direction vectors are mathematically identical in PCA optimization, and different PCA implementations may yield PCs with different signs. Accordingly, we calculated the absolute value of the cross product ranging from 0 to 1 for the all-to-all comparison and evaluated whether higher PCs, which correspond to lower eigenvalues, are accurately calculated. Figure 5a and Additional file 7 show that the higher PCs based on downsampling, orthiter/gd/sgd (OnlinePCA.jl), and PCA (dask-ml [115]) become inaccurate as the dimensionality of a PC increases. The higher PCs of these implementations also appear noisy and unclear in pair plots of PCs between each implementation and seem uninformative (Additional files 8, 9, 10, and 11). In particular, the higher PCs calculated by downsampling and sgd (OnlinePCA.jl) are sometimes influenced by the existence of outlier cells (Additional file 8 and Additional file 9). When performing some clustering methods, such as k-means and Gaussian mixture model (GMM [126]) methods, such outlier cells are also detected as singleton clusters having only a single cell as their cluster member (Additional file 12). Contrary to these results, all the implementations of IRLBA and IRAM, as well as the randomized SVD approaches except for PCA (dask-ml), are surprisingly accurate regardless of the language in which they are written or their developers. Although PCA (dask-ml) is based on Halko’s method and is nearly identical to the other implementations of Halko’s method, this function uses the direct tall-and-skinny QR algorithm [127] (https://github.com/dask/dask/blob/a7bf545580c5cd4180373b5a2774276c2ccbb573/dask/array/linalg.py#L52), and this characteristic might be related to the inaccuracy of the implementations. Because there is no gold standard in the case of the BrainSpinalCord and Brain datasets, we compared the eigenvectors of the PCA implementations in all possible combinations (Additional file 13) and found that the higher PCs of downsampling and sgd differed from those of the other PCA implementations.
Fig. 5.

(a) Comparison of all combinations of eigenvectors. Absolute values of the cross products of all combinations between the eigenvectors of the gold standard methods and those of the other principal component analysis (PCA) implementations were calculated. The closer the value is to 1 (yellow), the closer the two corresponding eigenvectors are to each other. If two PCA results are equal without considering differences in sign, the matrix in this figure becomes an identity matrix. (b) Comparison of all combinations of loading vectors. Number of intersection elements between the top 500 largest absolute value elements of the gold standard methods and those of the other PCA implementations were calculated. The closer the value is to 500 2 (yellow), the closer the two corresponding loading vectors are to each other. If two PCA results are equal without considering differences in their sign, all the diagonal elements of the matrix in this figure become 500
Because gene-wise eigenvectors (i.e., loading vectors) are also retrieved from the data matrix and cell-wise eigenvectors (i.e., PCs), we also compared the loading vectors (Fig. 5b and Additional file 14). We extracted the top 500 genes in terms of the largest absolute values of loading vectors and calculated the number of genes in common between the two loading vectors. As is the case with the eigenvectors, even for loading vectors, downsampling, orthiter/gd/sgd (OnlinePCA.jl), and PCA (dask-ml [115]) become inaccurate as the dimensionality of the PC increases. Because the genes with large absolute values for loading vectors are used as feature values in some studies [43–48], inaccurate PCA implementations may lower the accuracy of such an approach.
The distributions of the eigenvalues of downsampling, IncrementalPCA (sklearn), and sgd (OnlinePCA.jl) also differ from those of the other implementations (Fig. 6).
Fig. 6.

Comparison of eigenvalues. Distribution of eigenvalues of four real datasets used in this work ((a) PBMCs, (b) Pancreas, (c) BrainSpincalCord, and (d) Brain). The x-axis means the principal components of each principal component analysis (PCA) implementation, and the y-axis means the corresponding eigenvalues
Calculation time, memory usage, and scalability
We compared the computational time and memory usage of all the PCA implementations (Fig. 7). For the BrainSpinalCord dataset, downsampling itself was faster than most of the PCA implementations, but other preprocessing steps, such as matrix transposition and multiplication of the transposed data matrix and loading vectors to calculate PCs, were slow and had high memory space requirements (Additional file 3). For the Brain dataset, downsampling became slower than most of the PCA implementations, and such a tendency is noticeable as the size of the data matrix increases, because downsampling is based on the full-rank SVD in LAPACK.
Fig. 7.

Comparison of the elapsed time and maximum memory usage for empirical datasets. a The elapsed time of preprocessing steps such as binalization and normalization (orange bar) and the elapsed time of each PCA calculation itself (green bar). Only when performing the PCA implementations to the Brain dataset, we used our in-house Julia script to preprocess. This is because this dataset cannot be loaded to the memory space as a data.frame of R language. b The memory usage of all principal component analysis (PCA) implementations calculated for each empirical dataset (blue bar)
We also found that the calculation time of PCA (dask-ml) was not as fast in spite of its out-of-core implementation; for the BrainSpinalCord and Brain datasets, this implementation could not finish the calculation within 3 days in our computational environment. The other out-of-core PCA implementations, such as IncrementalPCA (sklearn), orthiter/gd/sgd/halko/algorithm971 (OnlinePCA.jl), and oocPCA_CSV (oocRPCA), were able to finish those calculations.
We also systemically estimated the calculation time, memory usage, and scalability of all the PCA implementations using 18 synthetic datasets consisting of {102, 103, 104} gene ×{102, 103, 104, 105, 106, 107} cell matrices (see the “Materials and methods” section). We evaluated whether the calculations could be finished or were interrupted by out-of-memory errors (Fig. 1b). We also manually terminated a PCA process that was unable to generate output files within 3 days (i.e., dask-ml). All the terminated jobs are summarized in Additional file 15. To evaluate only the scalability and computability, we set the number of epochs (also known as passes) in orthiter/gd/sgd (OnlinePCA.jl) to one. However, in actual data analysis, a value several times larger should be used.
Additional files 16 and 17 show the calculation time and the memory usage of all the PCA implementations, which can be scaled to a 104×107 matrix. IncrementalPCA (sklearn) and oocPCA_CSV (oocRPCA) were slightly slower than the other implementations (Additional file 16), and this was probably because the inputs of these implementations were CSV files while the other implementations used compressed binary files (Zstd). The memory usage of all the implementations was almost the same, except for IncrementalPCA (sklearn) and oocPCA_CSV (oocRPCA). oocPCA_CSV (oocRPCA) has a parameter that controls the maximum memory usage (mem), and we set the value to 10 GB (Additional file 3). Indeed, the memory usage had converged to around 10 GB (Additional file 17). This property is considered an advantage of this implementation; users can specify a different value to suit their computational environment.
The relationship between file format and performance
We also counted the passes of the Brain matrix in the out-of-core implementations such as oocPCA_CSV (R, oocRPCA), IncrementalPCA (Python, sklearn), and orthiter/gd/sgd/halko/algorithm971 (Julia, OnlinePCA.jl) (Additional file 18a). In the oocPCA_CSV (R, oocRPCA) and IncrementalPCA (Python, sklearn), the data matrix was passed to these function as the CSV format, and in the other out-of-core implementations, the data matrix was firstly binarized and compressed in the Zstd file format. We found that the calculation time was correlated with the number of passes of the implementation. Furthermore, binarizing and data compression substantially accelerated the calculation time. This suggests that the data loading process is very critical for out-of-core implementation and that the overhead for this process has a great effect on the overall calculation time and memory usage.
Accordingly, using different data formats, such as CSV, Zstd, Loom [93], and hierarchical data format 5 (HDF5), provided by the 10X Genomics (10X-HDF5) for the Brain dataset, we evaluated the calculation time and the memory usage for the simple one-pass orthogonal iteration (qr(XW)), where qr is the QR decomposition, X is the data matrix, and W represents the 30 vectors to be estimated as the eigenvectors (Additional file 18b). For this algorithm, incremental loading of large block matrices (e.g., 5000 rows) from a sparse matrix was faster than incremental loading of row vectors from a dense matrix, although the memory usage of the former was lower.
While it is not obvious that the usage of a sparse matrix accelerates the PCA with scRNA-seq datasets because scRNA-seq datasets are not particularly sparse compared with data from other fields (cf. recommender systems or social networks [128, 129]), we showed that it has the potential to speed up the calculation time for scRNA-seq datasets.
When all row vectors stored in 10X-HDF5 are loaded at once, the calculation is fastest, but the memory usage is also highest. Because the calculation time and the memory usage have a trade-off and the user’s computational environment is not always high-spec, the block size should be optionally specified as a command argument. For the above reasons, we also developed tenxpca, which is a new implementation that performs Li’s method for a sparse matrix stored in the 10X-HDF5 format. Using all the genes in the CSC matrix incrementally, tenxpca was able to finish the calculation in 1.3 h with a maximum memory usage of 83.0 GB. This is the fastest analysis of the Brain dataset in this study.
In addition to tenxpca, some algorithms used in this benchmarking, such as orthogonal iteration, GD, SGD, Halko’s method, and Li’s method, are implemented as Julia functions and command line tools, which have been published as a Julia package OnlinePCA.jl (Additional file 19). When data are stored as a CSV file, they are binarized and compressed in the Zstd file format (Additional file 19a), and then, some out-of-core PCA implementations are performed. When data are in 10X-HDF5 format, Li’s method is directly performed with the data by tenxpca (Additional file 19b). We also implemented some functions and command line tools to extract row-wise/column-wise statistics such as mean and variance as well as highly variable genes (HVGs) [130] in an out-of-core manner. Because such statistics are saved as small vectors, they can be loaded by any programming language without out-of-core implementation and used for QC, and the users can select only informative genes and cells. After QC, the filtering command removes low-quality genes/cells and generates another Zstd file.
Discussion
Guidelines for users
Based on all the benchmarking results and our implementation in this work, we propose some user guidelines (Fig. 8). Considering that bioinformatics studies combine multiple tools to construct a user’s specific workflow, the programming language is an important factor in selecting the right PCA implementation. Therefore, we categorized the PCA implementations according to language (i.e., R [111], Python [112], and Julia [113]; Fig. 8, column-wise). In addition to the data matrix size, we also categorized implementations according to the way they load data (in-memory or out-of-core) as well as their input matrix format (dense or sparse, Fig. 8, row-wise). Here, we define the GC value of a data matrix as the number of genes × the number of cells.
Fig. 8.

User guidelines. Recommended PCA implementations categorized based on written language and matrix size. The recommended parameter of each PCA implementation is also described (red)
If the data matrix is not too large (e.g., GC ≤107), the data matrix can be loaded as a dense matrix, and full-rank SVD in LAPACK is then accurate and optimal (in-memory and dense matrix). In such a situation, the wrapper functions for the full-rank SVD written in each language are suitable. However, if the data matrix is much larger (e.g., GC ≥108), an alternative to the full-rank SVD is needed. Based on the benchmarking results, we recommend IRLBA, IRAM, Halko’s method, and Li’s method as alternatives to the full-rank SVD. For intermediate GC values (108≤ GC ≤1010), if the data matrix can be loaded into memory as a sparse matrix, some implementations for these algorithms are available (in-memory and sparse matrix). In particular, such implementations are effective for large data matrices stored in 10X-HDF5 format using CSC format. Seurat2 [49] also introduces this approach by combining the matrix market format (R, Matrix) and irlba function (R, irlba). When the data matrix is dense and cannot be loaded into memory space (e.g., GC ≥1010), the out-of-core implementations, such as oocPCA_CSV (R, oocRPCA), IncrementalPCA (Python, sklearn), and algorithm971 (Julia, OnlinePCA.jl), are useful (dense matrix and out-of-core). If the data matrix is extremely large and cannot be loaded into memory even if the data are formatted as a sparse matrix, out-of-core PCA implementations for sparse matrix are needed. Actually, R cannot load the Brain dataset, even if the data is formatted as a sparse matrix (https://github.com/satijalab/seurat/issues/1644). Hence, in such a situation, tenxpca can be used if the data is stored in the 10X-HDF5 format.
The PCA implementations examined in this work are affected by various parameters. For example, in gd and sgd (OnlinePCA.jl), the result is sensitive to the value of learning parameters and the number of epochs. Therefore, a grid-search of such parameters is necessary (Additional file 20). When using IncrementalPCA (sklearn), the user specifies the chunk size of the input matrix, and a larger value slightly improves the accuracy of PCA (Additional file 21) and the calculation time (Additional file 16), although there is a trade-off between these properties and memory usage (Additional file 17). Both Halko’s method and Li’s method have a parameter for specifying the number of power iterations (niter), and this iteration step sharpens the distribution of eigenvalues and enforces a more rapid decay of singular values ([114] and Additional file 3). In our experiments, the value of niter is critical for achieving accuracy, and we highly recommend a niter value of three or larger (Additional file 22). In some implementations, the default values of the parameters are specified as inappropriate values or cannot be accessed as a function parameter. Therefore, users should carefully set the parameter or select an appropriate implementation.
Guidelines for developers
We have also established guidelines for developers. Many technologies such as data formats, algorithms, and computational frameworks and environments are available for developing fast, memory-efficient, and scalable PCA implementations (Additional file 23). Here, we focus on two topics.
The first topic is “loss of sparsity.” As described above, the use of a sparse matrix can effectively reduce memory space and accelerate calculation, but developers must be careful not to destroy the sparsity of a sparse matrix. PCA with a sparse matrix is not equivalent to SVD with a sparse matrix; in PCA, all sparse matrix elements must be centered by the subtraction of gene-wise average values. Once the sparse matrix X is centered (X−Xmean), where Xmean has gene-wise average values as column vectors, it becomes a dense matrix and the memory usage is significantly increased. Obviously, the explicit calculation of the subtraction described above should be avoided. In such a situation, if multiplication of this centered matrix and a dense vector/matrix is required, the calculation should be divided into two parts, such as (X−Xmean)W=XW−XmeanW, where W represents the vectors to be estimated as eigenvectors, and these parts should be calculated separately. If one or both parts require more than the available memory space, such parts should be incrementally calculated in an out-of-core manner. There are actually some PCA implementations that can accept a sparse matrix, but they may require very long calculation times and large memory space because of a loss of sparsity (cf. rpca of rsvd https://github.com/cran/rsvd/blob/7a409fe77b220c26e88d29f393fe12a20a5f24fb/R/rpca.R#L158https://github.com/cran/rsvd/blob/7a409fe77b220c26e88d29f393fe12a20a5f24fb/R/rpca.R#L158). To our knowledge, only prcomp_irlba in irlba (https://github.com/bwlewis/irlba/blob/8aa970a7d399b46f0d5ad90fb8a29d5991051bfe/R/irlba.R#L379https: //github.com/bwlewis/irlba/blob/8aa970a7d399b46f0d5ad90fb8a29d5991051bfe/R/irlba.R#L379), irlb in Cell Ranger (https://github.com/10XGenomics/cellranger/blob/e5396c6c444acec6af84caa7d3655dd33a162852/lib/python/cellranger/analysis/irlb.py#L118), safe_sparse_dot in sklearn (https://scikit-learn.org/stable/modules/generated/sklearn.utils.extmath.safe_sparse_dot.htmlhttps://scikit-learn.org/stable/modules/generated/sklearn.utils.extmath.safe_sparse_dot.html), and tenxpca in OnlinePCA.jl (https://github.com/rikenbit/OnlinePCA.jl/blob/c95a2455acdd9ee14f8833dc5c53615d5e24b5f1/src/tenxpca.jl#L183https://github.com/rikenbit/OnlinePCA.jl/blob/c95a2455acdd9ee14f8833dc5c53615d5e24b5f1/src/tenxpca.jl#L183https://github.com/rikenbit/OnlinePCA.jl/blob/c95a2455acdd9ee14f8833dc5c53615d5e24b5f1/src/tenxpca.jl#L183) deal with this issue. Likewise, as an alternative to the centering calculation, MaxAbsScaler in sklearn (https://scikit-learn.org/stable/modules/generated/sklearn.preprocessing.MaxAbsScaler.html) introduces a scaling method in which the maximum absolute value of each gene vector becomes one, thereby avoiding the loss of sparsity.
The second topic is “lazy loading.” The out-of-core PCA implementations used in this benchmarking explicitly calculate centering, scaling, and all other relevant arithmetic operations from the extracted blocks of the data matrix. However, to reduce the complexity of the source code, it is desirable to calculate such processes as if the matrix was in memory and only when the data are actually required, so the processes are lazily evaluated on the fly. Some packages, such as DeferredMatrix in BiocSingular (R/Bioconductor, https://bioconductor.org/packages/devel/bioc/html/BiocSingular.html), CenteredSparseMatrix (Julia, https://github.com/jsams/CenteredSparseMatrix), Dask [115] (Python, https://dask.org), and Vaex (Python, https://vaex.io/), support lazy loading.
Future perspective
In this benchmarking study, we found that PCA implementations based on full-rank SVD are accurate but cannot be scaled for use with high-throughput scRNA-seq datasets such as the BrainSpinalCord and Brain datasets, and alternative implementations are thus required. Some methods approximate this calculation by using truncated SVD forms that are sufficiently accurate as well as faster and more memory-efficient than full-rank SVD. The actual memory usage highly depends on whether an algorithm is implemented as out-of-core and whether sparse matrix can be specified as input. Some sophisticated implementations, including our OnlinePCA.jl, can handle such issues. Other PCA algorithms, such as downsampling and SGD, are actually not accurate, and their use risks overlooking cellular subgroups contained within scRNA-seq datasets. These methods commonly update eigenvectors with small fractions of the data matrix, and this process may overlook subgroups or subgroup-related gene expression, thereby causing the observed inaccuracy. Our literature review, benchmarking, special implementation for scRNA-seq datasets, and guidelines provide important resources for new users and developers tackling the UML of high-throughput scRNA-seq.
Although the downstream analyses of PCA vary widely, and we could not examine all the topics of scRNA-seq analyses, such as rare cell-type detection [59, 60] and pseudotime analysis [13, 62–66], differences among PCA algorithms might also affect the accuracy of such analyses. Butler et al. showed batch effect removal can be formalized as canonical correlation analysis (CCA) [49], which is mathematically very similar to PCA. The optimization of CCA is also formalized in various ways, including randomized CCA [131] or SGD of CCA [132].
This work also sheds light on the effectiveness of randomized SVD. This algorithm is popular in population genetic studies[110]. In the present study, we also assessed its effectiveness with scRNA-seq datasets with high heterogeneity. This algorithm is relatively simple, and some studies have implemented it from scratch (Table 1). Simplicity may be the most attractive feature of this algorithm.
There are also many focuses of recent PCA algorithms (Additional file 23). The randomized subspace iteration algorithm, which is a hybrid of Krylov and Rand methodologies, was developed based on randomized SVD [133, 134]. In pass-efficient or one-pass randomized SVD, some tricks to reduce the number of passes have been considered [135, 136]. TeraPCA, which is a software tool for use in population genetics studies, utilizes the Mailman algorithm to accelerate the expectation–maximization algorithms for PCA [137, 138]. Townes et al. recently proposed the use of PCA for generalized linear models (GLM-PCA) and unified some PCA topics, such as log-transformation, size factor normalization, non-normal distribution, and feature selection, in their GLM framework [139, 140]. Although such topics are beyond the scope of the present work, the current discussion will be useful for the development and application of such methods above.
Materials and methods
Benchmarking procedures
Assuming digital expression matrices of unique molecular identifier (UMI) counts, all the data files, including real and synthetic datasets, were in CSV format. When using the Brain dataset, the matrix stored in 10X-HDF5 format was converted to CSV using our in-house Python script [141].
After being loaded by each PCA implementation, the raw data matrix Xraw was converted to normalized values by count per median (CPMED [142–144]) normalization according to the formula , where M is the number of columns and Libsize is the column-wise sum of counts of X. After normalization, Xcpmed was transformed to X by the logarithm-transformation X= log10(Xcpmed+1), where log10 is the element-wise logarithm. In all the randomized PCA implementation, random seed was fixed.
When Xraw was extremely large and could not be loaded into the memory space all at once, we prepared two approaches to perform PCA with X. When PCA implementations are orthiter, gd, sgd, halko, or algorithm971 (OnlinePCA.jl), each row vector of Xraw is normalized using the pre-calculated Libsize by the sumr command, then log-transformed, and finally used for each of the PCA algorithms. When using other out-of-core PCA implementations such as IncrementalPCA (sklearn), oocPCA_CSV (oocRPCA), or PCA (dask-ml), there is no option to normalize and log-transform each row vector of Xraw, so we first calculated Xcpmed using our in-house Python script [141], which was then used for the input matrix of the PCA implementations.
We also investigated the effect of differences in normalization methods on the PCA results (Additional file 25). When performing each PCA implementation based on the truncated SVD, the number of PCs was specified in advance (Table 2).
Although it is unclear how many cells should be used in downsampling, one empirical analysis [94] suggests that 20,000 to 50,000 cells are sufficient for clustering and detecting subpopulations in the Brain dataset. Thus 50,000/1,300,000×100=3.8% of cells were sampled from each dataset and used for the downsampling method. When performing IncrementalPCA (sklearn), the row vectors, which match the number of PCs, were extracted until the end of the lines of the files. When performing irlb (Cell Ranger), the loaded dataset was first converted to a scipy sparse matrix and passed to it because this function supports sparse matrix data stored in 10X-HDF5 format. When performing the benchmark, conversion time and memory usage were also recorded. When performing all the functions of OnlinePCA.jl, including orthiter/gd/sgd/halko/algorithm971, we converted the CSV data to Zstd format, and the calculation time and the memory usage were recorded in the benchmarking for fairness. For orthiter, gd, and sgd (OnlinePCA.jl), calculations were performed until they converged (Additional file 20). For all the randomized SVD implementations, the niter parameter value was set to 3 (Additional file 22). When performing oocPCA_CSV, the users can also use oocPCA_BIN, which performs PCA with binarized CSV files. The binarization is performed by the csv2binary function, which is also implemented in the oocRPCA package. Although data binarization accelerates the calculation time for PCA itself, we confirmed that csv2binary is based on in-memory calculation, and in our computing environment, csv2binary was terminated by an out-of-memory error. Accordingly, we only used oocPCA_CSV, and the CSV files were directly loaded by this function.
Computational environment
All computations were performed on two-node machines with Intel Xeon E5-2697 v2 (2.70 GHz) processors and 128 GB of RAM, four-node machines with Intel Xeon E5-2670 v3 (2.30 GHz) processors and 96 GB of RAM, and four-node machines with Intel Xeon E5-2680 v3 (2.50 GHz) processors and 128 GB of RAM. Storage among machines was shared by NFS, connected using InfiniBand. All jobs were queued by the Open Grid Scheduler/Grid Engine (v2011.11) in parallel. The elapsed time and maximum memory usage were evaluated using the GNU time command (v1.7).
Reproducibility
All the analyses were performed on the machines described above. We used R v3.5.0, Python v3.6.4, and Julia v1.0.1 in the benchmarking; for t-SNE and CSV conversion of the Brain dataset, we used Python v2.7.9. The Sklearn (Python) package was used to perform k-means and GMM clustering methods. The igraph (R), nn2 (R), and Matrix (R) packages were used to perform Louvain clustering (Additional file 6). The hdbscan (Python) package was used to perform HDBScan clustering. The bhtsne (Python) package was used to perform t-SNE. Lastly, the umap (Python) package was used to perform UMAP. All the programs used to perform the PCA implementations in the benchmarking are summarized in Additional file 3.
Supplementary information
Additional file 1 Review of existing pCA algorithms and implementations.
Additional file 2 Pseudo-code of all the pCA algorithms.
Additional file 3 Source code of all the pCA implementations.
{kind=link}
Additional file 4 Results of t-SNE of all the pCA implementations.
{kind=link}
Additional file 5 Results of uMAP of all the pCA implementations.
Additional file 6 Results of clustering methods of all the pCA implementations.
{kind=link}
Additional file 7 Eigenvectors of all the pCA implementations (PBMCs and pancreas).
Additional file 8 Pair plots of all the pCA (PBMCs) implementations.
Additional file 9 Pair plots of all the pCA (Pancreas) implementations.
Additional file 10 Pair plots of all the pCA (BrainSpinalCord) implementations.
Additional file 11 Pair plots of all the pCA (Brain) implementations.
{kind=link}
Additional file 12 Number of singleton clusters.
{kind=link}
Additional file 13 Eigenvectors of all the pCA implementations (BrainSpinalCord and brain).
{kind=link}
Additional file 14 Loading vectors of all the pCA implementations (PBMCs and pancreas).
Additional file 15 Crashed jobs caused by out-of-memory errors.
{kind=link}
Additional file 16 Comparison of the elapsed time for simulated datasets.
{kind=link}
Additional file 17 Comparison of the maximum memory usage for simulated datasets.
{kind=link}
Additional file 18 Relationships of the algorithms/implementations, the number of passes, and the file format with the elapsed time for performing principal component analysis (PCA) with the brain dataset.
{kind=link}
Additional file 19 OnlinePCA.jl schematic.
Additional file 20 Parameter tuning of the orthogonal iteration, gradient descent, and stochastic gradient descent implementations.
Additional file 21 Parameter tuning of the IncrementalPCA implementations.
Additional file 22 Parameter tuning of the randomized sVD implementations.
{kind=link}
Additional file 23 Developer guidelines.
Additional file 24 Effect of feature selection on clustering accuracy.
Additional file 25 Comparison of normalizing size factors.
Acknowledgements
We thank Mr. Akihiro Matsushima and Mr. Manabu Ishii for their assistance with the IT infrastructure for the data analysis. We are also grateful to all members of the Laboratory for Bioinformatics Research, RIKEN Center for Biosystems Dynamics Research, for their helpful advice. Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Peer review information: Yixin Yao was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Authors’ contributions
KT and HS surveyed the PCA algorithms and implementations. KT and IN designed the benchmarking test. KT and KS implemented the Julia program and performed all the analyses. KT retrieved and preprocessed the test dataset to evaluate the proposed method. All the authors have written, read, and approved the manuscript.
Funding
This work was supported by MEXT KAKENHI Grant Number 19K20406. This work was partially supported by the Japan Science and Technology Agency (JST), CREST grant numbers JPMJCR16G3 and JPMJCR1926, and the Projects for Technological Development, Research Center Network for Realization of Regenerative Medicine by Japan (18bm0404024h0001), the Japan Agency for Medical Research and Development (AMED).
Availability of data and materials
The script file used to perform the benchmarking is available at https://github.com/rikenbit/onlinePCA-experiments [145]. We published our original out-of-core PCA implementations such as orthiter (Orthogonal iteration), gd (GD), sgd (SGD), halko (Halko’s method), and algorithm971 (Li’s method), which are implemented as a Julia package OnlinePCA.jl [146].
The gene expression matrix and cell type labels for the PBMCs dataset and the Brain dataset [39] were downloaded from the 10X Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets/pbmc_1k_protein_v3 and https://support.10xgenomics.com/single-cell/datasets/1M_neurons, respectively). The gene expression matrix and cell type labels for the Pancreas dataset [40] and the BrainSpinalCord dataset [41] were retrieved from the GEO database (GSE84133 and GSE110823, respectively). For the Pancreas dataset, only the sample of GSM2230759 was used. The genes of all matrices with zero variance were removed because such genes are meaningless for PCA calculation. We also removed the ERCC RNA Spike-Ins, and the number of remaining genes and cells is summarized in Table 2. Additionally, we investigated the effect of feature selection on clustering accuracy (Additional file 24).
All count datasets were generated by the R rnbinom (random number based on a negative binomial distribution) function with shape and rate parameters of 0.4 and 0.3, respectively. Matrices of {102, 103, 104} genes ×{102, 103, 104, 105, 106, 107} cells were generated.
All the real and simulated datasets described above are available from our server [147] (the hash values of md5sum have been saved, last modified 18 November 2019, 18:00 (JST)).
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Koki Tsuyuzaki, Email: koki.tsuyuzaki@gmail.com.
Hiroyuki Sato, Email: hsato@amp.i.kyoto-u.ac.jp.
Kenta Sato, Email: bicycle1885@gmail.com.
Itoshi Nikaido, Email: itoshi.nikaido@riken.jp.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13059-019-1900-3.
References
- 1.Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25(10):1491–8. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly parallel genome-wide expression profiling of individual cells using nanoliter dropltes. Cell. 2015;161:1202–14. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis Z, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166:1308–23. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AMJ, Resch JM, McCarroll SA, Rosen ED, Lowell BB, Tsai LT. A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neurosci. 2017;20(3):484–96. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, Melton DA, Yanai I. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3(4):346–60. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, sasaki N, Clevers H, Oudenaarden A. Single-cell messenger rna sequencing reveals rare intestinal cell types. Nature. 2015;525:251–5. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 8.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell rna-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–60. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 9.Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, Heller S. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014;157:1–15. doi: 10.1016/j.cell.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC. High-throughput spatial mapping of single-cell rna-seq data to tissue of origin. Nat Comput Biol. 2015;33(5):503–9. doi: 10.1038/nbt.3209. [DOI] [PubMed] [Google Scholar]
- 11.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495–508. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapnell C, Cacchiarelli D, Grimsby J, Pokhare P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–82. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svensson V, Tormo RV, Teichmann SA. Exponential scaling of single-cell rna-seq in the past decade. Nat Protoc. 2017;13(4):599–604. doi: 10.1038/nprot.2017.149. [DOI] [PubMed] [Google Scholar]
- 15.Sasagawa Y, Danno H, Takada H, Ebisawa M, Tanaka K, Hayashi T, Kurisaki A, Nikaido I. Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. BMC Genome Biology. 2018;19(29). 10.1186/s13059-018-1407-3. [DOI] [PMC free article] [PubMed]
- 16.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single cell rna-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–9. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KL, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I. Cel-seq2: sensitive highly-multiplexed single-cell RNA-seq. BMC Genome Biol. 2016;17(77). 10.1186/s13059-018-1407-3. [DOI] [PMC free article] [PubMed]
- 18.Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, Manno GL, Juréus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S. Cell types in the mouse cortex and hippocampus revealed by single-cell rna-seq. Science. 2015;347(6226):1138–42. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 19.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I. Cel-seq2: sensitive highly-multiplexed single-cell rna-seq. Genome Biol. 2016;17(77). 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed]
- 20.Shalek Alex K., Satija Rahul, Shuga Joe, Trombetta John J., Gennert Dave, Lu Diana, Chen Peilin, Gertner Rona S., Gaublomme Jellert T., Yosef Nir, Schwartz Schraga, Fowler Brian, Weaver Suzanne, Wang Jing, Wang Xiaohui, Ding Ruihua, Raychowdhury Raktima, Friedman Nir, Hacohen Nir, Park Hongkun, May Andrew P., Regev Aviv. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510(7505):363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Pary S, Parry C, Lee C, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19(2):335–46. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwai R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(14049):1–12. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao Junyue, Spielmann Malte, Qiu Xiaojie, Huang Xingfan, Ibrahim Daniel M., Hill Andrew J., Zhang Fan, Mundlos Stefan, Christiansen Lena, Steemers Frank J., Trapnell Cole, Shendure Jay. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566(7745):496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consortium TH. The human cell atlas white paper. 2017.
- 25.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The human cell atlas: from vision to reality. Nature. 2017;550:451–3. doi: 10.1038/550451a. [DOI] [PubMed] [Google Scholar]
- 26.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, Clevers H, Deplancke B, Dunham I, Eberwine J, Eils R, Enard W, Farmer A, Fugger L, Göttgens B, Hacohen N, Haniffa M, Hemberg M, Kim S, Klenerman P, Kriegstein A, Lein E, Linnarsson S, Lundberg E, Lundeberg J, Majumder P, Marioni JC, Merad M, Mhlanga M, Nawijn M, Netea M, Nolan G, Pe’er D, Phillipakis A, Ponting CP, Quake S, Reik W, Rozenblatt-Rosen O, Sanes J, Satija R, Schumacher TN, Shalek A, Shapiro E, Sharma P, Shin JW, Stegle O, Stratton M, Stubbington MJT, Theis FJ, Uhlen M, van Oudenaarden A, Wagner A, Watt F, Weissman J, Wold B, Xavier R, Yosef N, Participants HCAM. Science forum: the human cell atlas. eLife. 2017;:e37041.
- 27.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, Huang D, Xu Y, Huang W, Jiang M, Jiang X, Mao J, Chen Y, Lu C, Xie J, Fang Q, Wang Y, Yue R, Li T, Huang H, Orkin SH, Yuan GC, Chen M, Guo G. Mapping the mouse cell atlas by microwell-seq. Cell. 2018;172(5):1091–107. doi: 10.1016/j.cell.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Consortium TTM. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature. 2018;562(7727):367–72. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol. 2017;34(11):1145–160. doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet. 2015;16(3):133–45. doi: 10.1038/nrg3833. [DOI] [PubMed] [Google Scholar]
- 31.Bacher R, Kendziorski C. Design and computational analysis of single-cell rna-sequencing experiments. BMC Genome Biol. 2016; 17(63). 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed]
- 32.Poulin JF, Tasic B, Hjerling-Leffler J, Trimarchi JM, Awatramani R. Disentangling neural cell diversity using single-cell transcriptomics. Nat Neurosci. 2016;19(9):1131–41. doi: 10.1038/nn.4366. [DOI] [PubMed] [Google Scholar]
- 33.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell rna sequencing. Mol Cell. 2015;58(4):610–20. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Chen G, Ning B, Shi T. Single-cell rna-seq technologies and related computational data analysis. Front Genet. 2019; 10(317). [DOI] [PMC free article] [PubMed]
- 35.Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20(5):257–72. doi: 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- 36.Pearson K. On lines and planes of closest fit to systems of points in space. Phil Mag. 1901;2(11):559–72. [Google Scholar]
- 37.Hotelling H. Analysis of a complex of statistical variables into principal components. J Educ Psychol. 1933;24:417–41. [Google Scholar]
- 38.Broa R, K SA. Principal component analysis. R Soc Chem. 2014;6(2812):2812–31. [Google Scholar]
- 39.Genomics X. 1.3 million brain cells from E18 mice. https://support.10xgenomics.com/single-cell/datasets/1M_neurons.
- 40.Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, Melton DA, Yanai I. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3(4):346–60. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, Pun SH, Sellers DL, Tasic B, Seelig G. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360(6385):176–82. doi: 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole MB, Risso D, Wagner A, DeTomaso D, Ngai J, Purdom E, Dudoit S, Yosef N. Performance assessment and selection of normalization procedures for single-cell rna-seq. Cell Syst. 2019;8(4):315–28. doi: 10.1016/j.cels.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taguchi Y-H. Principal component analysis-based unsupervised feature extraction applied to single-cell gene expression analysis. In: 14th International Conference, ICIC 2018. China: 2018. p. 816–26.
- 44.Lin Z, Yang C, Zhu Y, Duchi J, Fu Y, Wang Y, Jiang B, Zamanighomi M, Xu X, Li M, Sestan N, Zhao H, Wong WH. Simultaneous dimension reduction and adjustment for confounding variation. PNAS. 2016;113(51):14662–7. doi: 10.1073/pnas.1617317113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lasrado R, Boesmans W, Kleinjung J, Pin C, Bell D, Bhaw L, McCallum S, Zong H, Luo L, Clevers H, Vanden BP, Pachnis V. Lineage-dependent spatial and functional organization of the mammalian enteric nervous system. Science. 2017;356(6339):722–6. doi: 10.1126/science.aam7511. [DOI] [PubMed] [Google Scholar]
- 46.Wagner F. Go-pca: an unsupervised method to explore gene expression data using prior knowledge. PLoS ONE. 2015;10(11):e0143196. doi: 10.1371/journal.pone.0143196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerosaletti K, Barahmand-Pour-Whitman F, Yang J, DeBerg HA, Dufort MJ, Murray SA, Israelsson E, Speake C, Gersuk VH, Eddy JA, Reijonen H, Greenbaum CJ, Kwok WW, Wambre E, Prlic M, Gottardo R, Nepom GT, Linsley PS. Single-cell rna sequencing reveals expanded clones of islet antigen-reactive cd4+ t cells in peripheral blood of subjects with type 1 diabetes. J Immunol. 2017;199(1):323–5. doi: 10.4049/jimmunol.1700172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Klughammer J, Farlik M, Penz T, Spittler A, Barbieux C, Berishvili E, Bock C, Kubicek S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Reports. 2016;17(2):178–87. doi: 10.15252/embr.201540946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butler HPA, Smibert P, Papalexi E, Satija R. Integrated analysis of single cell transcriptomic data across conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lun Aaron T.L., McCarthy Davis J., Marioni John C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, Teichmann SA. Classification of low quality cells from single-cell rna-seq data. BMC Genome Biol. 2016;17(29). 10.1186/s13059-016-0888-1. [DOI] [PMC free article] [PubMed]
- 52.Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174(3):716–29. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li WV, Li JJ. An accurate and robust imputation method scimpute for single-cell rna-seq data. Nat Commun. 2018; 9(997). 10.1038/s41467-018-03405-7. [DOI] [PMC free article] [PubMed]
- 54.Gong W, Kwak IY, Pota P, Koyano-Nakagawa N, Garry DJ. Drimpute: imputing dropout events in single cell rna sequencing data. BMC Bioinformatics. 2018;19(220). 10.1186/s12859-018-2226-y. [DOI] [PMC free article] [PubMed]
- 55.Büttner M, Miao Z, Wolf FA, Teichmann SA, Theis FJ. A test metric for assessing single-cell rna-seq batch correction. Nat Methods. 2019;16(1):43–9. doi: 10.1038/s41592-018-0254-1. [DOI] [PubMed] [Google Scholar]
- 56.Shaham U, Stanton KP, Zhao J, Li H, Raddassi K, Montgomery R, Kluger Y. Removal of batch effects using distribution-matching residual networks. Bioinformatics. 2017;33(16):2539–46. doi: 10.1093/bioinformatics/btx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korsunsky I, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, Raychaudhuri S. Fast, sensitive, and accurate integration of single cell data with harmony. bioRxiv. 2018. 10.1101/461954. [DOI] [PMC free article] [PubMed]
- 58.Scialdone A, Natarajan KN, Saraiva LR, Proserpio V, Teichmann SA, Stegle O, Marioni JC, Buettner F. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015;85:54–61. doi: 10.1016/j.ymeth.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 59.Tsoucas D, Yuan GC. Giniclust2: a cluster-aware, weighted ensemble clustering method for cell-type detection. BMC Genome Biol. 2018; 19(1). 10.1186/s13059-018-1431-3. [DOI] [PMC free article] [PubMed]
- 60.Herman JS, Sagar. Grün D. Fateid infers cell fate bias in multipotent progenitors from single-cell rna-seq data. Nat Methods. 2018;15:379–86. doi: 10.1038/nmeth.4662. [DOI] [PubMed] [Google Scholar]
- 61.Sato K, Tsuyuzaki K, Shimizu K, Nikaido I. Cellfishing.jl: an ultrafast and scalable cell search method for single-cell rna sequencing. BMC Genome Biol. 2019; 20(1). 10.1186/s13059-019-1639-x. [DOI] [PMC free article] [PubMed]
- 62.Diaz A, Liu SJ, Sandoval C, Pollen A, Nowakowski TJ, Lim DA, Kriegstein A. Scell: integrated analysis of single-cell rna-seq data. Bioinformatics. 2016;32(14):2219–20. doi: 10.1093/bioinformatics/btw201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ji Z, Ji H. Tscan: pseudo-time reconstruction and evaluation in single-cell rna-seq analysis. Nucleic Acids Res. 2016;44(13):e117. doi: 10.1093/nar/gkw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming GL, Song H. Single-cell rna-seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 2015;17(3):360–72. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018; 19(477). 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed]
- 66.Campbell Kieran R, Yau Christopher. Probabilistic modeling of bifurcations in single-cell gene expression data using a Bayesian mixture of factor analyzers. Wellcome Open Research. 2017;2:19. doi: 10.12688/wellcomeopenres.11087.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maaten L, Hinton G. Visualizing data using t-sne. J Mach Learn Res. 2008;:2579–605.
- 68.Maaten L. Accelerating t-sne using tree-based algorithms. J Mach Learn Res. 2014;:3221–45.
- 69.Linderman GC, Rachh M, Hoskins JG, Steinerberger S, Kluger Y. Fast interpolation-based t-sne for improved visualization of single-cell rna-seq data. Nat Methods. 2019;16:243–5. doi: 10.1038/s41592-018-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawrence ND. Gaussian process latent variable models for visualisation of high dimensional data. In: NIPS: 2003. p. 2004.
- 71.McInnes L, Healy J, Saul N, Großberger L. Umap: uniform manifold approximation and projection for dimension reduction. J Open Source Softw. 2018;3(29):861. [Google Scholar]
- 72.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, Newell EW. Dimensionality reduction for visualizing single-cell data using umap. Nat Biotechnol. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 73.Weinreb C, Wolock S, Klein AM. Spring: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics. 2018;34(7):1246–8. doi: 10.1093/bioinformatics/btx792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR, Hemberg M. Sc3: consensus clustering of single-cell rna-seq data. Nat Methods. 2017;14(5):483–6. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang B, Zhu J, Pierson E, Ramazzotti D, Batzoglou S. Visualization and analysis of single-cell rna-seq data by kernel-based similarity learning. Nat Methods. 2017;14(4):414–6. doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- 76.Yang Y, Huh R, Culpepper HW, Lin Y, Love MI, Li Y. Safe-clustering: single-cell aggregated (from ensemble) clustering for single-cell rna-seq data. Bioinformatics. 2018. [DOI] [PMC free article] [PubMed]
- 77.Zurauskiene J, Yau C. pcareduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinformatics. 2016; 17(140). 10.1186/s12859-016-0984-y. [DOI] [PMC free article] [PubMed]
- 78.Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol. 2016;34(11):1145–60. doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andrews TS, Hemberg M. Identifying cell populations with scrnaseq. Mol Asp Med. 2018;59:114–22. doi: 10.1016/j.mam.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 80.Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell rna-seq data. Nat Rev. 2019;20(5):273–82. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- 81.Oskolkov N. How to cluster in high dimensions. https://towardsdatascience.com/how-to-cluster-in-high-dimensions-4ef693bacc6.
- 82.McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell rna-seq data in r. Bioinformatics. 2017;33(8):1179–86. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jenkins D, Faits T, Khan MM, Briars E, Carrasco PS, Johnson WE. singleCellTK: interactive analysis of single cell RNA-Seq data. 2018. https://bioconductor.org/packages/release/bioc/html/singleCellTK.html.
- 84.Tian L, Su S, Dong X, Amann-Zalcenstein D, Biben C, Seidi A, Hilton DJ, Naik SH, Ritchie ME. scpipe: a flexible r/bioconductor preprocessing pipeline for single-cell rna-sequencing data. PLoS Comput Biol. 2018;14(8):e1006361. doi: 10.1371/journal.pcbi.1006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yip SH, Wang P, Kocher JA, Sham PC, Wang J. Linnorm: improved statistical analysis for single cell rna-seq expression data. Nucleic Acids Res. 2017;45(22):179. doi: 10.1093/nar/gkx828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R. Mast: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell rna sequencing data. BMC Genome Biol. 2015; 16(278). 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed]
- 87.Demsar J, Curk T, Erjavec A, Gorup C, Hocevar T, Milutinovic M, Mozina M, Polajnar M, Toplak M, Staric A, Stajdohar M, Umek L, Zagar L, Zbontar J, Zitnik M, Zupan B. Orange: data mining toolbox in python. J Mach Learn Res. 2013;:2349–53.
- 88.Zhu X, Wolfgruber TK, Tasato A, Arisdakessian C, Garmire DG, Garmire LX. Granatum: a graphical single-cell rna-seq analysis pipeline for genomics scientists. BMC Genome Med. 2017;9(108). 10.1186/s13073-017-0492-3. [DOI] [PMC free article] [PubMed]
- 89.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, Choi K, Fromme RM, Dao P, McKenney PT, Wasti RC, Kadaveru K, Mazutis L, Rudensky AY, Pe’er D. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;5(23):1293–308. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Golub GH, Loan CFV. Matrix computations (Johns Hopkins Studies in the Mathematical Sciences), fourth edition. Baltimore: Johns Hopkins University Press; 2012. [Google Scholar]
- 91.Senabouth A, Lukowski S, Alquicira J, Andersen S, Mei X, Nguyen Q, Powell J. ascend: R package for analysis of single cell rna-seq data. GigaScience. 2019;8(8):giz087. doi: 10.1093/gigascience/giz087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondl M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–30. [Google Scholar]
- 93.Wolf FA, Angerer P, Theis FJ. Scanpy: large-scale single-cell gene expression data analysis. BMC Genome Biol. 2018; 19(15). 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed]
- 94.Bhaduri A, Nowakowski TJ, Pollen AA, Kriegstein AR. Identification of cell types in a mouse brain single-cell atlas using low sampling coverage. BMC Biol. 2018. [DOI] [PMC free article] [PubMed]
- 95.Levy A, M K. Sequential Karhunen-Loeve basis extraction and its application to images. IEEE Trans Image Process. 2000;9(8):1371–4. doi: 10.1109/83.855432. [DOI] [PubMed] [Google Scholar]
- 96.Bai Z, Demmel J, Dongarra J, Ruhe A, Vorst HVD. Templates for the solution of algebraic eigenvalue problems, a practical guide. Philadelphia: Society for Industrial and Applied Mathematics; 1987. [Google Scholar]
- 97.Lehoucq R, Maschhoff K, Sorensen D, Yang C. ARPACK SOFTWARE. https://www.caam.rice.edu/software/ARPACK/.
- 98.Qiu Y. Spectra: C++ library for large scale eigenvalue problems. https://spectralib.org.
- 99.Larsen RM. PROPACK homepage. http://sun.stanford.edu/~rmunk/PROPACK/.
- 100.Baglama J, Reichel L. Augmented implicitly restarted lanczos bidiagonalization methods. SIAM J Sci Comput. 2005;27(1):19–42. [Google Scholar]
- 101.Lehoucq RB, Sorensen DC, Yang C. Arpack users’ guide: solution of large-scale eigenvalue problems with implicitly restarted arnoldi methods. 1997.
- 102.Chen J, Noack A, Edelman A. Fast computation of the principal components of genotype matrices in julia. arXiv. 2018. arXiv:1808.03374v1.
- 103.Balzano L, Chi Y, Lu YM. Streaming pca and subspace tracking: the missing data case. Proc IEEE. 2018;106(8):1293–310. doi: 10.1109/JPROC.2018.2847041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oja E. A simplified neuron model as a principal component analyzer. J Math Biol. 1982;15:267–73. doi: 10.1007/BF00275687. [DOI] [PubMed] [Google Scholar]
- 105.Oja E, Karhunen J. On stochastic approximation of the eigenvectors and eigenvalues of the expectation of a random matrix author links open overlay panel. J Math Anal Appl. 1985;106(1):69–84. [Google Scholar]
- 106.Oja E. Principal components, minor components, and linear neural networks. Neural Netw. 1992;5:927–35. [Google Scholar]
- 107.Halko N, Martinsson PG, Tropp JA. Finding structure with randomness: probabilistic algorithms for constructing approximate matrix decompositions. SIAM Rev Surv Rev. 2011;53(2):217–88. [Google Scholar]
- 108.Halko N, Martinsson PG, Shkolnisky Y, Tygert M. An algorithm for the principal component analysis of large data sets. SIAM J Sci Comput. 2011;33(5):2580–94. [Google Scholar]
- 109.Li Huamin, Linderman George C., Szlam Arthur, Stanton Kelly P., Kluger Yuval, Tygert Mark. Algorithm 971. ACM Transactions on Mathematical Software. 2017;43(3):1–14. doi: 10.1145/3004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Abraham G, Inouye M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE. 2014;9(4):93766. doi: 10.1371/journal.pone.0093766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5(3):299–314. [Google Scholar]
- 112.Rossum G. Python reference manual. Technical Report. 1995.
- 113.Perkel JM. Julia: come for the syntax, stay for the speed. Nature. 2019;572(7767):141–2. doi: 10.1038/d41586-019-02310-3. [DOI] [PubMed] [Google Scholar]
- 114.Erichson NB, Voronin S, Brunton SL, Kutz JN. Randomized matrix decompositions using r. J Stat Softw. 2019; 89(11). 10.18637/jss.v089.i11.
- 115.Rocklin M. Dask: parallel computation with blocked algorithms and task scheduling In: Huff K, Bergstra J, editors. Proceedings of the 14th Python in Science Conference: 2015. p. 130–6.
- 116.Lacono G, Mereu E, Guillaumet-Adkins A, Corominas R, Cusco I, Rodriguez-Esteban G, Gut M, Perez-Jurado LA, Gut I, Heyn H. bigscale: an analytical framework for big-scale single-cell data. Genome Res. 2018;28(6):878–90. doi: 10.1101/gr.230771.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, Oord J, Atak ZK, Wouters J, Aerts S. Scenic: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kisekev VY, Yiu A, Hemberg M. scmap: projection of single-cell rna-seq data across data sets. Nat Methods. 2018;15:359–62. doi: 10.1038/nmeth.4644. [DOI] [PubMed] [Google Scholar]
- 119.Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray JI, Raj A, Li M, Zhang NR. Saver: gene expression recovery for single-cell rna sequencing. Nat Methods. 2018;15:539–42. doi: 10.1038/s41592-018-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang D, Gu J. Vasc: Dimension reduction and visualization of single-cell rna-seq data by deep variational autoencoder. Genom Proteomics Bioinforma. 2018;16(5):320–31. doi: 10.1016/j.gpb.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ding J, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun. 2018; 2002. 10.1038/s41467-018-04368-5. [DOI] [PMC free article] [PubMed]
- 122.Pliner HA, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods. 2019. [DOI] [PMC free article] [PubMed]
- 123.Weber LM, Saelens W, Cannoodt R, Soneson C, Hapfelmeier A, Gardner PP, Boulesteix A-L, Saeys Y, Robinson MD. Essential guidelines for computational method benchmarking. BMC Genome Biol. 2019; 20(125). [DOI] [PMC free article] [PubMed]
- 124.Hubert L, Arabie P. Comparing partitions. J Classif. 1985;2(1):193–18. [Google Scholar]
- 125.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. arXiv. 2008. arXiv:0803.0476v2.
- 126.Bishop CM. Pattern recognition and machine learning (information science and statistics) New York City: Springer; 2006. [Google Scholar]
- 127.Benson AR, Gleich DF, Demmel J. Direct qr factorizations for tall-and-skinny matrices in mapreduce architectures. Proc IEEE Int Conf Big Data. 2013. 10.1109/BigData.2013.6691583.
- 128.Koren Y, Bell R, Volinsky C. Matrix factorization techniques for recommender systems. IEEE Comput. 2009;42(8):30–37. [Google Scholar]
- 129.Davis T. University of Florida Sparse Matrix Collection. https://sparse.tamu.edu.
- 130.Yip SH, Sham PC, J W. Evaluation of tools for highly variable gene discovery from single-cell rna-seq data. Brief Bioinforma. 2018;:bby011. [DOI] [PMC free article] [PubMed]
- 131.Mineiro P, Karampatziakis N. A randomized algorithm for cca. arXiv. 2014. arXiv:1411.3409v1.
- 132.Arora R, Cotter A, Livescu K, Srebro N. Stochastic optimization for pca and pls. In: 2012 50th Annual Allerton Conference on Communication, Control, and Computing (Allerton): 2012. p. 861–8.
- 133.Bose Aritra, Kalantzis Vassilis, Kontopoulou Eugenia-Maria, Elkady Mai, Paschou Peristera, Drineas Petros. TeraPCA: a fast and scalable software package to study genetic variation in tera-scale genotypes. Bioinformatics. 2019;35(19):3679–3683. doi: 10.1093/bioinformatics/btz157. [DOI] [PubMed] [Google Scholar]
- 134.Musco C, Musco C. Randomized block krylov methods for stronger and faster approximate singular value decomposition. arXiv. 2015. arXiv:1504.05477.
- 135.Wang S. A practical guide to randomized matrix computations with matlab implementations. arXiv. 2015. arXiv:1505.07570v6.
- 136.Yu W, Gu Y, Li J, Liu S, Li Y. Single-pass pca of large high-dimensional data. In: Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence. Hong Kong: 2017. p. 3350–6.
- 137.Agrawal A, Chiu AM, Halperin MLE, Sankararaman S. Scalable probabilistic pca for large-scale genetic variation data. bioRxiv. 2019. 10.1101/729202. [DOI] [PMC free article] [PubMed]
- 138.Liberty E, Zucker SW. The mailman algorithm: a note on matrix–vector multiplication. Inf Process Lett. 2009;109(3):179–82. [Google Scholar]
- 139.Townes FW, Hicks SC, Aryee MJ, Irizarry RA. Feature selection and dimension reduction for single cell rna-seq based on a multinomial model. bioRxiv. 2019. 10.1101/574574. [DOI] [PMC free article] [PubMed]
- 140.Chen M, Li W, Zhang W, Wang X. Dimensionality reduction with generalized linear models. In: Proceedings of the Twenty-Third International Joint Conference on Artificial Intelligence. Beijing: 2013. p. 1267–72.
- 141.Tsuyuzaki K. Gist onlinepca-data. 2019. https: //gist.github.com/kokitsuyuzaki/5b6cebcaf37100c8794bdb89c7135fd5/revisions\#diff-99790d5a16a30380f17bd9d396670acd.
- 142.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166(5):1306–23. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Peer D. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174(3):716–29. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(14049). 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed]
- 145.Tsuyuzaki K. GitHub onlinePCA-experiments. 2019. 10.5281/zenodo.3341871. https://github.com/rikenbit/onlinePCA-experiments.
- 146.Tsuyuzaki K. GitHub OnlinePCA.jl. 2019. 10.5281/zenodo.3367116. https://github.com/rikenbit/OnlinePCA.jl.
- 147.Tsuyuzaki K. onlinepca-data. 2019. https://bioinformatics.riken.jp/onlinepca-data/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Review of existing pCA algorithms and implementations.
Additional file 2 Pseudo-code of all the pCA algorithms.
Additional file 3 Source code of all the pCA implementations.
Additional file 4 Results of t-SNE of all the pCA implementations.
Additional file 5 Results of uMAP of all the pCA implementations.
Additional file 6 Results of clustering methods of all the pCA implementations.
Additional file 7 Eigenvectors of all the pCA implementations (PBMCs and pancreas).
Additional file 8 Pair plots of all the pCA (PBMCs) implementations.
Additional file 9 Pair plots of all the pCA (Pancreas) implementations.
Additional file 10 Pair plots of all the pCA (BrainSpinalCord) implementations.
Additional file 11 Pair plots of all the pCA (Brain) implementations.
Additional file 12 Number of singleton clusters.
Additional file 13 Eigenvectors of all the pCA implementations (BrainSpinalCord and brain).
Additional file 14 Loading vectors of all the pCA implementations (PBMCs and pancreas).
Additional file 15 Crashed jobs caused by out-of-memory errors.
Additional file 16 Comparison of the elapsed time for simulated datasets.
Additional file 17 Comparison of the maximum memory usage for simulated datasets.
Additional file 18 Relationships of the algorithms/implementations, the number of passes, and the file format with the elapsed time for performing principal component analysis (PCA) with the brain dataset.
Additional file 19 OnlinePCA.jl schematic.
Additional file 20 Parameter tuning of the orthogonal iteration, gradient descent, and stochastic gradient descent implementations.
Additional file 21 Parameter tuning of the IncrementalPCA implementations.
Additional file 22 Parameter tuning of the randomized sVD implementations.
Additional file 23 Developer guidelines.
Additional file 24 Effect of feature selection on clustering accuracy.
Additional file 25 Comparison of normalizing size factors.
Data Availability Statement
The script file used to perform the benchmarking is available at https://github.com/rikenbit/onlinePCA-experiments [145]. We published our original out-of-core PCA implementations such as orthiter (Orthogonal iteration), gd (GD), sgd (SGD), halko (Halko’s method), and algorithm971 (Li’s method), which are implemented as a Julia package OnlinePCA.jl [146].
The gene expression matrix and cell type labels for the PBMCs dataset and the Brain dataset [39] were downloaded from the 10X Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets/pbmc_1k_protein_v3 and https://support.10xgenomics.com/single-cell/datasets/1M_neurons, respectively). The gene expression matrix and cell type labels for the Pancreas dataset [40] and the BrainSpinalCord dataset [41] were retrieved from the GEO database (GSE84133 and GSE110823, respectively). For the Pancreas dataset, only the sample of GSM2230759 was used. The genes of all matrices with zero variance were removed because such genes are meaningless for PCA calculation. We also removed the ERCC RNA Spike-Ins, and the number of remaining genes and cells is summarized in Table 2. Additionally, we investigated the effect of feature selection on clustering accuracy (Additional file 24).
All count datasets were generated by the R rnbinom (random number based on a negative binomial distribution) function with shape and rate parameters of 0.4 and 0.3, respectively. Matrices of {102, 103, 104} genes ×{102, 103, 104, 105, 106, 107} cells were generated.
All the real and simulated datasets described above are available from our server [147] (the hash values of md5sum have been saved, last modified 18 November 2019, 18:00 (JST)).