

Abstract
INTRODUCTION:
Crofelemer, the active compound purified from latex of Croton lechleri, has been shown to improve HIV and traveler's diarrhea and improve pain in women with irritable bowel syndrome–diarrhea (IBS-D). This trial evaluated the effect of crofelemer on abdominal pain in women with IBS-D.
METHODS:
Women with IBS-D were randomized to crofelemer (125 mg) or placebo twice daily for 12 weeks. The primary efficacy endpoint was overall change in percentage of abdominal pain/discomfort-free days. Post hoc analysis for Food and Drug Administration (FDA) monthly responders was performed for stool consistency, abdominal pain, and combined stool consistency and abdominal pain.
RESULTS:
A total of 240 women were enrolled. There was no significant difference in overall percentage of pain/discomfort-free day between the groups. In post hoc analysis, FDA abdominal pain monthly responders were significantly more likely during months 1 through 2 (58.3% vs 45.0%, P = 0.030) as well as during the entire 3 months (54.2% vs 42.5%, P = 0.037) in the crofelemer group when compared with placebo. However, there was no significant difference in the percentage of FDA stool consistency monthly responders or combined stool consistency and pain monthly responders between the groups. Crofelemer had a safety profile similar to placebo.
DISCUSSION:
Crofelemer did not significantly improve abdominal pain over placebo by the primary endpoint. However, it did based on the FDA abdominal pain monthly responder endpoint. This suggests that crofelemer may have a role in the treatment of abdominal pain associated with IBS-D. Further studies are warranted to evaluate the potential of crofelemer as a visceral analgesic.
INTRODUCTION
Irritable bowel syndrome (IBS) is a gastrointestinal condition defined by abdominal pain and altered bowel habits in the absence of another disease that can account for these symptoms (1). IBS is the most commonly diagnosed gastrointestinal condition and has a population prevalence of up to 12% in North America (1–3) and is more prevalent in women than in men (4). Currently, IBS is a clinical diagnosis based on abdominal pain associated with a change in bowel habits. Specifically IBS—diarrhea (IBS-DO) is one of the subtypes characterized by the presence of softer stool (Bristol Stool type 6 or 7) in 25% of bowel movements (5). Patients with IBS, but particularly those with IBS-D, report significantly reduced quality of life, higher indirect costs, and greater impairments in daily and work activities (6–10). Although there are currently 3 Food and Drug Administration (FDA)-approved medications for IBS-D (i.e., alosetron, eluxadoline, and rifaximin), there is still a significant unmet clinical need for effective and safe treatments particularly for the symptom of abdominal pain associated with IBS-D (11).
Crofelemer is an active compound extracted from the latex of the Western South American plant Croton lechleri (12). The latex, whose active properties have been predominantly attributable to the crofelemer compound, has been used for centuries by indigenous peoples for medicinal purposes including diarrhea, inflammation, insect bites, viral infections, and wounds (13). Crofelemer's large molecular weight (∼2000 Da) coupled with high aqueous solubility allows for limited absorption when given orally, and it has been observed to have a partial inhibitory effect on the cAMP-mediated secretion of chloride ions in T84 and Caco-2 epithelial cells (12–14). Crofelemer's partial antagonism of chloride ion secretion through the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel and the calcium-dependent chloride channel (CaCC) in intestinal T84 and Fisher rat thyroid cells may be useful in the treatment of diarrhea and related symptoms (15). Crofelemer was approved by the FDA in 2012 for the treatment of HIV-associated noninfectious diarrhea in adults (16) at an oral dose of 125 mg twice daily (b.i.d.) (17). Crofelemer also improves traveler's diarrhea, reducing its duration by 8.1, 8.4, and 6.1 hours at dosages of 125, 250, and 500 mg 4 times a day, respectively (18).
In a previous phase II dose-ranging trial, crofelemer was evaluated in 244 men and women with IBS-D at dosages of 125, 250, or 500 mg or placebo b.i.d. for 12 weeks (19). This study did not meet the primary endpoint of altering stool consistency or the secondary endpoints of stool frequency, pain/discomfort scores, and pain/discomfort-free days (PFDs). Post hoc analyses were performed specifically in women with IBS-D because there was no effect of crofelemer in men with IBS-D. After excluding 5 outliers defined as >11 stools per day which was >3 SDs from the mean stool frequency of all randomized women, crofelemer 125 mg b.i.d. exhibited a significantly increased number of PFDs (21.6% vs 8.9%, P = 0.03) and reduced abdominal pain/discomfort scores compared with placebo (−0.96 vs −0.54 on a 5-point Likert scale, P = 0.02) (20).
In the interim, Salix Pharmaceuticals had licensed the rights to crofelemer from Napo Pharmaceuticals and pursued the development and commercialization of the drug for treatment of noninfectious HIV-associated diarrhea. After its approval by the FDA for this indication, the decision to pursue crofelemer in IBS-D was deferred. Thus, a more than 10-year delay has occurred between the study and the publication of its findings.
After initiation and completion of the study, the US FDA issued revised recommendations for outcome measures in IBS clinical trials in 2010. Post hoc analysis of the previous study showed that crofelemer 125 mg b.i.d. demonstrated significant improvement over placebo in the FDA coprimary endpoints. Thus, the current trial was designed to investigate the effects of crofelemer 125 mg b.i.d. on abdominal pain in women with IBS-D.
METHODS
Study design
This was a multicenter, phase II, randomized, double-blind, placebo-controlled trial of crofelemer in women with IBS-D at 46 sites in the United States between 17 October 2006 (first patient screened) and 14 December 2007 (last patient visit). The trial was designed, conducted, and reported in compliance with the principles of Good Clinical Practice guidelines. At each center, an informed consent form approved by an institutional review board was reviewed and signed by all patients before their participation in the trial (Clinicaltrial.gov number NCT00461526).
This trial consisted of a 2-week screening period followed by a 12-week double-blind treatment period in which patients received either twice daily crofelemer 125 mg or matched placebo orally. Afterward, there was a 4-week follow-up period. At the screening visit, inclusion and exclusion criteria were reviewed. A medical history, physical examination, electrocardiogram, and laboratory evaluations were performed. From the start of the screening period through the end of the follow-up period, outcome data were collected on the interactive voice response system daily and weekly. At visit 2, patients who still met the eligibility criteria were randomized 1:1 into either the crofelemer 125 mg b.i.d. or the placebo b.i.d. group. Subjects were randomized according to a computer-generated central randomization schema. Study assessment visits were conducted every 4 weeks throughout the treatment period. Patient compliance was continually monitored. After the 4-week treatment-free follow-up period, patients were contacted by phone for a final assessment.
Study population
Women were considered eligible if they were 18 years old or older, met the Rome II definition of IBS-D, and met the following criteria during the 2-week screening period: ≥70% compliance with daily assessments, mean daily stool frequency of ≥2 and <8 stools per day, mean stool consistency score of ≥3 (scale: 1 = very hard; 2 = hard; 3 = formed; 4 = loose; and 5 = watery), and mean daily score of ≥1 for abdominal pain/discomfort (scale: 0 = none; 1 = mild; 2 = moderate; 3 = intense; and 4 = severe).
Patients were excluded from the trial if they had a history of alcohol or substance abuse within 1 year of the screening visit, had been hospitalized for a major psychiatric condition or suicide attempt within the past 3 years, were pregnant or lactating, had previous surgery on the small intestine or colon, had participated in an investigational study within 30 days of the screening visit, or had a history or presence of a malignancy or clinically significant medical disease that might compromise the trial or be detrimental to the patient including but not limited to diverticulosis, duodenal ulcer, erosive esophagitis, gastric ulcer, cholelithiasis, amyloidosis, ileus, noncontrolled gastroesophageal reflux disease, or pancreatitis within 12 months of screening. Patients were also excluded if they had constipation as defined by less than 3 bowel movements per week for at least 12 weeks in the previous 12 months. Patients with a history of laxative abuse were excluded. If a patient was taking any other treatment for IBS such as laxatives, antidiarrheals, narcotic-containing analgesics, or macrolide antibiotics, the patient was required to undergo a 7-day washout period before being allowed to enter the screening period. As for antidepressants and antipsychotics, patients were allowed to continue usage if they had been on a 30-day stable dose before the screening.
Study outcomes
The primary endpoint was the overall change from baseline in the percentage of abdominal PFDs during the 12-week treatment period. Secondary endpoints included weekly, monthly, and overall percentage of PFDs; change in weekly and monthly percentage of PFDs from baseline; weekly and monthly average abdominal pain/discomfort scores; and weekly and monthly changes in abdominal pain/discomfort scores from baseline. Other secondary endpoints included weekly and monthly average daily stool consistency, daily stool frequency, daily urgency, as well as weekly, monthly, and overall adequate relief of IBS symptoms.
Patients rated their abdominal pain/discomfort on a daily basis using a five-point Likert scale: 0 = none; 1 = mild; 2 = moderate; 3 = intense; and 4 = severe. A PFD was defined as any day with a pain/discomfort score of 0. Stool consistency was rated daily as 1 = very hard; 2 = hard; 3 = formed; 4 = loose; and 5 = watery. Stool frequency was rated as the number of daily bowel movements passed. Patients rated urgency daily based on whether or not they had satisfactory control of their bowel urgency (yes or no). Patients were asked weekly whether or not they had adequate relief of their IBS symptoms over the previous 7 days (yes or no).
After the trial was conducted, the US FDA issued recommendations for outcome measures in IBS clinical trials. Therefore, post hoc analyses were conducted based on the FDA responder definitions. An FDA stool consistency and abdominal pain responder is defined as a patient with improvement in the weekly average abdominal pain/discomfort score of ≥30% compared with baseline and weekly average stool consistency score of <4 (4 = loose). An FDA abdominal pain responder was defined as weekly average of ≥30% abdominal pain improvement compared with baseline. An FDA stool consistency responder was defined as a weekly average stool consistency score <4 (4 = loose stool) (16). Monthly responders were defined as patients who were weekly responders for stool consistency, abdominal pain, or both for ≥2 weeks per month. These supplementary FDA responder endpoints were assessed over each individual month of the treatment period.
Statistical analysis
The primary analysis was based on the observed case data and was performed on the modified intention-to-treat (mITT) population (defined as any patient who was randomized and took at least 1 dose of the study medication and entered at least 1 week of efficacy assessment data). Any patient randomized was included in the safety population. This analysis was performed using a main-effect analysis of covariance model with fixed-effect terms for study center cluster and a treatment group with the baseline abdominal pain as a covariable. These terms were fitted and remained in the model regardless of their statistical significance. The sample size was determined based on results from a previous phase II trial (19). Per the phase 2 clinical trial, an estimate of SD of 27.5% for the monthly percentage of PFDs was assumed (based on a 5-point Likert scale) between the 2 treatment groups. A sample size of 120 subjects per group would allow detection of an 11% difference between 2 groups in a 2-sided t test at alpha 0.050 and power 80%. The test for treatment effect from this model was performed using the F test with 1 degree of freedom from the type III sums of squares using a significance level of 0.050. The secondary efficacy analyses were analyzed using the main-effect analysis of covariance model described above. Categorical secondary efficacy variables were analyzed using a Cochran-Mantel-Haenszel test stratifying by study center cluster. In addition, post hoc analyses of FDA stool consistency and abdominal pain monthly responders, FDA stool consistency monthly responders, and FDA abdominal pain monthly responders were performed, also using the Cochran-Mantel-Haenszel test, and using the last observation carried forward principle. Categorical variables were summarized by number and percentage of patients for each category. Continuous variables were summarized by total number, mean, SD, minimum, and maximum values (21).
RESULTS
Demographics and disposition
Of the 240 women randomized for this trial, 237 were included in the mITT population as 3 patients withdrew before receiving study medication; in total, 181 patients completed the trial. The reasons for withdrawal are outlined in Figure 1. The 2 treatment groups were similar with respect to demographics and baseline characteristics (Table 1). All patients were female, and most patients were white. The mean age was 49 years. Nearly all patients (99%) reported >3 bowel movements per day. Patients in the crofelemer group reported 3.2% ± 7.6 PFDs during the 2-week screening period. Patients in the placebo group reported 2.6% ± 6.7 PFDs during the 2-week screening period.
Figure 1.

Patient flow through the trial.
Table 1.
Baseline demographics of patients with IBS-D (mITT)

Efficacy results
The primary efficacy endpoint, overall increase from baseline in percentage of PFDs, for patients in the crofelemer group was not statistically significant when compared with patients in the placebo group (15.9% ± 23.8% vs 13.8% ± 22.2%, P = 0.51) (Table 2). The secondary efficacy endpoints of pain/discomfort score, stool consistency, stool frequency, urgency, and report of adequate relief also did not show any significant difference between the crofelemer and placebo groups (Tables 2 and 3). There also was no change in weekly abdominal pain/discomfort scores or in weekly stool consistency between the crofelemer and placebo groups.
Table 2.
Abdominal pain efficacy results (mITT)

Table 3.
Change from baseline in secondary efficacy results (observed case; mITT)

FDA responder analyses
In post hoc analysis of the FDA abdominal and stool consistency responder endpoint, there was no significant difference between women receiving crofelemer compared with placebo for any of the individual 3 months (Figure 2a). Although not statistically significant, a proportion of patients who received crofelemer tended to have a higher FDA stool consistency and abdominal pain response across each of the 3 months (month 1: 38.3% vs 31.7%, P = 0.32; month 2: 40.8% vs 33.3%, P = 0.28; month 3: 40.8% vs 34.2%, P = 0.36). In addition, more women in the crofelemer group compared with the placebo group were FDA stool consistency monthly responders for each of the 3 months, despite these differences not meeting statistical significance (month 1: 52.5% vs 47.5%, P = 0.62; month 2: 55.0% vs 51.7%, P = 0.76; month 3: 59.2 vs 54.2%, P = 0.51) (Figure 2c).
Figure 2.

There were no significant differences with respect to FDA stool consistency and abdominal pain monthly responders between women in the crofelemer and placebo groups (P = 0.32, 0.28, 0.36 for months 1, 2, and 3). There were no significant differences in stool consistency monthly responders between women in the crofelemer and placebo treatment groups (P = 0.62, 0.76, 0.51 for months 1, 2, and 3). In months 1 and 3, there were no significant differences in percentage of FDA abdominal pain monthly responders between the crofelemer and placebo treatment groups (P = 0.23 for month 1 and P = 0.07 for month 3). In month 2, there were significantly more FDA abdominal pain monthly responders in the crofelemer group when compared with the placebo group. (a) There was no significant differences with respect to FDA stool consistency and abdominal pain monthly responders between the crofelemer and placebo groups (P = 0.32, 0.28, 0.36 for months 1, 2, and 3). (b) In months 1 and 3, there were no significant differences in percentage of FDA abdominal pain monthly responders between the crofelemer and placebo treatment groups (P = 0.23 for month 1 and P = 0.07 for month 3). (c) In month 2, there were significantly more abdominal pain monthly responders in the crofelemer group compared with the placebo group. FDA, Food and Drug Administration.
For the FDA abdominal pain responder endpoint, there was a numerical increase in each of the 3 months in the crofelemer group over placebo, although this only met statistical significance in month 2 (month 1: 62.5% vs 55.0%, P = 0.23; month 2: 68.3% vs 50.8%, P = 0.006; month 3: 65.8% vs 54.2%, P = 0.07). In addition, there were significantly more FDA abdominal pain monthly responders during months 1 and 2 (58.3% vs 45.0%, P = 0.030) and during months 1, 2, and 3 (54.2% vs 42.5%, P = 0.037) (Figure 3).
Figure 3.

A significantly greater percentage of women in the crofelemer group were FDA abdominal pain responders during months 1, 2, and 3 when compared with the placebo group (54.2% vs 42.5%, P = 0.037). A greater percentage of women were also FDA abdominal pain responders for 2 of 3 months. By contrast, a greater percentage of women in the placebo group were FDA abdominal pain responders for 0 of 3 months and 1 of 3 months. *Indicates statistical significance. FDA, Food and Drug Administration.
Safety and tolerability
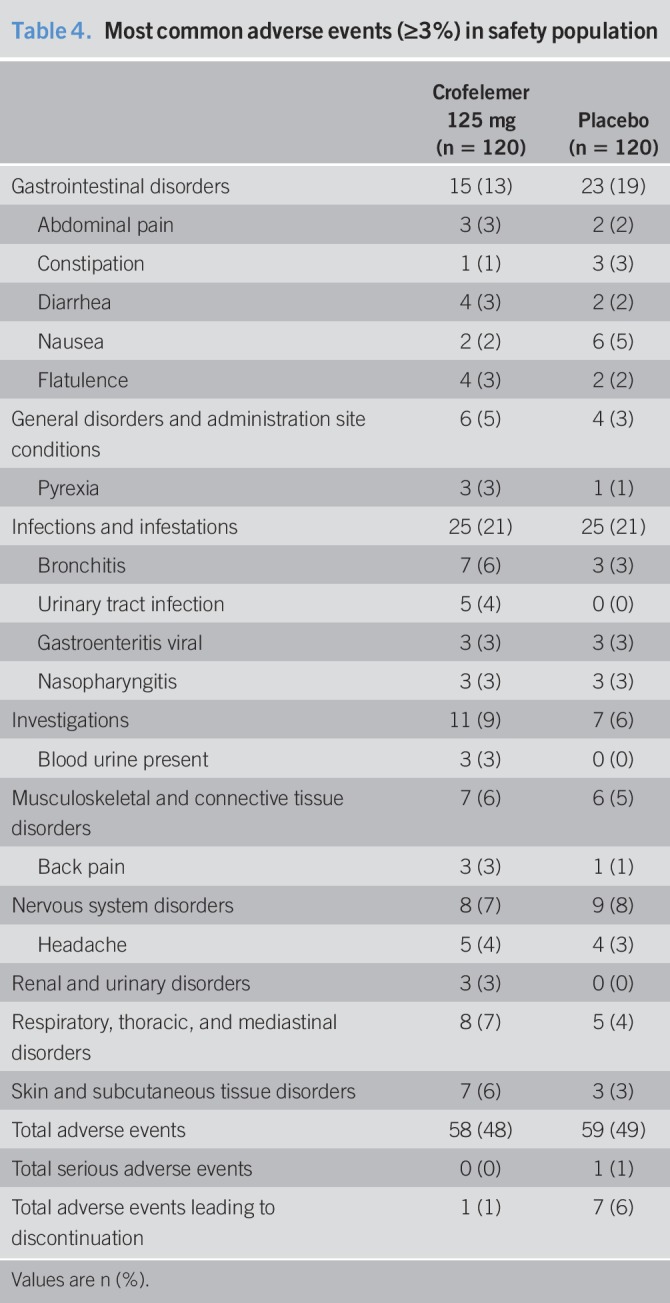
Overall, the occurrence of treatment-emergent adverse events (TEAEs) in patients randomized into the crofelemer and placebo groups was similar (Table 4). Fifty-eight of 120 patients (48%) in the crofelemer group experienced TEAEs, whereas 59 of 120 patients (49%) in the placebo group experienced TEAEs. The most common TEAEs, occurring at a greater percentage in the crofelemer group, were bronchitis and headache. Bronchitis was experienced by 7 (6%) patients in the crofelemer group vs 3 (3%) patients in the placebo group, and headache was experienced by 5 (4%) patients in the crofelemer group and 4 (3%) patients in the placebo group. Occurrence of constipation was less in the crofelemer group where 1 (1%) patient compared with 3 (3%) patients in the placebo group. One patient in the placebo group experienced a serious TEAE of chest pain while no patients in the crofelemer group experienced a serious TEAE. One patient (1%) in the crofelemer group and 7 (6%) patients in the placebo group experienced TEAEs leading to discontinuation from the trial. There were no deaths in the trial.
Table 4.
Most common adverse events (≥3%) in safety population
DISCUSSION
Post hoc analysis removing outliers from a previous dose ranging trial of crofelemer in women with IBS-D showed improvement in multiple endpoints, specifically in terms of pain for crofelemer 125 mg b.i.d. (22). Therefore, the current trial of crofelemer 125 mg b.i.d. was performed to investigate the effects of crofelemer on abdominal pain. There was no significant difference in the primary endpoint of an overall increase in abdominal PFDs in women with IBS-D or secondary endpoints (i.e., stool consistency, frequency, urgency, and adequate relief). However, a supplementary analysis demonstrated a significant improvement in the FDA abdominal pain responder endpoint for those responding during the entire 3 months of the trial in the crofelemer group compared with placebo.
There are multiple reasons for which crofelemer failed to meet the primary endpoint. In this population of women with IBS-D, pain was a prominent symptom as shown by the high prevalence of daily pain. The mean percentages of PFD were 3.2% and 2.6% (less than 1 PFD per month) for the crofelemer and placebo groups, respectively. PFDs as a primary endpoint is were difficult to achieve because of the requirement that discomfort, a vague symptom that could encompass even relatively minor abdominal related IBS symptoms, be absent. Unlike the PFDs, FDA abdominal pain responder measures only change in abdominal pain/discomfort, as opposed to the absence of pain/discomfort. Therefore, the FDA responder endpoints may be a more sensitive assessment of patients' symptoms, making them clinically relevant to IBS (23).
The lack of any significant effects could additionally be attributed to the overall milder symptoms in this selected population. The mean stool consistency scores were formed (3.9 ± 0.4 and 3.9 ± 0.4 for the crofelemer and placebo groups) and the mean stool frequency was only slightly above the normal range of three bowel movements per day to three bowel movements per week (3.6 ± 1.3 and 3.8 ± 1.2 for the crofelemer and placebo groups), both of which were less severe compared to other IBS-D study populations (21,22). Furthermore, crofelemer is a nonconstipating, nonopiate drug and only modulates the CFTR and CaCC chloride ion channels to normalize the chloride and fluid content in the gut lumen, and it would be difficult to obtain a significant lowering of stool consistency on the five-point stool consistency scale with crofelemer treatment.
The similar response rates between the crofelemer and placebo groups may be attributed to the high placebo effect, which is known to be prominent in IBS (24). The placebo response rate for FDA abdominal pain monthly responders by month was 55.0%, 50.8%, and 54.2% for months 1, 2, and 3, respectively, comparable with the placebo response to abdominal pain in other studies, such as with eluxadoline (41.9%, 59.7%, and 56.2%) (23). The percent differences between monthly responders in the crofelemer and placebo groups of 7.5%, 17.5%, and 11.6% for months 1, 2, and 3, respectively, are also comparable with those in a similar trial with eluxadoline that were 2.9% to 4.8% to 14.8% (23). A higher-powered trial may reveal significance in more endpoints.
Thus, despite the fact that the primary endpoint was not met, a significantly higher percentage of women in the crofelemer group were found to be FDA abdominal pain monthly responders during months 1 and 2 but not 3 (P = 0.030) as well as during months 1, 2, and 3 when compared with women in the placebo group (P = 0.037) in post hoc analysis. In each of the 3 months, more women in the crofelemer group were FDA abdominal pain monthly responders when compared with women in the placebo group. When comparing women in the crofelemer and placebo groups, the difference of 17.5% was statistically significant in month 2 (P = 0.006) and the difference of 11.6% approached significance in month 3 (P = 0.07). Using a similar responder endpoint (i.e., <25% of days in a given week with a stool consistency score of ≥4 [4 = loose] and >30% improvement in abdominal pain scores in a given week), post hoc analyses from Mangel and Chaturvedi similarly found women receiving crofelemer 125 mg were more likely to be responders during months 1 and 2 (30.4% vs 15.2% P = 0.046) as well as during months 1, 2, and 3 (26.1% vs 10.9% P = 0.034) compared with those receiving placebo. These observed trends toward pain improvement suggest crofelemer may have a role in the treatment pain in IBS-D but not for changes to bowel movements.
In patients with HIV and traveler's diarrhea, the antidiarrheal effects of crofelemer have been linked to crofelemer's inhibitory effect on the CFTR chloride ion and CaCC channels, reducing the chloride ion and water secretions into the gastrointestinal lumen (18,19). Stool consistency in this population of women with IBS-D was not significantly changed in the crofelemer group compared with the placebo group. The reason for this is not clear; however, IBS-D is multifactorial disorder, and the role of CFTR in IBS-D is not known. Although the mechanism for its potential effects on abdominal pain is not known, a nonconstipating medication may be a helpful agent in reducing abdominal pain without inducing constipation. Only 1 patient in the crofelemer group experienced constipation, compared with 3% of patients in the placebo group.
Crofelemer at a dose of 125 mg had a safety profile that was similar to placebo for women with IBS-D in this trial. The occurrence of adverse events in the crofelemer and placebo groups was similar, 48% and 49%, respectively. No serious adverse events occurred in women receiving crofelemer. During the trial, only 1 adverse event led to discontinuation of treatment due to an exacerbation of IBS, which was deemed to be possibly related to the drug. Gastrointestinal-related side effects were experienced by more patients in the placebo group, 19%, compared with the crofelemer group, 13%.
Currently, there are few treatments for pain associated with IBS-D (11). Although laxatives and antidiarrheals can be used to modulate bowel function in patients with IBS-C and IBS-D, they do not improve visceral pain associated with IBS. Crofelemer may have a potential use for treatment in abdominal pain in IBS-D patients without having significant changes to bowel habits. The observed improvement in abdominal pain suggests that crofelemer is a well-tolerated drug that may warrant further investigation that be considered for further treatment for pain in patients with IBS-D.
CONFLICTS OF INTEREST
Guarantor of the article: Judy Nee, MD.
Specific author contributions: The manuscript was drafted by K.S. and A.G.L and J.N. The drafted manuscript was critically reviewed by T.S., S.B., E.T., S.D., P.S., J.I., J.K., H.-N.L., V.R., and A.J.L. All authors reviewed and approved the final version of the manuscript.
Financial support: This trial was funded by Trine Pharmaceuticals and Napo Pharmaceuticals.
Potential competing interests: None to report.
Clinicaltrial.gov number NCT00461526.
Study Highlights.
WHAT IS KNOWN
✓ Crofelemer, a nonabsorbed, nonopioid, chloride ion-channel modulator, is currently FDA approved for the treatment of HIV associated noninfectious diarrhea.
✓ In a post hoc analysis of a phase II trial of crofelemer compared with placebo in IBS-D, crofelemer 125 mg b.i.d. exhibited a significantly increased number of PFDs and reduced abdominal pain/discomfort scores in women compared with placebo.
WHAT IS NEW HERE
✓ Crofelemer 125 mg b.i.d. did not significantly increase the percentage of abdominal PFDs in women with IBS-D compared with placebo.
✓ However, in post hoc analysis, the croflemer group vs placebo showed significantly greater improvement in FDA-specified abdominal pain monthly responder analysis for months 1 and 2 but not 3. The crofelemer group demonstrated significant improvement compared with placebo for those responding during all 3 months of the trial.
✓ Crofelemer did not have significant effects on stool consistency over placebo.
TRANSLATIONAL IMPACT
✓ Crofelemer could be a potential treatment option for abdominal pain associated with IBS-D.
References
- 1.Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: A clinical review. JAMA 2015;313:949–58. [DOI] [PubMed] [Google Scholar]
- 2.Lovell RM, Ford AC. Global prevalence of and risk factors for irritable bowel syndrome: A meta-analysis. Clin Gastroenterol Hepatol 2012;10:712–21.e4. [DOI] [PubMed] [Google Scholar]
- 3.Mearin F, Badía X, Balboa A, et al. Irritable bowel syndrome prevalence varies enormously depending on the employed diagnostic criteria: Comparison of Rome II versus previous criteria in a general population. Scand J Gastroenterol 2001;36:1155–61. [DOI] [PubMed] [Google Scholar]
- 4.Lovell RM, Ford AC. Effect of gender on prevalence of irritable bowel syndrome in the community: Systematic review and meta-analysis. Am J Gastroenterol 2012;107:991–1000. [DOI] [PubMed] [Google Scholar]
- 5.Lacy BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016;150:1393–407.e5. [DOI] [PubMed] [Google Scholar]
- 6.Buono JL, Carson RT, Flores NM. Health-related quality of life, work productivity, and indirect costs among patients with irritable bowel syndrome with diarrhea. Health Qual Life Outcomes 2017;15:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nellesen D, Yee K, Chawla A, et al. A systematic review of the economic and humanistic burden of illness in irritable bowel syndrome and chronic constipation. J Manag Care Pharm 2013;19:755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paré P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: Baseline results from logic (longitudinal outcomes study of gastrointestinal symptoms in Canada), a naturalistic study. Clin Ther 2006;28:1726–35. [DOI] [PubMed] [Google Scholar]
- 9.Hulisz D. The burden of illness of irritable bowel syndrome: Current challenges and hope for the future. J Manag Care Pharm 2004;10:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dean BB, Aguilar D, Barghout V, et al. Impairment in work productivity and health-related quality of life in patients with IBS. Am J Manag Care 2005;11:S17–26. [PubMed] [Google Scholar]
- 11.Munjal A, Dedania B, Cash B. Update on pharmacotherapy for irritable bowel syndrome. Curr Gastroenterol Rep 2019;21:25. [DOI] [PubMed] [Google Scholar]
- 12.Fischer H, Machen TE, Widdicombe JH, et al. A novel extract SB-300 from the stem bark latex of Croton lechleri inhibits CFTR-mediated chloride secretion in human colonic epithelial cells. J Ethnopharmacol 2004;93:351–7. [DOI] [PubMed] [Google Scholar]
- 13.Jones K. Review of sangre de drago (Croton lechleri)—A South American tree sap in the treatment of diarrhea, inflammation, insect bites, viral infections, and wounds: Traditional uses to clinical research. J Altern Complement Med 2003;9:877–96. [DOI] [PubMed] [Google Scholar]
- 14.Gabriel SE, Davenport SE, Steagall RJ, et al. A novel plant-derived inhibitor of cAMP-mediated fluid and chloride secretion. Am J Physiol 1999;276:G58–63. [DOI] [PubMed] [Google Scholar]
- 15.Hornby PJ. Drug discovery approaches to irritable bowel syndrome. Expert Opin Drug Discov 2015;10:809–24. [DOI] [PubMed] [Google Scholar]
- 16.Irritable bowel syndrome—Clinical evaluation of drugs for treatment. Irrit Bowel Syndr 2012;17:1–17. [Google Scholar]
- 17.MacArthur RD, Hawkins TN, Brown SJ, et al. Efficacy and safety of crofelemer for noninfectious diarrhea in HIV-seropositive individuals (ADVENT trial): A randomized, double-blind, placebo-controlled, two-stage study. HIV Clin Trials 2013;14:261–73. [DOI] [PubMed] [Google Scholar]
- 18.DiCesare D, DuPont HL, Mathewson JJ, et al. A double blind, randomized, placebo-controlled study of SP-303 (Provir) in the symptomatic treatment of acute diarrhea among travelers to Jamaica and Mexico. Am J Gastroenterol 2002;97:2585–8. [DOI] [PubMed] [Google Scholar]
- 19.Mangel AW, Chaturvedi P. Evaluation of crofelemer in the treatment of diarrhea-predominant irritable bowel syndrome patients. Digestion 2008;78:180–6. [DOI] [PubMed] [Google Scholar]
- 20.Lembo A, Rosenbaum D, Chey W, et al. Safety and efficacy of crofelemer in patients with diarrhea-predominant irritable bowel syndrome (D-IBS). Gastroenterology 2007;132:A141. [Google Scholar]
- 21.Macdougall JE, Johnston JM, Lavins BJ, et al. An evaluation of the FDA responder endpoint for IBS-C clinical trials: Analysis of data from linaclotide phase 3 clinical trials. Neurogastroenterol Motil 2013;25:481–6. [DOI] [PubMed] [Google Scholar]
- 22.Walter SA, Kjellström L, Nyhlin H, et al. Assessment of normal bowel habits in the general adult population: The popcol study. Scand J Gastroenterol 2010;45:556–66. [DOI] [PubMed] [Google Scholar]
- 23.Lembo AJ, Lacy BE, Zuckerman MJ, et al. Eluxadoline for irritable bowel syndrome with diarrhea. N Engl J Med 2016;374:242–53. [DOI] [PubMed] [Google Scholar]
- 24.Ballou S, Beath A, Kaptchuk TJ, et al. Factors associated with response to placebo in patients with irritable bowel syndrome and constipation. Clin Gastroenterol Hepatol 2018;16:1738–44.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]