

Abstract
Platelets are key mediators of hemostasis and thrombosis and can be inhibited by nonsteroidal anti‐inflammatory drugs (NSAIDs). As a result, platelet donors are temporarily deferred from donating if they have recently taken NSAIDs such as aspirin or ibuprofen. Despite these measures, a proportion of platelet donations show exposure to these drugs; however, little is known about the effect of NSAIDs and their metabolites on platelet quality in vivo and during storage. In this review, the effect of NSAIDs on platelet function is summarized, with a focus on the widely consumed over‐the‐counter (OTC) medications aspirin, ibuprofen, and the non‐NSAID paracetamol. Aspirin and ibuprofen have well‐defined antiplatelet effects. In comparison, studies regarding the effect of paracetamol on platelets report variable findings. The timing and order of NSAID intake is important, as concurrent NSAID use can inhibit or potentiate platelet activation depending on the drug taken. NSAID deferral periods and maximum platelet shelf‐life is set by each country and are revised regularly. Reduced donor deferral periods and longer platelet storage times may affect the quality of platelet products, and it is therefore important to identify the possible impact of NSAID intake on platelet quality before and after storage.
Keywords: acetaminophen, anti‐inflammatory agents, aspirin, blood platelets, ibuprofen, nonsteroidal
Essentials.
Nonsteroidal anti‐inflammatory drugs (NSAIDs) can inhibit platelet aggregation and secretion by inhibiting cyclo‐oxygenase 1.
The effect of paracetamol (acetaminophen) on platelets is poorly studied.
Little is known about the effect of NSAIDs and their metabolites on stored platelets.
Postponing platelet donation after NSAID intake may affect platelet quality following storage.
1. INTRODUCTION
Transfusion of platelet products is lifesaving for trauma, surgical and hemato‐oncology patients, yet they are often in short supply and high demand due to their short shelf life. Although the inhibitory effect of aspirin and ibuprofen on platelets is well established, only a small number of studies have investigated the effect of NSAID metabolites and paracetamol on platelet function. There is even less known about the impact of NSAIDs, paracetamol, and metabolites on platelet quality during storage and after transfusion. In this review, we provide an overview of the effect of NSAIDs on platelet function. This review focuses on the over‐the‐counter (OTC) medications aspirin, ibuprofen, and paracetamol, as these medications are widely consumed and consumer knowledge about these medications is limited.1
The popularity and accessibility of NSAIDs presents a challenge to blood services that collect and store platelet products. Prior to blood donation, donors are asked to complete a health questionnaire, which asks donors about recent NSAID use. Based on their disclosure, donors may be prevented from donation for a period of time (deferral). Each country sets its own deferral periods and maximum platelet shelf life (Table 1). There are currently no deferrals in place for paracetamol.
Table 1.
NSAID deferrals and maximum platelet shelf‐life
2. NSAIDs: MECHANISM OF ACTION
Cyclo‐oxygenase (COX) enzymes are the primary targets of NSAIDs and exist in 2 isoforms: COX‐1 and COX‐2. COX‐1 is constitutively expressed, in contrast to COX‐2, which is inducible.2 COXs are glycosylated, bifunctional, membrane‐bound enzymes primarily located in the endoplasmic reticulum and catalyse the conversion of arachidonic acid (AA) to prostaglandin G2 (PGG2) and PGG2 to prostaglandin G2 (PGH2) by peroxidase (POX) activity. COX is integrated into only 1 leaflet of the lipid bilayer, and due to its hydrophobicity, the COX substrate AA remains near the enzyme once it is released from the membrane phospholipids by cytosolic phospholipase A2 (cPLA2).3 AA is converted by COX‐1 into thromboxane A2 (TxA2), which promotes platelet activation and hemostatic clot formation.
Inhibition of COX is the primary mechanism of action of NSAIDs, which can be broadly classified according to their binding (reversible or irreversible) and selectivity. Common OTC NSAIDs such as aspirin and ibuprofen are cross‐selective inhibitors of both COX isoforms. Aspirin irreversibly acetylates a key residue within COX‐1 that blocks the access of AA to the active site,4, 5 whereas ibuprofen reversibly blocks AA access.6, 7 Paracetamol is an OTC drug, and although not categorized as an NSAID, it reversibly inhibits COX by acting as a reducing cosubstrate at the POX site.8 The metabolites of NSAIDs may also inhibit COX activity. In vivo, aspirin is rapidly deacetylated to form the active metabolite salicylic acid,9 which is less potent than aspirin itself and binds to COX in a reversible manner.10
3. ROLE OF TxA2 IN PLATELET ACTIVATION
Upon vascular injury, collagen fibers become exposed, bridging von Willebrand factor (VWF) to the platelet adhesion receptor glycoprotein Ibα (GPIbα), thereby activating cPLA2 leading to release of polyunsaturated fatty acids including AA (Figure 1).11 Binding of other platelet agonists to their respective receptors also induces AA release. The majority of released AA is metabolized by COX‐1 to form endoperoxides and converted by thromboxane (Tx) synthase to TxA2. AA is also metabolized further into eicosanoids including leukotrienes and related bioactive lipid mediators (Figure 2). Platelets synthesize long‐chain fatty acids, such as hydro(pero)xyeicosatetraenoic acids (HPETEs) by 12‐lipoxygenase (12‐LOX), which are unstable precursors of hydroxyeicosatetraenoic acids (HETEs). The final products of cytochrome P450 (CYP450) activity are 14‐ and 15‐epoxyeicosatrienoic acids (EETs) (Figure 2).12, 13 A wide variety of potential AA metabolites are produced by platelet COX, LOX, and CYP450 enzymes (Figure 2), which exhibit both anti‐ and pro‐platelet activation properties.14 Formation of other AA metabolites is of great importance in circumstances when platelet COX activity is blocked by the presence of NSAIDs.
Figure 1.
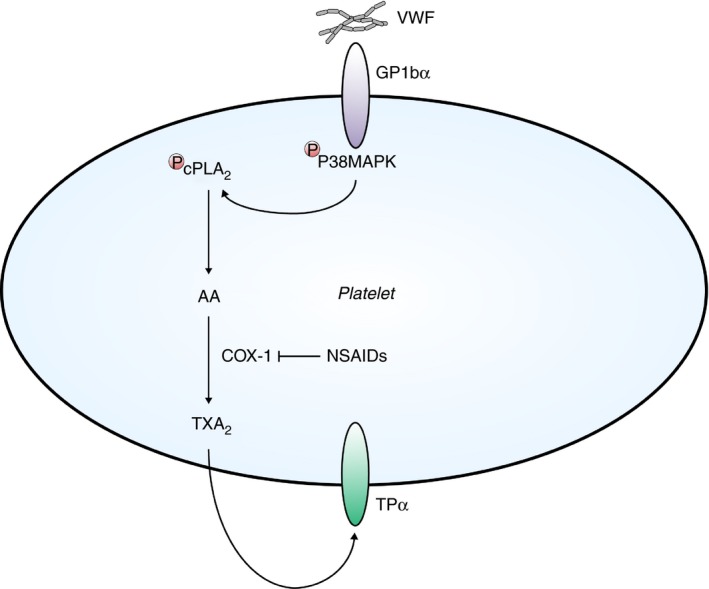
Thromboxane‐mediated platelet activation. At sites of vascular injury, von Willebrand factor (VWF) binds platelet adhesion receptor glycoprotein (GP)Ibα. Subsequent phosphorylation of P38 mitogen activated protein kinase (MAPK) and cytosolic phospholipase A2 (cPLA2) leads to the liberation of arachidonic acid (AA) from the phospholipid bilayer. Similar to VWF binding, AA is also released following stimulation of platelets by other agonists. AA is converted into thromboxane (Tx)A2 by cyclo‐oxygenase 1 (COX‐1) activity. TxA2 amplifies platelet aggregation by binding to its receptor TPα in a positive feedback loop. NSAID including aspirin inhibit COX‐1 activity and thereby inhibit platelet aggregation
Figure 2.
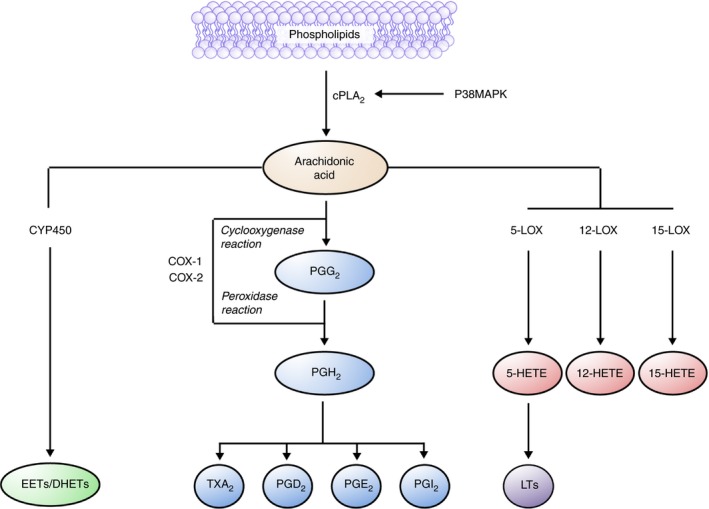
Role of cyclooxygenase (COX)‐1 and COX‐2 in platelet activation. Arachidonic acid (AA) is liberated from the phospholipid bilayer and metabolized into various prostanoids. COX‐1 and 2 contain distinct COX and peroxidase (POX) sites. Within platelets, COX‐1 is present in higher amounts than COX‐2 and converts AA to prostaglandin (PG)G2 by COX and prostaglandin H2 by POX activity. The other COX‐1 metabolite, PGH2, is further metabolized into the platelet activator thromboxane A2 (TxA2) as well as other prostaglandins. Cytochrome P450 monooxygenase (CYP450) activity results in the conversion of AA into epoxy‐eicosatrienoic acids (EETs) and dihydroxyeicosatrienoic acids (DHETs). Lipoxygenase (LOX) enzymes (5‐, 12‐, 15‐LOX) also metabolize AA resulting in the formation of various hydro(peroxy)‐eicosatetraenoic acids (HETEs) and leukotrienes (LTs)
The other COX‐1 product, PGH2, is converted to PGI2, PGE2, PGD2, and TxA2 by PG‐ and TxA2‐synthase, respectively.15, 16 Upon binding to the G protein–coupled receptors for prostaglandin and thromboxane (TPα) respectively, PGI2 and TxA2 have opposing roles in vascular hemostasis. PGI2 and PGD2 inhibit platelet secretion and aggregation by activation of the enzyme adenylyl cyclase, thereby increasing cyclic adenosine monophosphate levels and dampening the activation induced calcium rise.17 TxA2 triggers calcium mobilization, platelet shape change, aggregation, and secretion, finally promoting hemostasis plug formation. TxA2 acts in a positive feedback loop by binding to TPα, and more platelets are recruited upon TxA2 release. TPα‐mediated platelet shape change depends mainly on G12/13, whereas aggregation depends primarily on Gαq.17, 18 TxA2‐induced platelet aggregation appears to depend exclusively on ADP and other released platelet granule contents.19
4. EFFECT OF ASPIRIN ON PLATELETS
Acetylation of the COX active site by aspirin blocks binding of the substrate AA over the lifetime of platelets in vivo (8‐9 days); as they are anucleated, they are unable to synthesize new COX‐1.20, 21 Overall platelet function starts to recover 48‐72 hours after aspirin treatment and fully recovers after 96 hours, as new platelets are continuously formed from megakaryocytes.22 Complete inhibition of TxA2 production is achieved for at least 24 hours after a single 100‐mg dose of oral aspirin.23
Aspirin interferes with platelet activation, aggregation, and secretion by inhibiting the production of TxA2.24, 25 Aspirin has also been shown to stimulate the production of the platelet inhibitor nitric oxide (NO) in vitro and in vivo, which may contribute to its antiplatelet activity.26 Aspirin impairs α granule secretion and VWF binding in response to weak platelet activators such as ADP and epinephrine. However, VWF binding is unaffected by aspirin when platelets are stimulated with potent agonists such as thrombin.27 Timing of aspirin intake may be linked to platelet inhibition, as one study demonstrated that TxA2 inhibition was suboptimal when 80 mg of aspirin was taken in the morning when compared to the same dose in the evening.28
In a recent analysis of the platelet lipidome,29 resting platelets were found to contain over 5000 unique lipid species, of which thrombin stimulation increased 900. Aspirin treatment (75 mg/day for 7 days) blocked the formation of TxA2 and inhibited the formation of thrombin‐induced lipid species by 50%. This indicates that COX‐1 is crucial for platelet activation–dependent changes in the lipidome. Among other lipids, aspirin also increased formation of AA in resting platelets in some but not all donors, highlighting that this process is donor specific.29
Inhibition of TxA2 formation is not the only mechanism by which aspirin acts on platelets. The degree of platelet inhibition following 75 mg of oral aspirin is proportional to decreases in 12‐HETE, a metabolite of 12‐LOX.30 Inhibition of 12‐HETE production ex vivo has been observed with aspirin doses as low as 20 mg.31
Aspirin resistance can occur if aspirin is unable to inhibit platelets and has been linked to an increase in the expression of the β3 domain of fibrinogen receptor αIIbβ3 integrin, thereby rescuing urinary dehydrothromboxane B2 and AA‐induced platelet aggregation.32 Moreover, platelet multidrug resistance protein 4, an ATP‐binding cassette membrane transporter associated with aspirin resistance, can be upregulated following chronic aspirin treatment, leading to incomplete COX‐1 inhibition.33, 34 Ethnic variations in aspirin efficacy have also been recorded and are associated with the thrombin receptor protease‐activated receptor‐4.35, 36 Varied aspirin efficacy in different donor populations complicates the process of determining optimal aspirin deferral periods.
5. THE EFFECT OF ASPIRIN ON PLATELET‐DERIVED VESICLES
As platelets are highly activated or become procoagulant following stimulation by collagen and thrombin (known as COATED platelets), platelet‐derived extracellular vesicles (EVs) are shed.37 EVs are also shed into the storage medium during platelet storage.38 COX‐1 and ‐2 are present in EVs; however, their role is unclear. 39 EVs also contain 12‐LOX, which converts AA into 12‐HPETE. 12‐HETE within EVs promotes their internalization into activated neutrophils, characterizing EVs as potentially important mediators of intercellular communication and inflammation.40
The effect of aspirin on EV release and phenotype is poorly studied, and findings are contradictory. Addition of aspirin to platelets in vitro (50 µM) has been shown to inhibit EV release.41 However, in another study, 150 mg of aspirin for 3 days did not alter the number of EVs released in healthy subjects.42
PLA2 is present in platelet‐derived EVs and released (free) mitochondria, which are also released during platelet storage.43 The potential AA accumulation due to the presence of aspirin or other NSAIDs might be further metabolized by this enzyme. As AA is also present in EVs, increasing various signaling proteins including protein kinase C and p38 mitogen‐activated protein kinases (P38MAPK) and ultimately COX‐2 upregulation in monocytes and endothelial cells.44
Smaller platelet‐derived EVs, known as exosomes (50‐100 nm in size), contain cytokines, chemokines, growth factors, coagulation factors, lipoproteins, and other lipids, as well as several types of RNA. In one study, low‐dose aspirin treatment for 1 week (dose not specified) suppressed a variety of these cargo proteins including the α‐granule protein platelet factor 4, as well as platelet cytoplasmic proinflammatory protein high‐mobility group box 1.45 Aspirin had no effect on the total number of exosomes shed.
6. THE EFFECT OF ASPIRIN ON PLATELET DEATH AND CLEARANCE
Platelets are able to undergo cell death via the intrinsic apoptosis pathway involving Bcl‐2 family proteins.46 Platelet apoptosis may be induced by NSAID treatment. When COX‐1 is inhibited by the NSAID indomethacin in washed platelets, AA accumulates and induces apoptosis through interactions with GPIbα and the scaffolding protein 14‐3‐3ζ.47 Accumulation of AA also triggers changes in the Bcl‐2‐associated agonist of cell death protein, inducing activation of the proapoptotic protein Bax and subsequently platelet apoptosis.47 Cold storage of platelets also induces accumulation of AA by activation of P38MAPK and cPLA2, as TxA2 formation by COX‐1 is prevented at this low temperature.47 When AA is depleted from platelets, their life span in vivo is rescued in mice, indicating that AA plays a role in platelet apoptosis and clearance.
Aspirin can also reduce platelet life span by upregulation of proapoptotic Bax, leading to increased clearance by phagocytosis.48 However, the involvement of AA accumulation has not been investigated. These findings show a potential link between the administration of NSAIDs, platelet death, and clearance. It is unclear whether this might become important following transfusion of platelet products if NSAIDs and their metabolites were to be present.
7. EFFECT OF IBUPROFEN ON PLATELETS
Ibuprofen is administered as a racemic mixture of S‐ and R‐ enantiomers, but only S‐ibuprofen inhibits COX‐1 and ‐2.49 Addition of ibuprofen in vitro (80 µM) inhibits ADP‐induced aggregation and prostaglandin synthesis and also stimulates the production of NO. Unlike aspirin, the inhibition is transient in nature due to reversible binding of S‐ibuprofen to COX‐1.26 Ibuprofen has also been shown to inhibit AA, epinephrine, and collagen‐induced platelet aggregation in a dose‐dependent manner in vitro. 50, 51
The platelet function analyzer (PFA‐100) is used to assess platelet activation under high shear stress and stimulation by collagen and ADP or epinephrine. In healthy individuals completing a 7‐day course of ibuprofen (600 mg orally every 8 h), PFA‐100 scores indicated platelet dysfunction in 63% of participants, which normalized after 24 hours due to reversible COX‐1 inhibition.52
8. EFFECT OF PARACETAMOL ON PLATELETS
Due to limited anti‐inflammatory activity, paracetamol is not categorized as an NSAID. However, paracetamol is commonly taken for the relief of pain and fever.53 Metabolism occurs primarily in the liver, but also in the gut and kidneys.54 Important metabolites include paracetamol glucuronide, paracetamol sulfate and the hepatotoxic N‐acetyl‐p‐benzoquinone imine (NAPQI).54, 55 Paracetamol does not bind to the aspirin‐binding site on COX‐1, as paracetamol treatment prior to aspirin does not protect platelets from irreversible COX‐1 inhibition.56 Instead, paracetamol inactivates the POX site within COX‐1 and ‐2 by acting as a reducing cosubstrate.8 Paracetamol is a much less potent COX inhibitor in platelets when compared to other cell types. After platelet activation, 12‐HPETE is formed by platelet 12‐LOX, which activates COX‐1 and reverses the action of paracetamol on COX‐1.8
To date, few studies have attempted to characterize the effect of paracetamol on platelets, and those published report variable findings. The prevailing dogma is that paracetamol has limited or no antiplatelet effects when compared to aspirin.57 Some studies have shown that paracetamol has antiplatelet effects, while other studies suggest that it does not.
In healthy volunteers, 500 mg of oral paracetamol had no effect on TxA2 synthesis as determined by gas chromatography–mass spectrometry (GC‐MS); however, only 2 individuals were included in this study.57 Paracetamol failed to inhibit TxA2 synthesis following a single oral dose of 3 g.58 In a cohort of 35 surgery patients, 3 g of paracetamol administered intravenously failed to inhibit AA, ADP, or thrombin receptor–activating peptide (TRAP)‐induced platelet aggregation.59 In a comparison of 1‐g oral aspirin with paracetamol, paracetamol was found to have no effect on collagen‐induced platelet aggregation or bleeding time after 24 hours,60 which is expected, given the short plasma half‐life of paracetamol (1.5‐2.5 h).54
Other studies have shown that paracetamol has antiplatelet effects and is able to inhibit TxA2 synthesis.61, 62, 63 During whole blood clotting in vitro, addition of 100 µM of paracetamol was found to inhibit both PGE2 and TxA2 production.61 Administration of intravenous paracetamol (15‐30 mg/kg) inhibited AA, ADP, and epinephrine‐induced platelet aggregation for up to 90 minutes after infusion in a dose‐dependent manner, accompanied by a reduction in TxA2 synthesis.62 When paracetamol tablets (650‐1000 mg) were taken orally, paracetamol inhibited AA‐, collagen‐, and epinephrine‐induced platelet aggregation (performed ex vivo 1 h after ingestion), and reduced AA‐induced TxA2 formation by 40% to 99%.63 Further, the paracetamol metabolite NAPQI inhibited AA‐induced aggregation but had no effect on AA‐induced TxA2 formation or collagen‐induced platelet aggregation.64 Of interest, paracetamol‐mediated inhibition of platelet aggregation was influenced by plasma glucose levels in patients with diabetes, an effect not observed after ibuprofen treatment.65 The influence of plasma glucose levels on platelet aggregation has not been studied in healthy subjects.
Differences in study design, in particular the concentration of platelet agonists used in aggregation experiments, with or without plasma, may play a role in the mixed results regarding the effect of paracetamol on platelets. The methods used to measure TxA2 synthesis varied as well, as some studies that measured the urinary metabolites of TxB2 using GC‐MS did not show inhibition, whereas studies that used radioimmunoassay techniques measured a significant reduction in TxA2 synthesis after paracetamol treatment. More standardized studies are required to determine the effect of paracetamol on platelet aggregation and TxA2 formation.
9. CONCURRENT NSAID USE AND DRUG‐DRUG INTERACTIONS
There are a variety of NSAIDs that have the potential to affect platelet function. The 2 main classes are nonselective NSAIDs, such as diclofenac, naproxen, aspirin, and ibuprofen, and selective NSAIDs (coxibs), such as celecoxib and rofecoxib. Selective NSAIDs selectively inhibit COX‐2, which is upregulated in response to inflammation. COX‐2 inhibitors are selective, as they bind to a channel in the active site of COX‐2 that is not present in COX‐1.66
Multiple types of NSAIDs may be taken within a short period or combined in a single formulation. When aspirin is taken after ibuprofen, platelet function recovers after 26 hours, which is similar to platelets inhibited with ibuprofen only.67 This suggests that aspirin and ibuprofen may compete for COX‐1 binding and that ibuprofen can protect COX‐1 against irreversible acetylation by aspirin.66, 67 Concurrent administration of ibuprofen and aspirin results in potential loss of aspirin’s cardioprotective effects, as patients have a higher risk of all‐cause mortality compared to patients taking aspirin only.68 Paracetamol binds to the POX site, and does not compete for the aspirin‐binding site. As a result, paracetamol does not reduce the inhibitory effect of aspirin on platelet function.56, 66, 69
Timing and order of drug intake also influences the extent of inhibitory effects on platelets. Naproxen, similar to ibuprofen, appears to prevent the acetylation of COX‐1 by aspirin by competitively binding to COX‐1. Naproxen reduces the inhibitory effect of aspirin on TXA2 production as well as AA‐induced aggregation when administered simultaneously, but not when they are taken 2 hours apart.70 Interestingly, naproxen has been shown to potentiate increases in PFA‐100 closure time after concurrent administration with aspirin,71 indicating reduced platelet function. The clinical implications of the naproxen‐aspirin interaction remain unclear.
Studies into the concurrent administration of other NSAIDs such as celecoxib and diclofenac report conflicting results. As COX‐2 selective inhibitors do not introduce a compound that blocks COX‐1, it is expected that concurrent administration would not attenuate the inhibition of platelets by aspirin. Celecoxib did not interact with aspirin in 2 studies66, 69; however, celecoxib has also been shown to reduce the antiplatelet activity of aspirin.56, 72 Celecoxib may block aspirin inhibition despite being classified as a selective COX‐2 inhibitor, as it can still bind to COX‐1, albeit with lower potency than aspirin or ibuprofen. Diclofenac has been shown to interfere with the ability of aspirin to inhibit platelets, as measured by PFA‐10071; however, diclofenac has also failed to interfere with aspirin in other studies.56, 66
10. NSAIDS AND PLATELET STORAGE LESION
The main types of platelet products for transfusion are apheresis platelets and pooled platelets. Apheresis platelets are collected by plateletpheresis, and up to 3 units can be collected from a single donor.73 They may be stored in 100% autologous plasma, or a mixture of autologous plasma and platelet additive solution.74 Alternatively, pooled platelets are derived from the buffy coat fraction of whole blood. The buffy coats from 4 or 5 different donors are pooled, and the platelet components are then stored in plasma or a mixture of additive and plasma.
Platelets are stored for a maximum of 5 to 7 days, as they deteriorate rapidly during storage. The changes in platelet function and quality that take place during storage are known collectively as the platelet storage lesion (PSL).75 The PSL is linked with decreased platelet survival and function after transfusion and is characterized by (1) shape change,76 (2) reduced activation in response to agonists,76 (3) secretion of granule content,77 (4) externalization of phosphatidylserine,78 and (5) release of microparticles.38 A reduced response to the platelet agonist TxA2 and removal of extracellular GPIbα (shedding) are also characteristics of the PSL.79, 80 To date, there is very limited evidence on the direct effect of NSAIDs on the PSL. One study demonstrated that when platelets were stored in the presence of aspirin (1 mM), the storage‐induced shedding of GPIbα was unchanged.79
11. THROMBOXANE AND AA METABOLISM DURING PLATELET STORAGE
There are currently very few studies that have examined the direct effects of NSAIDs on stored platelets. However, the accumulation of various enzymes involved in AA release and metabolism has been demonstrated in platelets during their storage. Mitochondria, containing secreted PLA2‐IIA are released during storage, promoting leukocyte activation linked to acute adverse transfusion reactions.43 In situations where stored platelets have higher PLA2‐IIA activity, more AA might be formed, and if NSAIDs are also present, this could be further exacerbated, potentially leading to even higher AA concentrations.
Leukotriene B4 is a metabolite of AA, formed by 5‐LOX, which also accumulates in platelet concentrates during storage and has been implicated in transfusion‐related acute lung injury.81 In addition, a phospholipid, lysophosphatidylcholine accumulates during platelet storage and may result in the release of more AA.82 In the presence of NSAIDs, a reduction in COX‐1 activity could potentially lead to further AA production. Consequently, more AA could then be available to be metabolized into other reactive lipid metabolites and potentially contributing to the PSL. It remains unclear whether AA further exacerbates the PSL in stored platelets in cases when the donor has taken NSAIDs prior to donation.
In the 1980s, it was shown that the majority of AA is incorporated into phospholipids as part of the phospholipid bilayer in platelets. They found in platelet concentrates, isolated by the platelet‐rich plasma method, that 45% of AA is part of phosphatidylinositol (PI) phospholipids while ~ 16% is present in phosphatidylcholine.83 During 3 days of platelet storage, ~10% of total phospholipid was lost and released into the plasma storage media, while the content of individual phospholipids remained unchanged.83
A later study84 described that AA content as part of PS phospholipids was increased in platelets concentrates at day 3, but even higher at day 5 following storage. In contrast to AA incorporation into PI phospholipids, this was decreased following 3 days of platelet storage, but somewhat recovered on day 5. The total AA content in resting platelets was decreased.84 These findings indicate that AA incorporation into phospholipids is a highly dynamic process, especially during storage of platelets.
Various enzymatic pathways implicated in AA metabolism are also impaired during platelet storage. When stored platelets are stimulated by thrombin, TxA2 and 12‐HETE are decreased, indicating reduced activity of COX‐1 and 12‐LOX, respectively.84 When recalcified and clotted, platelets stored for 3 days and longer showed reduced TxA2 formation, indicating decreased COX‐1 activity. At days 3 and 5, TxA2 accumulates in unstimulated stored platelets.85 Furthermore, when platelets stored for 3 days are stimulated with collagen, TxA2 formation is reduced in the platelet supernatant by 20% to 30%.86 TxA2 release may be further reduced during storage in the presence of NSAIDs. As a result, platelets might be less likely to be preactivated during storage and therefore might respond better upon transfusion in vivo when activated by agonists to cease hemorrhage. However, to date, there is no direct evidence that alterations in AA metabolism and reduced TxA2 might lead to higher AA levels in platelets during storage.
12. NSAIDs AND IMPLICATIONS FOR TRANSFUSION OF PLATELET PRODUCTS
Platelet transfusion can be used to reverse the effects of antiplatelet drugs to prevent life‐threatening bleeding. As a model, aspirin‐treated platelets can be mixed with untreated platelets. The addition of 10% to 60% v/v untreated platelets to aspirin‐inhibited platelets in vitro ameliorates aspirin‐mediated inhibition of ADP and AA‐induced aggregation,87, 88 as only a fraction of the total platelet population must be replaced to restore full COX‐1 activity. This is of importance for transfused platelets, as they could affect the recipient’s COX‐1 activity if the donor had taken aspirin prior to donation. This is unlikely, as all potential donors are currently asked about their medication use and are deferred if they have taken aspirin, although the deferral times vary globally (Table 1).
To date, only a few studies have investigated the effect of aspirin intake on the function of donated platelet products. Intake of 500 mg of aspirin 12 hours prior to platelet donation decreased platelet aggregation but also TRAP‐6–induced release of the α granule protein P‐selectin, even after 3 days of storage.89 In contrast, lactate dehydrogenase, lactate, pH, morphology score, and fibrinogen binding were not affected by aspirin intake. Additionally, aspirin (10 μg/mL) does not prevent the release of proinflammatory CD40L from platelet granules following storage for 4 days.90 In a small cohort, ibuprofen intake prior to whole blood donation and subsequent platelet manufacture led to a variable plasma concentration of 0 to 5 mg/L. In vitro addition of ibuprofen (20 mg/L) fully abolished AA‐mediated aggregation of buffy coat platelets up to 8 days of storage; however, 5 mg/L showed reversible inhibition.90
In the future, platelets might be stored at low temperatures (2‐6°C), which might also induce accumulation of the COX‐1 substrate AA. In cold‐stored washed platelets, activation of p38MAPK and cPLA2 were induced, as TxA2 formation by COX‐1 was prevented at this low temperature.47 The presence of NSAIDs, together with cold storage, might further potentiate AA accumulation in platelet products during storage. It is currently unknown whether this affects platelet quality or transfusion effectiveness.
13. NSAID DEFERRALS FOR DONATION OF PLATELET PRODUCTS
Despite donor questionnaires and screening, some donated platelets show exposure to NSAIDs. It has been shown that up to 30% of platelet donations may show exposure to aspirin.91 However, in a different study, only 7% of donors disclosed NSAID intake prior to donation.92 Another study found 16% of platelet donors displayed a prolonged closure time over 3 donating days in response to collagen‐ADP and collagen‐epinephrine, when measured by the shear‐based PFA‐100, and 9% had a severe platelet function defect.93 While impaired platelet function may have causes other than NSAID intake, it is possible that some donors may not accurately disclose their use of these medications.
Consumer knowledge about OTC NSAIDs is limited, which may impact the ability of donors to disclose recent NSAID use. In a recent Australian survey of 262 consumers of Nurofen (ibuprofen) or Nurofen Plus (ibuprofen + codeine), one third of respondents could not correctly indicate the maximum daily dose, and the majority of respondents failed to recognize potential side effects.1 Further, a fifth of respondents did not correctly identify the active ingredient of Nurofen or Nurofen Plus.1 A failure to identify active ingredients is relevant to platelet donation, as donor screening forms do not feature brand names and assume that blood donors have basic knowledge about the medications they are taking.
Even when NSAIDs are not administered topically as eyedrops, they are able to affect platelet activity systemically by inhibiting AA‐induced aggregation, P‐selectin and TxA2 formation.94 Intraocular routes of administration might therefore need to be considered alongside oral NSAID use when screening prospective blood donors.
14. FUTURE DIRECTIONS
Securing a safe and efficient supply of platelet products is essential for any modern medical system. Blood services have to balance the requirement for donor screening and careful monitoring of risk factors with the high demand for transfusions. Despite a long history of research into the effects of aspirin on platelet function in vivo and in vitro, platelet function following treatment by other NSAIDs and related drugs such as paracetamol is not as well studied. Moreover, concurrent NSAID use, the effect of NSAID metabolites on platelet function, and the impact of these compounds on platelet quality during storage and after transfusion demands further research.
The common use of NSAID medications may support the dogma that these medications have negligible effects on platelet quality; however, it is unclear what the potential impact of NSAIDs is on platelets during storage. Some consumers are also unaware of the risks of OTC medications. Changes to donor deferral periods, inclusion of more donors, and longer platelet storage times may also affect platelet products, and it is therefore important to identify and study the possible impact of NSAID intake on platelet quality, death, and clearance following their transfusion.
RELATIONSHIP DISCLOSURES
The authors report nothing to disclose.
AUTHOR CONTRIBUTIONS
BD, DCM, and DEvdW wrote the paper. BD created all cartoons.
ACKNOWLEDGMENTS
The authors thank Robert Harley for reviewing the manuscript.
Driver B, Marks DC, van der Wal DE. Not all (N)SAID and done: Effects of nonsteroidal anti‐inflammatory drugs and paracetamol intake on platelets. Res Pract Thromb Haemost. 2020;4:36–45. 10.1002/rth2.12283
Handling Editor: Neil Zakai
Funding information
Australian governments fund the Australian Red Cross Blood Service to provide blood, blood products, and services to the Australian community.
Contributor Information
Ben Driver, https://twitter.com/bendriverr.
Dianne E. van der Wal, Email: divanderwal@redcrossblood.org.au, https://twitter.com/DianvanderwalDr.
REFERENCES
- 1. Mullan J, Weston KM, Bonney A, Burns P, Mullan J, Rudd R. Consumer knowledge about over‐the‐counter NSAIDs: they don't know what they don't know. Aust NZ J Publ Heal. 2017;41:210–4. [DOI] [PubMed] [Google Scholar]
- 2. Topper JN, Cai J, Falb D, Gimbrone MA. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase‐2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up‐regulated by steady laminar shear stress. Proc Natl Acad Sci U S A. 1996;93:10417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Picot D, Loll PJ, Garavito RM. The X‐ray crystal structure of the membrane protein prostaglandin H2 synthase‐1. Nature. 1994;367:243–9. [DOI] [PubMed] [Google Scholar]
- 4. Funk CD, Funk LB, Kennedy ME, Pong AS, Fitzgerald GA. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;5:2304–12. [PubMed] [Google Scholar]
- 5. Mancini JA, O'Neill GP, Bayly C, Vickers PJ. Mutation of serine‐516 in human prostaglandin G/H synthase‐2 to methionine or aspirin acetylation of this residue stimulates 15‐R‐HETE synthesis. FEBS Lett. 1994;342:33–7. [DOI] [PubMed] [Google Scholar]
- 6. Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time‐dependent and time‐independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40:5172–80. [DOI] [PubMed] [Google Scholar]
- 7. Orlando BJ, Lucido MJ, Malkowski MG. The structure of ibuprofen bound to cyclooxygenase‐2. J Struct Biol. 2015;189:62–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc Natl Acad Sci U S A. 2002;99:7130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nagelschmitz J, Blunck M, Kraetzschmar J, Ludwig M, Wensing G, Hohlfeld T. Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers. Clin Pharmacol. 2014;6:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol. 1995;2:637–43. [DOI] [PubMed] [Google Scholar]
- 11. Garcia A, Quinton TM, Dorsam RT, Kunapuli SP. Src family kinase–mediated and Erk‐mediated thromboxane A2 generation are essential for VWF/GPIb‐induced fibrinogen receptor activation in human platelets. Blood. 2005;106:3410–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maskrey BH, Bermúdez‐Fajardo A, Morgan AH, Stewart‐Jones E, Dioszeghy V, Taylor GW, et al. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem. 2007;282:20151–63. [DOI] [PubMed] [Google Scholar]
- 13. Zhu Y, Schieber Elimor B, McGiff John C, Balazy M. Identification of arachidonate P‐450 metabolites in human platelet phospholipids. Hypertension. 1995;25:854–9. [DOI] [PubMed] [Google Scholar]
- 14. Yeung J, Hawley M, Holinstat M. The expansive role of oxylipins on platelet biology. J Mol Med. 2017;95:575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chandrasekharan JA, Marginean A, Sharma‐Walia N. An insight into the role of arachidonic acid derived lipid mediators in virus associated pathogenesis and malignancies. Prostaglandins Other Lipid Mediat. 2016;126:46–54. [DOI] [PubMed] [Google Scholar]
- 16. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakahata N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008;118:18–35. [DOI] [PubMed] [Google Scholar]
- 18. Offermanns S. Activation of platelet function through G protein–coupled receptors. Circ Res. 2006;99:1293–304. [DOI] [PubMed] [Google Scholar]
- 19. Paul BZS, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A2‐induced platelet aggregation: essential role for p2t(ac) and α(2a) receptors. J Biol Chem. 1999;274:29108–14. [DOI] [PubMed] [Google Scholar]
- 20. Cohen JA, Leeksma CH. Determination of the life span of human blood platelets using labelled diisopropylfluorophosphonate. J Clin Invest. 1956;35:964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low‐dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–83. [DOI] [PubMed] [Google Scholar]
- 22. Lee J, Kim JK, Kim JH, Dunuu T, Park S‐H, Park SJ, et al. Recovery time of platelet function after aspirin withdrawal. Curr Ther Res Clin Exp. 2014;76:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low‐dose aspirin in healthy subjects. J Clin Invest. 1982;69:1366–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Gaetano G, Cerletti C, Dejana E, Latini R. Pharmacology of platelet inhibition in humans: implications of the salicylate‐aspirin interaction. Circulation. 1985;72:1185–93. [DOI] [PubMed] [Google Scholar]
- 25. Taylor ML, Misso NL, Stewart GA, Thompson PJ. The effects of varying doses of aspirin on human platelet activation induced by PAF, collagen and arachidonic acid. Br J Clin Pharmacol. 1992;33:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chakraborty K, Khan GA, Banerjee P, Ray U, Sinha AK. Inhibition of human blood platelet aggregation and the stimulation of nitric oxide synthesis by aspirin. Platelets. 2003;14:421–7. [DOI] [PubMed] [Google Scholar]
- 27. Parker RI, Gralnick HR. Effect of aspirin on platelet‐von Willebrand factor surface expression on thrombin and ADP‐stimulated platelets. Blood. 1989;74:2016–21. [PubMed] [Google Scholar]
- 28. Racca C, van Diemen JJK, Fuijkschot WW, Spit K, Bonten TN, Numans ME, et al. Aspirin intake in the morning is associated with suboptimal platelet inhibition, as measured by serum thromboxane B2, during infarct‐prone early‐morning hours. Platelets. 2018;22:1–29. [DOI] [PubMed] [Google Scholar]
- 29. Slatter D, Aldrovandi M, O’Connor A, Allen S, Brasher C, Murphy R, et al. Mapping the human platelet lipidome reveals cytosolic phospholipase A2 as a regulator of mitochondrial bioenergetics during activation. Cell Met. 2016;23:930–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maskrey BH, Rushworth GF, Law MH, Treweeke AT, Wei J, Leslie SJ, et al. 12‐hydroxyeicosatetraenoic acid is associated with variability in aspirin‐induced platelet inhibition. J Inflamm. 2014;11:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eynard AR, Galli G, Tremoli E, Maderna P, Magni F, Paoletti R. Aspirin inhibits platelet 12‐hydroxy‐eicosatetraenoic acid formation. J Lab Clin Med. 1986;107:66–72. [PubMed] [Google Scholar]
- 32. Floyd CN, Goodman T, Becker S, Chen N, Mustafa A, Schofield E, et al. Increased platelet expression of glycoprotein IIIa following aspirin treatment in aspirin‐resistant but not aspirin‐sensitive subjects. Br J Clin Pharmacol. 2014;78:320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Massimi I, Lotti L, Temperilli F, Mancone M, Sardella G, Calcagno S, et al. Enhanced platelet MRP4 expression and correlation with platelet function in patients under chronic aspirin treatment. Thromb Haemost. 2016;116:1100–10. [DOI] [PubMed] [Google Scholar]
- 34. La Rosa G, Biasucci LM, Mandolini C, Massimi I, Copponi G, Pulcinelli FM, et al. Platelet miRNA‐26b down‐regulates multidrug resistance protein 4 in patients on chronic aspirin treatment. J Cardiovasc Med. 2018;19:611–3. [DOI] [PubMed] [Google Scholar]
- 35. Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M. Mechanism of race‐dependent platelet activation through the protease‐activated receptor‐4 and Gq signaling axis. Arterioscler Thromb Vasc Biol. 2014;34:2644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tourdot BE, Stoveken H, Trumbo D, Yeung J, Kanthi Y, Edelstein LC, et al. Genetic variant in human PAR (protease‐activated receptor) 4 enhances thrombus formation resulting in resistance to antiplatelet therapeutics. Arterioscler Thromb Vasc Biol. 2018;38:1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dale GL, Remenyi G, Friese P. Quantitation of microparticles released from coated‐platelets. J Thromb Haemost. 2005;3:2081–8. [DOI] [PubMed] [Google Scholar]
- 38. Marcoux G, Duchez A‐C, Rousseau M, Lévesque T, Boudreau LH, Thibault L, et al. Microparticle and mitochondrial release during extended storage of different types of platelet concentrates. Platelets. 2017;28:272–80. [DOI] [PubMed] [Google Scholar]
- 39. Hu Q, Cho MS, Thiagarajan P, Aung FM, Sood AK, Afshar‐Kharghan V. A small amount of cyclooxygenase 2 (COX2) is constitutively expressed in platelets. Platelets. 2017;28:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duchez AC, Boudreau LH, Naika GS, Bollinger J, Belleannee C, Cloutier N, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12‐lipoxygenase and secreted phospholipase A2‐IIA. Proc Natl Acad Sci U S A. 2015;112:E3564–E3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Connor DE, Ly K, Aslam A, Boland J, Low J, Jarvis S, et al. Effects of antiplatelet therapy on platelet extracellular vesicle release and procoagulant activity in health and in cardiovascular disease. Platelets. 2016;27:805–11. [DOI] [PubMed] [Google Scholar]
- 42. Rosinska J, Maciejewska J, Narożny R, Osztynowicz K, Raczak B, Michalak S, et al. Effect of acetylsalicylic acid intake on platelet derived microvesicles in healthy subjects. Platelets. 2019;21:1–9. [DOI] [PubMed] [Google Scholar]
- 43. Boudreau LH, Duchez A‐C, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA‐secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barry OP, Kazanietz MG, Praticò D, FitzGerald GA. Arachidonic acid in platelet microparticles up‐regulates cyclooxygenase‐2‐dependent prostaglandin formation via a protein kinase C/mitogen‐activated protein kinase‐dependent pathway. J Biol Chem. 1999;274:7545–56. [DOI] [PubMed] [Google Scholar]
- 45. Goetzl EJ, Goetzl L, Karliner JS, Tang N, Pulliam L. Human plasma platelet‐derived exosomes: effects of aspirin. FASEB J. 2016;30:2058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. [DOI] [PubMed] [Google Scholar]
- 47. van der Wal DE, Gitz E, Du VX, Lo K, Koekman CA, Versteeg S, et al. Arachidonic acid depletion extends survival of cold‐stored platelets by interfering with the [glycoprotein Ibα – 14‐3‐3ζ] association. Haematologica. 2012;97:1514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nayak MK, Dash A, Singh N, Dash D. Aspirin delimits platelet life span by proteasomal inhibition. PLoS ONE. 2014;9:e105049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evans AM. Comparative pharmacology of S(+)‐ibuprofen and (RS)‐ibuprofen. Clin Rheumatol. 2001;20:S9–14. [DOI] [PubMed] [Google Scholar]
- 50. McIntyre BA, Philp RB, Inwood MJ. Effect of ibuprofen on platelet function in normal subjects and hemophiliac patients. Clin Pharmacol Ther. 1978;24:616–21. [DOI] [PubMed] [Google Scholar]
- 51. De la Cruz JP, Reyes JJ, Ruiz‐Moreno MI, Lopez‐Villodres JA, Jebrouni N, Gonzalez‐Correa JA. Differences in the in vitro antiplatelet effect of dexibuprofen, ibuprofen, and flurbiprofen in human blood. Anesth Analg. 2010;111:1341–6. [DOI] [PubMed] [Google Scholar]
- 52. Goldenberg NA, Jacobson L, Manco‐Johnson MJ. Duration of platelet dysfunction after a 7‐day course of Ibuprofen. Ann Intern Med. 2005;142:506–9. [DOI] [PubMed] [Google Scholar]
- 53. Flower RJ, Vane JR. Inhibition of prostaglandin synthetase in brain explains the anti‐pyretic activity of paracetamol (4‐acetamidophenol). Nature. 1972;240:410–1. [DOI] [PubMed] [Google Scholar]
- 54. Prescott LF. Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol. 1980;10:S291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen‐induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Ther. 1973;187:195–202. [PubMed] [Google Scholar]
- 56. Saxena A, Balaramnavar VM, Hohlfeld T, Saxena AK. Drug/drug interaction of common NSAIDs with antiplatelet effect of aspirin in human platelets. Eur J Pharmacol. 2013;721:215–24. [DOI] [PubMed] [Google Scholar]
- 57. Grèen K, Drvota V, Vesterqvist O. Pronounced reduction of in vivo prostacyclin synthesis in humans by acetaminophen (paracetamol). Prostaglandins. 1989;37:311–5. [DOI] [PubMed] [Google Scholar]
- 58. Trettin A, Böhmer A, Suchy M‐T, Probst I, Staerk U, Stichtenoth DO, et al. Effects of paracetamol on NOS, COX, and CYP activity and on oxidative stress in healthy male subjects, rat hepatocytes, and recombinant NOS. Oxid Med Cell Longev. 2014;2014:212576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Silvanto M, Munsterhjelm E, Savolainen S, Tiainen P, Niemi T, Ylikorkala O, et al. Effect of 3 g of intravenous paracetamol on post‐operative analgesia, platelet function and liver enzymes in patients undergoing tonsillectomy under local anaesthesia. Acta Anaesthesiol Scand. 2007;51:1147–54. [DOI] [PubMed] [Google Scholar]
- 60. Seymour RA, Williams FM, Oxley A, Ward A, Fearns M, Brighan K, et al. A comparative study of the effects of aspirin and paracetamol on platelet aggregation and bleeding time. Eur J Clin Pharmacol. 1984;26:567–71. [DOI] [PubMed] [Google Scholar]
- 61. Sciulli MG, Seta F, Tacconelli S, Capone ML, Ricciotti E, Pistritto G, et al. Effects of acetaminophen on constitutive and inducible prostanoid biosynthesis in human blood cells. Br J Pharmacol. 2003;138:634–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Munsterhjelm E, Munsterhjelm NM, Niemi TT, Ylikorkala O, Neuvonen PJ, Rosenberg PH. Dose‐dependent inhibition of platelet function by acetaminophen in healthy volunteers. Anesthesiology. 2005;103:712–7. [DOI] [PubMed] [Google Scholar]
- 63. Lages B, Weiss HJ. Inhibition of human platelet function in vitro and ex vivo by acetaminophen. Thromb Res. 1989;53:603–13. [DOI] [PubMed] [Google Scholar]
- 64. Shorr RI, Kao KJ, Pizzo SV, Rauckman EJ, Rosen GM. In vitro effects of acetaminophen and its analogues on human platelet aggregation and thromboxane B2 synthesis. Thromb Res. 1985;38:33–43. [DOI] [PubMed] [Google Scholar]
- 65. Kobzar G, Mardla V, Samel N. Short‐term exposure of platelets to glucose impairs inhibition of platelet aggregation by cyclooxygenase inhibitors. Platelets. 2011;22:338–44. [DOI] [PubMed] [Google Scholar]
- 66. Catella‐Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–17. [DOI] [PubMed] [Google Scholar]
- 67. Rao GH, Johnson GG, Reddy KR, White JG. Ibuprofen protects platelet cyclooxygenase from irreversible inhibition by aspirin. Arteriosclerosis. 1983;3:383–8. [DOI] [PubMed] [Google Scholar]
- 68. MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573–4. [DOI] [PubMed] [Google Scholar]
- 69. Li X, Fries S, Li R, Lawson JA, Propert KJ, Diamond SL, et al. Differential impairment of aspirin‐dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2014;111:16830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Capone ML, Sciulli MG, Tacconelli S, Grana M, Ricciotti E, Renda G, et al. Pharmacodynamic interaction of naproxen with low‐dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295–301. [DOI] [PubMed] [Google Scholar]
- 71. Galliard‐Grigioni KS, Reinhart WH. A randomized, controlled study on the influence of acetaminophen, diclofenac, or naproxen on aspirin‐induced inhibition of platelet aggregation. Eur J Pharmacol. 2009;609:96–9. [DOI] [PubMed] [Google Scholar]
- 72. Ouellet M, Riendeau D, Percival MD. A high level of cyclooxygenase‐2 inhibitor selectivity is associated with a reduced interference of platelet cyclooxygenase‐1 inactivation by aspirin. Proc Natl Acad Sci U S A. 2001;98:14583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lozano M, Cid J. Platelet components today . ISBT Sci Ser. 2007;2:216–9. [Google Scholar]
- 74. Nogawa M, Naito Y, Chatani M, Onodera H, Shiba M, Okazaki H, et al. Parallel comparison of apheresis‐collected platelet concentrates stored in four different additive solutions. Vox Sang. 2013;105:305–12. [DOI] [PubMed] [Google Scholar]
- 75. Devine DV, Serrano KS. The platelet storage lesion. Clin Lab Med. 2010;30:475–87. [DOI] [PubMed] [Google Scholar]
- 76. Murphy S, Gardner FH. Platelet storage at 22˚C; metabolic, morphologic, and functional studies. J Clin Invest. 1971;50:370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Reddoch KM, Pidcoke HF, Montgomery RK, Fedyk CG, Aden JK, Ramasubramanian AK, et al. Hemostatic function of apheresis platelets stored at 4˚C and 5˚C. Shock. 2014;41:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shapira S, Friedman Z, Shapiro H, Presseizen K, Radnay J, Ellis MH. The effect of storage on the expression of platelet membrane phosphatidylserine and the subsequent impact on the coagulant function of stored platelets. Transfusion. 2000;40:1257–63. [DOI] [PubMed] [Google Scholar]
- 79. Canault M, Duerschmied D, Brill A, Stefanini L, Schatzberg D, Cifuni SM, et al. p38 mitogen‐activated protein kinase activation during platelet storage: consequences for platelet recovery and hemostatic function in vivo. Blood. 2010;115:1835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen W, Liang X, Syed AK, Jessup P, Church WR, Ware J, et al. Inhibiting GPIbα shedding preserves post‐transfusion recovery and hemostatic function of platelets after prolonged storage. Arterioscler Thromb Vasc Biol. 2016;36:1821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Silliman CC, Bjornsen AJ, Wyman TH, Kelher M, Allard J, Bieber S, et al. Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion. 2003;43:633–40. [DOI] [PubMed] [Google Scholar]
- 82. Vlaar APJ, Hofstra JJ, Kulik W, van Lenthe H, Nieuwland R, Schultz MJ, et al. Supernatant of stored platelets causes lung inflammation and coagulopathy in a novel in vivo transfusion model. Blood. 2010;116:1360–8. [DOI] [PubMed] [Google Scholar]
- 83. Hamid MA, Kunicki TJ, Aster RH. Lipid composition of freshly prepared and stored platelet concentrates. Blood. 1980;55:124–30. [PubMed] [Google Scholar]
- 84. Cesar JM, Navarro JL. Arachidonic acid metabolism in platelets stored for 5 days. Br J Haematol. 1990;74:295–9. [DOI] [PubMed] [Google Scholar]
- 85. Wenzel F, Baertl A, Hohlfeld T, Zimmermann N, Weber AA, Lorenz H, et al. Determination of thromboxane formation, soluble CD40L release and thrombopoietin clearance in apheresis platelet concentrates. Platelets. 2012;23:150–6. [DOI] [PubMed] [Google Scholar]
- 86. Bock M, Glaser A, Pfosser A, Schleuning M, Heim MU, Mempel W. Storage of single‐donor platelet concentrates: metabolic and functional changes. Transfusion. 1993;33:311–5. [DOI] [PubMed] [Google Scholar]
- 87. O'Brien JR. Effects of salicylates on human platelets. Lancet. 1968;291:779–83. [DOI] [PubMed] [Google Scholar]
- 88. Zhang S, Xu KE, Mei L, Zhu H, Li J, Wang F, et al. Reversal of the antiplatelet effect of ticagrelor by simulated platelet transfusion. Transfusion. 2019;59:1850–6. [DOI] [PubMed] [Google Scholar]
- 89. Zeiler T, Gritzka D, Karger R, Kretschmer V. The effect of ASA on platelet activation during apheresis and on in‐vitro properties of stored platelet concentrates. Transfusion. 2004;44:1300–5. [DOI] [PubMed] [Google Scholar]
- 90. Kaufman J, Spinelli SL, Schultz E, Blumberg N, Phipps RP. Release of biologically active CD154 during collection and storage of platelet concentrates prepared for transfusion. J Thromb Haemost. 2007;5:788–96. [DOI] [PubMed] [Google Scholar]
- 91. Li X, Fries S, Sachais Bruce S, Cuker A, Blair Ian A, Grosser T. Abstract 15635: consumption of aspirin and naproxen by platelet donors and the quality of platelet transfusion. Circulation. 2015;132:A15635. [Google Scholar]
- 92. Curvers J, Dielis AW, Heeremans J, Van Wersch JW. Platelet function in whole‐blood donors is impaired: the effects of painkillers. Transfusion. 2007;47:67–73. [DOI] [PubMed] [Google Scholar]
- 93. Harrison P, Segal H, Furtado C, Verjee S, Sukhu K, Murphy MF. High incidence of defective high‐shear platelet function among platelet donors. Transfusion. 2004;44:764–70. [DOI] [PubMed] [Google Scholar]
- 94. Falcinelli E, Iannone A, Mezzasoma AM, Amato L, Fierro T, Guglielmini G, et al. Inhibition of platelet function after ocular administration of non‐steroidal anti‐inflammatory drugs. Thromb Res. 2019;175:1–5. [DOI] [PubMed] [Google Scholar]
- 95. Eligibility: Australian Red Cross Blood Service; [Accessed 2019 April 30]. Available from https://www.donateblood.com.au/eligibility.
- 96. Platelets: Australian Red Cross Blood Service; [Accessed 2019 April 30]. Available from https://www.transfusion.com.au/blood_products/components/platelets.
- 97. Blood donation FAQs: AABB; [Accessed 2019 April 30]. Available from http://www.aabb.org/tm/donation/Pages/donatefaqs.aspx.
- 98. Eligibility Criteria: Alphabetical: American Red Cross Blood Services; [Accessed 2019 April 30]. Available from https://www.redcrossblood.org/donate-blood/how-to-donate/eligibility-requirements/eligibility-criteria-alphabetical.html.
- 99. Blood & blood products: FDA U.S. Food & Drug administration; [Accessed 2019 April 30]. Available from https://www.fda.gov/vaccines-blood-biologics/blood-blood-products.
- 100. ABCs of eligibility: Canadian Blood Services; [Accessed 2019 April 30]. Available from https://blood.ca/en/blood/am-i-eligible/abcs-eligibility.
- 101. Serrano K, Schubert P, Devine D. [Accessed 2019 April 30]. Platelet product quality remains high after seven days of storage. Available from https://www.professionaleducation.blood.ca/en/transfusion/publications/platelet-product-quality-remains-high-after-seven-days-storage.
- 102. Nonsteroidal anti‐inflammatory drugs: JPAC; [Accessed 2019 April 30]. Available from https://www.transfusionguidelines.org/dsg/wb/guidelines/no006-nonsteroidal-anti-inflammatory-drugs.
- 103. Blood products: JPAC; [Accessed 2019 April 30]. Available from https://www.transfusionguidelines.org/transfusion-handbook/3-providing-safe-blood/3-3-blood-products.