

Abstract
Preterm infants are vulnerable to inflammation‐induced white matter injury (WMI), which is associated with neurocognitive impairment and increased risk of neuropsychiatric diseases in adulthood. Epigenetic mechanisms, particularly DNA methylation, play a role in normal development and modulate the response to pathological challenges. Our aims were to determine how WMI triggered DNA methylation alterations in brains of neonatal rats and if such changes persisted over time. We used a robust model of WMI by injecting lipopolysaccharide (LPS) or sterile saline in the corpus callosum of 3‐day‐old (P3) rat pups. Brains were collected 24 hours (P4) and 21 days post‐injection (P24). We extracted genomic DNA from the brain to establish genome‐wide quantitative DNA methylation profiles using reduced representation bisulfite sequencing. Neonatal LPS exposure induced a persistent increased methylation of genes related to nervous system development and a reduced methylation of genes associated with inflammatory pathways. These findings suggest that early‐life neuroinflammatory exposure impacts the cerebral methylation landscape with determining widespread epigenetic modifications especially in genes related to neurodevelopment.
Keywords: DNA methylation, epigenetics, lipopolysaccharide, periventricular leukomalacia
Abbreviations
- ASD
autism spectrum disorder
- CpG
cytosine followed by guanine
- DMR
differentially methylated region
- DOHaD
developmental origins of health and disease
- E. coli
Escherichia coli
- GO
gene ontology
- LPS
lipopolysaccharide
- P3
postnatal day 3
- P4
postnatal day 4
- P24
postnatal day 24
- PBS
phosphate‐buffered saline
- PFA
paraformaldehyde
- RRBS
reduced representation bisulfite sequencing
- WMI
white matter injury
1. INTRODUCTION
Exposure to infections during the postnatal period in preterm newborns has been shown to have a drastic effect on neurodevelopmental outcomes especially in the setting of early meningitis but also in cases of sepsis and necrotizing enterocolitis.1, 2, 3 Preterm infants are highly vulnerable to infection due to their immature immune system particularly the extreme‐born preterm (born 22‐ to 28‐weeks gestation) in which 25%‐60% develop at least one infectious episode prior to hospital discharge with risk of recurring infections.4 These infectious episodes are associated with drastic production of inflammatory cytokines known to be highly toxic to the vulnerable developing white matter and they are associated to increased risk of white matter injury (WMI). WMI in the preterm is characterized by microglial activation, astrogliosis, ventricular dilatation, hypomyelination, altered connectivity, and reduction of white and grey matter volumes5, 6, 7, 8 leading to a heightened risk of neurodevelopmental and neurocognitive impairments.5, 6, 8 To better understand the pathophysiological impact of inflammation on the premature infant’s brain, different animal models have been developed which includes intracerebral lipopolysaccharide (LPS) injection that mimics the effect of Gram‐negative infection. It was shown that intracerebral LPS injection in the corpus callosum of 3‐day‐old (P3) rats, at a brain developmental state equivalent to very preterm babies, induced microglia and astrocyte activation, ventricular dilatation, hypomyelination, and hippocampal atrophy.9, 10, 11, 12 This recognized animal model also exhibited behavioral impairments including hyperactivity, memory impairments, and motor deficits13, 14, 15 similar to alterations seen in preterm infants with WMI.5, 7, 8 This early‐life inflammatory insult has long‐term repercussion characterized by sustained inflammation and increased sensitivity to secondary insults at adulthood.
The developmental origins of health and disease hypothesis postulates that adaptation in response to early‐life injury alters the risk of disease during adulthood.16, 17 Animal exposed to inflammation, whether by systemic or intracerebral injection, during the perinatal period had sustained increase in pro‐inflammatory markers (IL‐1β, IL‐6, and iNOS) and higher number of microglia in the striatum, the substantia nigra, and the hippocampus at adulthood.14, 18 This early insult sensitized the brain to secondary insults at adulthood as seen with exacerbated impairments following induction of seizure and even of Parkinson disease.18, 19, 20 Furthermore, in humans, recent evidences showed how perinatal inflammatory exposure predisposes to delayed psychiatric manifestations such as depression and schizophrenia.21, 22, 23, 24, 25 In the Helsinki Birth Cohort Study, preterm birth was a significant factor for appearance of early signs of mild cognitive decline and poorer cognitive score at adulthood.26 The increased risk for neuropsychiatric pathologies and early signs of neurodegeneration identified later in life following perinatal inflammation begs to question the possible involvement of long‐lasting epigenetic changes.
Epigenetic mechanisms play an important developmental role in central nervous system. These mechanisms primarily involve DNA methylation, post‐translational histone modifications, and noncoding RNAs that together remodel chromatin to facilitate or suppress gene expression without changing the DNA sequence and are heritable during cell division.27 DNA methylation is the most stable and well‐studied epigenetic change and consists of the addition or removal of a methyl group on the CpG region (cytosine followed by guanine) which alters DNA interactions with transcriptions factors.28 Addition of methyl group, hypermethylation, decreases or stops transcription particularly when it occurs in CpG dense promoter regions, and removal of a methyl group, hypomethylation, inversely tends to increase transcription rates.29 Currently, there are two major paradigms on the effect of methylation alteration in pathologies: (i) methylation changes leading to clear gene activation or silencing which is highly found in oncogenes and (ii) subtle and complex accumulation of methylation changes that are associated to disease phenotype particularly in programming non‐malignant pathologies such as depression or schizophrenia.30, 31 Particularly, subtle methylation changes during the early‐life period have been associated to increased risk of developing neurodegenerative and neuropsychiatric disorders.30
In humans, preterm birth has been associated to short‐ and long‐term alterations of the epigenome landscape. Analysis of buccal swab at term equivalent age showed that very preterm with poorer neurobehavioral outcomes had altered methylation in genes relevant to neurodevelopment and neurodegenerative disease.32 Furthermore, DNA methylation in preterm saliva at term equivalent age, which has a higher correlation to the brain methylome compared to blood or buccal swab,33, 34 identified differential methylation of 10 protein coding genes playing a role in neural cell functions and behavioral traits development.35 As assessed through analysis of blood spot methylation, very preterm born infants displayed until 18 years old persistent methylation changes in pathways related to nervous system and hematological system development, antigen presentation, and embryonic development when compared to term born infants.36 This was corroborated by analysis of whole blood DNA methylation in preterm born pair of twins that showed methylation alterations in genes related to immune response at adulthood (30‐35 years) that persisted at older ages (56‐80 years).37 The persistent signs of altered methylation profile in genes related to immune response and neuronal development in preterm infants further corroborate the idea of the early establishment of altered inflammatory and developmental states that could facilitate neurological disorders at adulthood. Yet, a clear description of the short‐ and long‐term impact of neonatal inflammation on brain methylome is not available to date.
Using this robust animal model of inflammatory WMI,10, 11, 38, 39, 40 we hypothesized that postnatal inflammatory exposure induced significant cerebral epigenetics modifications similarly to recent epidemiological findings in former preterm infants.
2. MATERIALS AND METHODS
2.1. Animal preparation
Animal‐handling procedures were approved by the Animal Research Ethics Committee of the Montreal Heart Institute in accordance with the Canadian Council of Animal Care. The animals were given ad libitum access to food and water and were kept under 12 hours light/dark cycles. A total of 32 male and female Sprague‐Dawley rats at 3‐day old (P3) were randomly assigned to one of four groups (n = 8 in each group): (i) Sham at postnatal day 4 (P4), (ii) Sham at postnatal day 24 (P24), (iii) LPS at P4, and (iv) LPS at P24. P4 corresponds to the acute phase of inflammation and represent an age where the rat brain is equivalent to the brain of a preterm infant,9 and P24 rat brain is equivalent to the brain of 2‐3 years old human child.9, 41 Animals were injected in the corpus callosum, at a level equivalent to P‐7, c9,42 under ultrasound guidance using Vevo LAZR micro‐ultrasound system (FUJIFILM VisualSonics Inc., Toronto, ON, Canada). The LPS animals were injected with a suspension of 1 mg/kg LPS (Escherichia coli, serotype 055:B5, Sigma) in sterile saline solution. The LPS dosage was established in previous studies using this model of WMI.10, 40 An equivalent volume of sterile saline alone was injected to the Sham group pups. A micropipette mounted on a microprocessor‐controlled injector (Micro4 from World Precision Instruments, Sarasota, FL, USA) with a speed rate of 100 nL/min was used for the injections. All the injections were performed under isoflurane anesthesia. The animals were euthanized at P4 and P24 and the brains were collected for DNA methylation assessment (n = 4 per group) and for histology (n = 4 per group).
2.2. Histology
Rats at P4 and P24 were perfused through the heart with PBS followed by 4% paraformaldehyde (PFA) and brains were extracted and submerged in 4% PFA for 24 hours at 4°C and then cryoprotected by immersion in 30% sucrose for at least 48 hours. Brains were kept at 4°C until cutting. Coronal sections (50 µm thick) were cut on a cryostat and conserved at −20°C in cryoprotectant solution (30% ethylene glycol in 0.03 mol/L PBS and 15% sucrose) until staining. Brain sections at the level of the bregma were stained with cresyl violet and scanned at 20× in the slide scanner Axioscan Z1 (Carl Zeiss Inc, ON, Canada) to measure lateral ventricle size. Area of left and right ventricles and total brain section area were measured for each section using Fiji.39 The ventricle size index was calculated as the ratio between area of each ventricle and area of the whole brain section and ratio between left and right ventricles was also calculated. Comparisons between groups at each time point were performed using a Mann‐Whitney test with statistical significance set at P < .05. Analysis were performed using GraphPad Prism (GraphPad Software, La Jolla, CA).
2.3. DNA extraction
Rat pups used for DNA methylation were perfused through the heart with phosphate‐buffered saline solution (PBS), then, the ipsilateral hemispheres were snap‐frozen on dry ice and kept at −80°C. The ipsilateral brain hemisphere was coronally trimmed into a block of tissue, containing areas near the site of injection, from 2.80 to −0.40 mm from Bregma at P4 and 2.80 to −1.80 mm from Bregma at P24 and processed for DNA extraction. In brief, the block of tissue was reduced to powder by grinding it in liquid nitrogen and the powder was kept at −80°C until DNA extraction. Total DNA was extracted with the QIAGEN DNA mini kit (#51304) according to the manufacturer’s instructions. DNA quality and concentration were assessed by spectrophotometry using the Nanodrop apparatus (Thermoscientific, Wilmington, DE, USA).
2.4. Reduced representation bisulfite sequencing
Quantitative DNA methylation profiles were obtained by reduced representation bisulfite sequencing using a modified version of previously published protocols.43, 44, 45, 46, 47 Briefly, 600 ng of genomic DNA was digested over night with non‐methylation specific Msp1 enzyme (NEB # R0106M). Ligation of methylated adaptors (NEB # E7535L) was done on the digested DNA with T4 ligase enzyme (NEB #M0202L) before sodium bisulfite conversion (QIAGEN # 59826). Libraries were amplified by qPCR with Illumina indexing primers (NEB # E7535L) followed by clean up and size selection of the libraries using Agencourt AMPure XP magnetic beads (Beckman Coulter # A63881). Quality of the libraries was assessed by Bioanalyzer (Agilent # 5067‐4626) and eight individual libraries were multiplexed into one lane of 100bp paired‐end sequencing on Illumina HiSeq 2000 sequencer. Raw sequencing data were preprocessed for quality control and adapters removal with TrimGalore (version 0.3.3), and alignment to the rat reference genome (rn6) and methylation calls with BSMAP (version 2.9).48 X and Y chromosomes were removed from the rest of the analysis to avoid sex bias. R package Methylkit (version 0.5.3)49 was used to obtain differentially methylated regions (DMRs) applying specific parameters (P value threshold of q = 0.01 by the Benjamini‐Hochberg FDR procedure, 100 bp stepwise tiling windows, a minimum of 2 CpGs per tile, and a minimum of 10× CpG coverage of each tile per sample). The methylation score of each tile was the average of the methylation of each single CpGs within the tile. For each time points, DMRs corresponded to tiles with at least 10% of difference of methylation between the two conditions (Sham vs LPS) as previously published.50, 51, 52 DMRs were finally annotated using Homer (version 4.1).
2.5. Gene ontology and pathway analysis
The hypermethylated (gain of methylation) and hypomethylated (loss of methylation) genic DMRs at P4 and P24 were analyzed with the online software Metascape version 3.0 (http://metascape.org)53, 54 for functional enrichment using ontology terms collected from gene ontology (GO) for biological processes, cellular components, and molecular functions (http://geneontology.org), Reactome Gene Set pathways (http://portal.genego.com), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (http://www.genome.jp/kegg). Functional analyses for the datasets were run to display the 40 most significant biological functions across the dataset that meet a P value cutoff of .01. For ease of visualization, only the 10 functions with the lowest p value are displayed.
3. RESULTS
3.1. Neonatal LPS‐induced WMI resulted in ventricles dilatation
We first sought to determine the impact of WMI on animal weight and ventricles morphology. Animal’s body weight did not vary significantly between Sham and LPS groups at each time points (10.50 ± 0.45 vs 10.06 ± 0.41 g at P4; 70.24 ± 3.29 vs 68.69 ± 3.46 g at P24, Sham vs LPS, respectively). Knowing that lateral ventricle enlargement is the most common histopathological hallmark in LPS‐injected rats, we measured ventricle size index on brain sections stained with cresyl violet.10, 12 At P4, ventricle size did not have a significant difference between Sham and LPS animal for both left and right ventricles (Figure 1). On the opposite, at P24, LPS rats presented increased bilateral ventricle size index and the left ventricle, the side of injection, reached statistical significance (0.49 ± 0.11 vs 9.65 ± 4.75 P < .0286, Sham vs LPS, respectively) (Figure 1). Thus, neonatal LPS exposure resulted in bi‐hemispheric cerebral injury.
Figure 1.
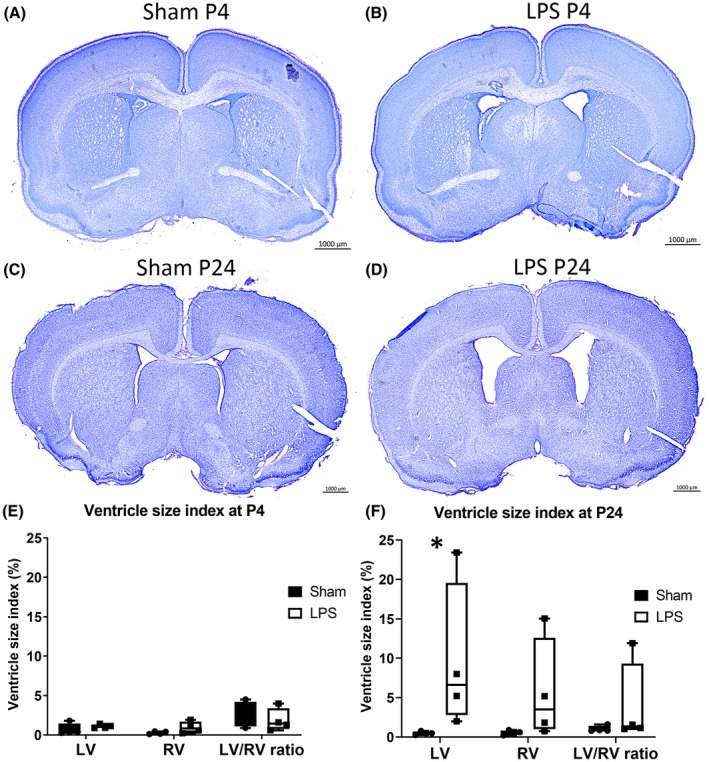
Lateral ventricles size index at P4 and P24. Representative image of lateral ventricle size for Sham and LPS animals at P4 (A and B) and at P24 (C and D). Bar graph of ventricle size at (E) P4 and (F) P24. *P < .05 compared to Sham. LV, left ventricle; RV, right ventricle
3.2. Changes in methylation following inflammation‐induced WMI
We then aimed at investigating the impact of LPS‐induced WMI on epigenetic marks by establishing methylation profiles of rat brains, at P4 and P24, by reduced representation bisulfite sequencing (RRBS). Following RRBS, DMRs were identified between Sham and LPS animals at each time point by comparing CGs methylation levels. Due to sex heterogeneity in the groups, DMRs on chromosomes X and Y were excluded (Figure S1). DMRs were distributed across all chromosomes studied at both time points (Figure S1).
First, DMRs at P4 (Figure 2) and P24 (Figure 3) were aligned to the reference genome (rn6) and 100 bp regions were compared between Sham and LPS animals. At P4, 183210 100bp regions were analyzed with a high proportion of them located within intergenic (46%), intronic (26%), and promoter (14%) regions (Figure 2B). Of the total 100 bp regions studied at P4, 1453 DMRs were identified following LPS exposure (696 hypermethylated and 757 hypomethylated DMRs) with the majority located in intergenic regions (59%) or in introns (30%) (Figure 2A,B). At P24, 168 667 100 bp regions were analyzed and they were mainly located in intergenic regions (45%), in introns (25%), and in promoters (15%) similarly to P4 (Figure 3B). From the 1502 DMRs identified at P24 (902 hypermethylated and 600 hypomethylated), the majority were either intergenic (58%) or located in introns (29%) (Figure 3A,B). In sum, neonatal inflammatory WMI increased the occurrence of methylation changes within the intergenic region and introns at both time points (Figures 2B and 3B).
Figure 2.
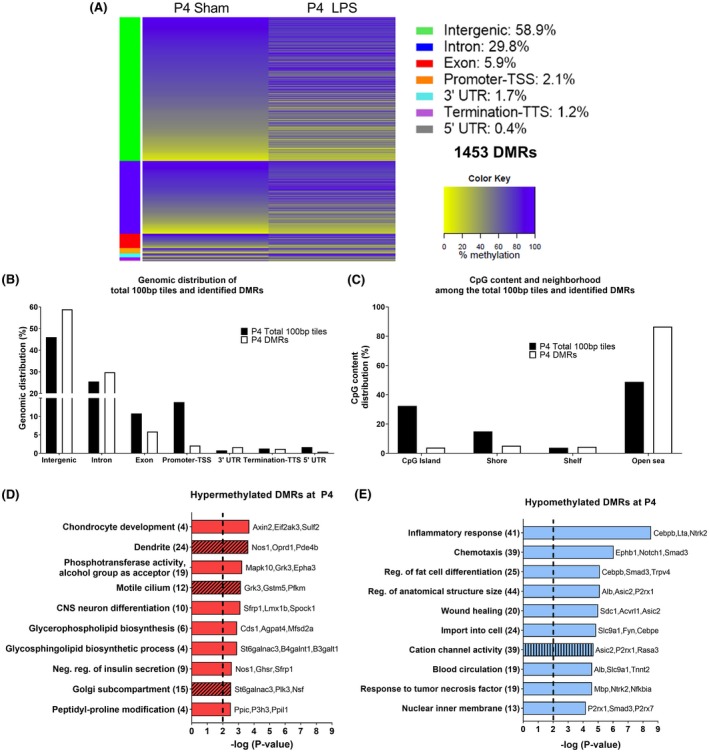
DNA methylation alteration at P4 in the brain of Sprague‐Dawley rats exposed to neonatal WMI. A, Heat map of mean methylation levels for 1453 DMRs with methylation alteration in the LPS group (n = 4) compared to Sham (n = 4) at 24‐h post‐injection. B, Genomic distribution of total 100 bp tiles and the 1453 DMRs. C, CpG content and neighborhood in total 100 bp tiles and DMRs (D‐E) Top 10 pathways identified following GO analysis without intergenic DMRs for (D) hypermethylated DMRs and (E) hypomethylated DMRs. GO enrichment was performed for biological processes (plain bar), molecular functions (bar with vertical lines), and cellular components (bar with oblique lines). The number of genes associated to each pathway is in parenthesis. LINE, long interspersed nuclear element; LTR, long terminal repeat element; SINE, short interspersed nuclear element
Figure 3.
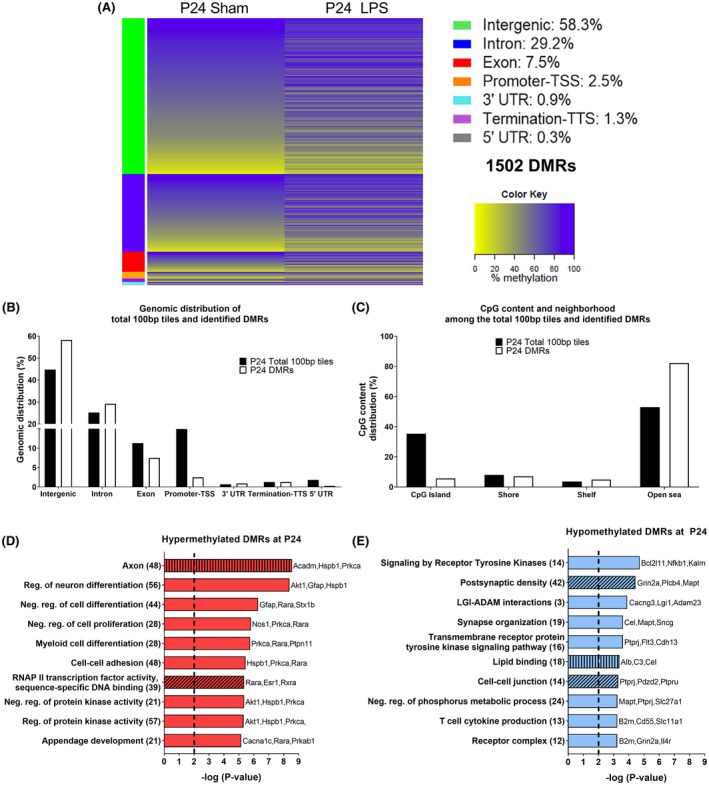
DNA methylation changes at P24 in the brain of Sprague‐Dawley rats subjected to neonatal WMI. A, Heat map of mean methylation levels for 1502 DMRs with methylation alteration in the LPS group (n = 4) compared to Sham (n = 4) 21 days post‐injection. B, Distribution of total 100 bp tiles and 1502 DMRs across the genome. C, CpG content and neighborhood in total 100 bp tiles and DMRs (D and E) Top 10 pathways identified following GO analysis without intergenic DMRs for (D) hypermethylated DMRs and (E) hypomethylated DMRs. GO enrichment was performed for biological processes (plain bar), molecular functions (bar with vertical lines), and cellular components (bar with oblique lines). The number of genes associated to each pathway is in parenthesis. LINE, long interspersed nuclear element; LTR, long terminal repeat element; SINE, short interspersed nuclear element
3.3. CpG content and neighborhood
Analogous to the characterization by genic region, DMRs distribution was also assessed based on their proximity to CpG content and neighborhood context (island, shore, shelf, and open sea). CpG shores are located up to 2 kb from CpG islands, shelves are 2‐4 kb away, and open sea regions are isolated CpG in the genome.55 We compared the relative proportion of DMRs CpG location to the proportion within the total 100bp regions at both time points.
At both P4 and P24, the majority of CpG of the total 100 bp tiles were located within the open sea and on CpG islands (Figures 2C and 3C). Within identified DMRs, the majority were in the open sea at both P4 and P24 with the lowest percentage of DMRs found on CpG islands at P4 and CpG shelves at P24 (Figures 2C and 3C). Compared to the distribution pattern seen in all 100 bp regions analyzed, there was a strong increase in DMRs in the open sea and a decrease in CpG islands at both time points (Figures 2C and 3C). Therefore, inflammation‐induced WMI resulted in a persistent increase in methylation alterations across CpG in the open sea. When taken together, the preferential methylation changes in the intergenic region and in the open sea could point toward alteration of genomic stability following neonatal WMI.
3.4. Gene enrichment analysis
GO analysis was performed with Metascape to characterize pathways associated to the genes with DMRs.53, 56 The intergenic regions were excluded, which leaves 597 genic DMRs (281 hypermethylated and 316 hypomethylated) at P4 and 626 genic DMRs (380 hypermethylated and 246 hypomethylated) at P24 (Table 1). The first analysis performed with the DMRs‐associated genes, regardless of their methylation status, showed that majority of the pathways were related to inflammatory response at P4 and to neurodevelopment at P24 (Figure S2). Knowing that the methylation status can influence differently gene expression, genes were separated in function of whether their DMRs were hyper‐ or hypomethylated for subsequent GO analysis. For ease of comprehension, only the 10 pathways with the strongest enrichment were depicted in Figures 2 and 3 with the complete set illustrated in Figures S3 and S4.
Table 1.
Distribution of the DMRs, excluding intergenic regions, and average percentage difference of methylation changes compared to sham at P4 and P24
| Condition | Number of DMRs | Avg % difference of methylation changes | Number of unique genes | Genes with DMRs located on promoter region |
|---|---|---|---|---|
| P4 hypermethylated | 281 | 14.08 ± 4.11 | 240 | ELMOD3, HORMAD2, KRTCAP3, MIR219‐2, MIR290, ODF3, TNS3 |
| P4 hypomethylated | 316 | −13.55 ± 3.68 | 283 | ABCC6, ACVRL1, ADAMTS18, APOBR, ARPP21, CAR13, F2RL3, FXN, GFER, IGBP1B, LAT, LTA, MCEMP1, MPDU1, NKAPL, P2RX1, PF4, SIPA1, SMIM3, TMEM255B, TNFRSF25, ZRSR1 |
| P24 hypermethylated | 380 | 14.74 ± 5.22 | 323 | CCKBR, CDC20, CXCL1, DPYS, EXOSC7, F8A1, FAM217A, FSTL3, HK2, MIR219‐2, MSH4, ODF3, PABPC4, PAQR7, RN5‐8S, SALL3, SGK2, SIPA1, STK3, TENM3, UPB1 |
| P24 hypomethylated | 246 | −14.42 ± 4.53 | 219 | ALOX12E, CD55, COX6A2, DOK3, MIR671, ODF3, OTOP3, RMND1, SLAMF8, SNCG, SOAT2, ST13, TRIM17 |
At P4, 240 unique genes had hypermethylated DMRs and 283 genes had hypomethylated DMRs (Table 1). Hypermethylated DMRs within genes showed multiple pathways associated with nervous system development such as the dendritic compartment and neuron differentiation, and also with protein and lipid synthesis pathways (Figures 2D and S3A). The 283 genes with hypomethylated DMRs at P4 were highly enriched for immune and inflammatory response pathways and pathways associated to synaptic and axonal compartments (Figures 2E and S3B). Furthermore, regulation of synapse assembly was associated to both hypermethylated and hypomethylated DMRs within genes (Table S1). Thus, at P4, neonatal WMI altered methylation within genes associated to innate immune response and to nervous system.
At P24, 323 genes had hypermethylated DMRs and 219 had hypomethylated DMRs (Table 1). Although there were more genes with hypermethylated than hypomethylated genic DMRs, the absolute values of average percentage difference of methylation change were similar (Table 1). Hypermethylated DMRs within genes at P24 were highly enriched in neurodevelopmental pathways such as regulation of neuronal differentiation, axonal compartment, and ensheathment of neurons (Figures 3D and S4A). Hypermethylated DMRs within genes were also associated to immune system regulatory pathways which included myeloid cell differentiation and CTLA4 inhibitory signal (Figures 3D and S4A). At P24, hypomethylated DMRs within genes still showed an increased representation of inflammatory and innate immune response pathways like T‐cell cytokine production and interleukin 12 production and were also associated to neurodevelopmental pathways particularly synaptic organization and to receptor tyrosine kinase signaling pathways (Figures 3E and S4B). At P24, lipid binding, regulation of GTPase activity and phosphoric ester hydrolase activity pathways were associated to both hypermethylated and hypomethylated DMRs within genes (Table S1). Even at P24, neonatal WMI changed DNA methylation in genes associated to nervous system and inflammatory pathways.
In light of global alteration of pathways associated to neurodevelopment and immune response at P4 and P24, and the fact that DNA methylation is a dynamic process, we compared the GO analysis results to identify identical pathways between both time points. No identical pathway was found for hypermethylated DMRs within genes at P4 and P24. Cell projection membrane, innate immune system, and regulation of cell adhesion pathways were identified at P4 and P24 for hypomethylated DMRs within genes (Table S1). Furthermore, regulation of fat cell differentiation, import into cell and the axonal compartment pathways were associated to hypomethylated DMRs at P4 and to hypermethylated at P24 (Table S1). CNS neuron differentiation pathway was identified at P4 to hypermethylated DMRs within genes and to hypomethylated DMRs within genes at P24 (Table S1).
Next, we investigated, with GO analysis, pathways that could be associated to altered methylation in promoter regions at P4 and P24 (Table 1). No pathways were identified for hypermethylated DMRs‐associated genes at P4 and hypomethylated DMRs at P24. At P4, hypomethylated DMRs in the promoter region were associated to inflammatory processes (Figure S3C). At P24, promoter region hypermethylated DMRs were linked to cellular nitrogen compound catabolic process and gland development (Figure S4C).
Taken together, neonatal inflammatory WMI induced a persistent methylation alteration in genes relevant to nervous system developments, inflammatory response, and alteration in the synaptic compartment pathways.
3.5. Methylation difference in genes related to inflammation and myelination
As interindividual variabilities are present in the histological examination (Figure 1), we looked at occurrence of individual variability in methylation changes for genes related to inflammatory response, and to myelination and oligodendrocytes. These pathways were chosen as inflammation and hypomyelination are two established pathological hallmarks in this animal model. At P4 and P24, the genes DLX1, SDC1, and UNC13D had multiple methylation changes as noted with the presence of at least two DMRs for each gene (Figure 4A,D). For inflammatory response, all DMRs were hypomethylated at P4 and both hyper‐ and hypomethylated DMRs were present at P24 (Figure 4A,B). At both P4 and P24, hypermethylated and hypomethylated DMRs were present for myelination/oligodendrocyte pathway (Figure 4C,D). For both hypermethylated and hypomethylated DMRs‐associated genes in each pathway, LPS‐exposed animals had similar methylation changes on some genes while there was individual variability for other genes (Figure 4). Furthermore, the genes ABCD2 and NRTK2, identified at P24, were associated to both pathways (Figure 4B,D). Consequently, LPS‐exposed animals had variable methylation changes in some genes while other genes had more conserved alteration between animals.
Figure 4.
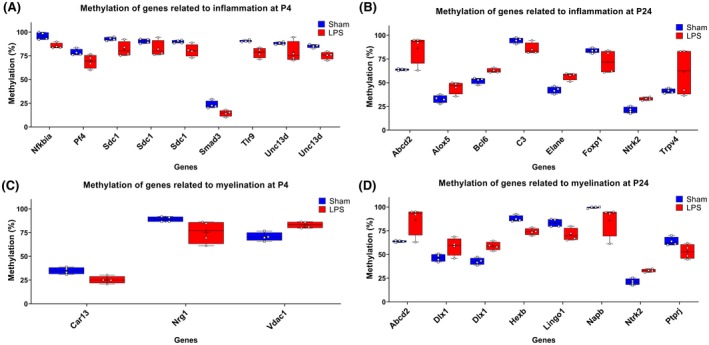
Methylation levels of genes of interest associated to inflammatory response (A and B) and myelination (C and D) at P4 and P24 in brains of Sham rats compared to LPS‐exposed animals. Individual values are represented by square (Sham) and dots (LPS) for each gene and box plot represent minimal to maximal value with line at median
3.6. Common DMRs at P4 and P24
Considering the presence of similar genes and pathways between P4 and P24 (Table S1), we looked at the possibility of finding identical DMRs at both time points. Over the 1453 and 1502 DMRs identified at P4 and P24 respectively, 102 DMRs were identified on the exact same 100bp region at both time points. Of the 102 DMRs, 50% (n = 51) kept similar methylation status between P4 and P24 and 50% (n = 51) had a shift to the opposite methylation status (Figure 5A). Similar to results with all identified DMRs, common DMRs at both time points had enhanced methylation changes in the intergenic region and in the open sea (Figure S5). Following exclusion of intergenic DMRs, GO analysis performed with Metascape, regardless of DMRs methylation status evolution, showed an enrichment in genes related to negative regulation of fat cell differentiation, receptor‐mediated endocytosis, ion channel transport, and cilium compartment (Figure 5B), thus pointing toward persistent alterations in these pathways following early‐life inflammation.
Figure 5.
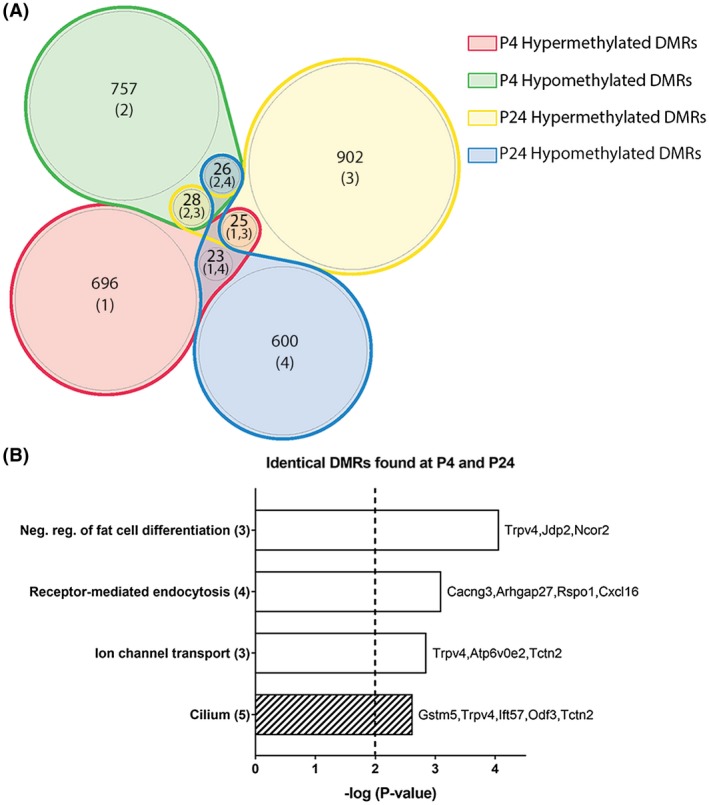
Analysis of similar DMRs found at P4 and P24 in rats subjected to inflammatory WMI. A, Venn diagram of the methylation status changes occurring between P4 and P24 of identical DMRs identified at both time points. B, Gene ontology (GO) enrichment analysis of the DMRs on all regions excluding intergenic. GO enrichment was performed for biological processes (plain bar), molecular functions (bar with vertical lines), and cellular components (bar with oblique lines). The number of genes associated to each pathway is in parenthesis
4. DISCUSSION
In the present study, we investigated methylation profile changes in rat brains subjected to neonatal LPS‐induced moderate‐to‐severe WMI at 24 hours and 21 days following exposure. We observed that WMI induced methylation perturbation in a period highly important for neurodevelopment that persisted up to 21 days post‐injury. We identified increased rate of methylation changes occurring in intergenic regions and CpG open‐sea, which could point toward altered genomic stability. Moreover, GO analysis showed that neonatal inflammation induced persistent methylation alterations in genes associated to neurodevelopmental and immune response pathways particularly in axonal and synaptic compartments, and in inflammatory response and leukocyte‐mediated immunity. Furthermore, we observed methylation alteration on genes enriched in pathways related to protein and lipid metabolism and receptor tyrosine kinase signaling.
DNA methylation is an important regulatory mechanism that partakes in neurodevelopmental processes, different neurological functions such as learning and memory and maintenance of homeostasis at adulthood.57, 58, 59, 60 Alteration of DNA methylation has been implicated in many neurodevelopmental and neurodegenerative diseases including autism spectrum disorder, Down syndrome, Alzheimer disease, and schizophrenia. In our study, neonatal inflammation‐induced WMI resulted in DNA methylation alteration that persisted up to 21 days post‐injury. In accordance to our results, animal studies showed that adverse events during the perinatal period can lead to sustained alteration of the brain methylome up to 1 year after the initial insult.61, 62 For example, exposure to maternal maltreatment during the first days of life in rats induced a global alteration of DNA methylation in the amygdala and hippocampus at adolescence (P30).61 Our datasets revealed that major portion of methylation changes occurred in intergenic and intronic regions and on CpG within the open sea. The susceptibility of these regions to methylation changes was also seen in models of maternal separation and in prenatal alcohol exposure.62, 63 It was suggested that methylation in intergenic and introns could target regulatory sequences that play a role in gene expression.63, 64, 65, 66
One major group of pathways that emerged from our methylome analysis following neonatal inflammation‐induced WMI was related to the axon. We noted that genes associated to axonal compartment had a shift from a hypomethylated to a hypermethylated state which could be associated to the presence of axonal injury as it has been confirmed in this model by diffusion tensor imaging and by immunohistochemistry using the apoptotic marker fractin.38, 40 Furthermore, using the microtubule‐associated protein 1 expressed in axons, Fan et al have confirmed that axonal fragmentation, already present 3 days after LPS exposure, was still there 70 days later.13, 67 In postmortem preterm infants brains, diffuse axonal injury has been shown in the white matter by immunostaining for fractin and other axonal damage markers.68 Surviving preterm infants had impaired hippocampal and cortical growth seen 7 years later69, 70 and even signs of increasing atrophy over time.71 Moreover, hypermethylation of synaptic processes and axonal compartment was associated to aging and cognitive decline in aging rat prefrontal cortices.72 Thus, the hypermethylation of genes related to axonal compartment, seen in our study, could reflect the onset of long‐lasting changes in neurodevelopment.
The enrichment of genes in pathways relevant to inflammation and immune response at P4 and P24 supports the presence of a sustained inflammatory state that could be caused in part by methylome alteration. As stated earlier, perinatal inflammation is associated to increased vulnerability to secondary insults at later time points in life particularly in the context of microglia priming. It was shown that microglia of adult rats exposed to E. coli bacteria in the neonatal period had a stronger response to a secondary LPS challenge compared to naïve rats’ microglia.18 Furthermore, neonatal infection in mice led to heightened susceptibility to amyloid beta‐induced neuroinflammation at adulthood with increased microglial activation and higher memory impairment in the novel object recognition test.73 Indeed, the methylation alterations in inflammatory pathways seen in this study could be a key component of a chronic low‐level inflammation state, which is being increasingly recognized as central to cognitive decline, neuropsychiatric, and neurodegenerative diseases.74, 75, 76, 77, 78 Several world leaders in neonatal neurobiology, actively studying the inflammation‐induced sensitization paradigm, are expressing grave concern over the increased risk of neurodegeneration following subsequent insults.79, 80, 81
In all, 102 identical DMRs were identified between P4 and P24 for which half preserved their methylation status and the other half shifted to the opposite status. DNA methylation is a dynamic process that evolves with age and in pathological conditions.82, 83, 84 During normal aging, GO analysis of peripheral blood samples from pediatric and aged population revealed that methylation changes were highly related to developmental and inflammatory pathways.85, 86 It was shown that aging was associated to decreased DNA methylation of pro‐inflammatory cytokines genes in both the brain and blood and these changes could render them more susceptible to be expressed following a secondary adverse insult.87 Furthermore, alteration of DNA methylation by adverse events can lead to discrepancy between chronological age and methylome age, which is associated to accelerated aging processes and age‐related pathologies including neurodegenerative diseases and decreased white matter integrity.88, 89, 90, 91, 92 Although the DNA methylation changes seen in this study were mostly subtle compared to changes occurring during development or in cancer, their accumulation could have the potential to impact the normal neurodevelopmental course.30 Therefore, it is possible that the changes in the epigenome seen in this study could play a major role in potentiating brain injury after neuroimmune challenges later in life.
A limitation of this study is that methylation data were derived from brain homogenates that are composed of different cell types. It was previously established that epigenetic difference could be seen within same tissue due to cell heterogeneity.93 Although a single‐cell methylome analysis would have allowed a better understanding of inflammation effect on different type of brain cells, it would have been less impactful to study the broad effect of early‐life inflammation on the brain methylome. Another limitation is the presence of pathways not related to the central nervous system like prostate morphogenesis or chondrocyte differentiation, this points toward the presence of gene networks not characterized in regard to brain development and/or brain‐related functions.94
In conclusion, we showed how postnatal inflammatory exposure caused a vast array of cerebral epigenomic alterations in genes relevant to inflammatory response and neurodevelopment with significant modifications still present 3 weeks after the injury. Given the long‐lasting impact of neonatal inflammation on methylation profile, we believe that the inclusion of epigenetic analysis will become mandatory in neonatal neuroprotection studies in the future and whether future preclinical studies should (i) consider an epigenomic intervention as done in psychiatric and cancer research and (ii) seek a protective role on the epigenetic signature by specific anti‐inflammatory therapies.
AUTHOR CONTRIBUTIONS
W.C. Pierre participated in data acquisition, data analysis, and manuscript draft, reviewed and revised the manuscript; L.‐M. Legault has contributed to the data acquisition, data analysis, and manuscript preparation and revision; I. Londono has contributed to the study preparation, data acquisition, and manuscript revision; G.A. Lodygensky and S. McGraw conceptualized and designed the study, contributed to data acquisition, supervised the data analysis, and revised the manuscript.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Supporting information
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes of Health Research (http://www.cihr-irsc.gc.ca/)—Institute of Human Development, Child and Youth Health (IHDCYH) (136908 to G.A.L.). G.A.L. and S.M are supported by a start‐up grant from the Research Center of CHU Sainte‐Justine. Salary support to L.M.L is provided by the Canadian Institutes of Health Research and to S.M. by the Fonds de Recherche Québec Santé.
Pierre WC, Legault L‐M, Londono I, McGraw S, Lodygensky GA. Alteration of the brain methylation landscape following postnatal inflammatory injury in rat pups. The FASEB Journal. 2020;34:432–445. 10.1096/fj.201901461R
Serge McGraw and Gregory A. Lodygensky contributed equally to this study.
REFERENCES
- 1. van Vliet EO, de Kieviet JF, Oosterlaan J, van Elburg RM. Perinatal infections and neurodevelopmental outcome in very preterm and very low‐birth‐weight infants: a meta‐analysis. JAMA Pediatr. 2013;167:662‐668. [DOI] [PubMed] [Google Scholar]
- 2. Thornton C, Rousset CI, Kichev A, et al. Molecular mechanisms of neonatal brain injury. Neurol Res Int. 2012;2012:506320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hagberg H, Mallard C, Ferriero DM, et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol. 2015;11:192‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strunk T, Inder T, Wang X, Burgner D, Mallard C, Levy O. Infection‐induced inflammation and cerebral injury in preterm infants. Lancet Infect Dis. 2014;14:751‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Volpe JJ. Cerebral white matter injury of the premature infant‐more common than you think. Pediatrics. 2003;112:176‐180. [DOI] [PubMed] [Google Scholar]
- 7. Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008;93:F153‐F161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble‐Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurogibol. 2013;106‐107:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pang Y, Cai Z, Rhodes PG. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Dev Brain Res. 2003;140:205‐214. [DOI] [PubMed] [Google Scholar]
- 11. Cai Z, Pang Y, Lin S, Rhodes PG. Differential roles of tumor necrosis factor‐alpha and interleukin‐1 beta in lipopolysaccharide‐induced brain injury in the neonatal rat. Brain Res. 2003;975:37‐47. [DOI] [PubMed] [Google Scholar]
- 12. Lodygensky GA, West T, Stump M, Holtzman DM, Inder TE, Neil JJ. In vivo MRI analysis of an inflammatory injury in the developing brain. Brain Behav Immun. 2010;24:759‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang KC, Fan LW, Kaizaki A, Pang Y, Cai Z, Tien LT. Neonatal lipopolysaccharide exposure induces long‐lasting learning impairment, less anxiety‐like response and hippocampal injury in adult rats. Neuroscience. 2013;234:146‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fan LW, Tien LT, Zheng B, et al. Dopaminergic neuronal injury in the adult rat brain following neonatal exposure to lipopolysaccharide and the silent neurotoxicity. Brain Behav Immun. 2011;25:286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan LW, Tien LT, Mitchell HJ, Rhodes PG, Cai Z. Alpha‐phenyl‐n‐tert‐butyl‐nitrone ameliorates hippocampal injury and improves learning and memory in juvenile rats following neonatal exposure to lipopolysaccharide. Eur J Neurosci. 2008;27:1475‐1484. [DOI] [PubMed] [Google Scholar]
- 16. Langley‐Evans SC, McMullen S. Developmental origins of adult disease. Med Princ Pract. 2010;19:87‐98. [DOI] [PubMed] [Google Scholar]
- 17. Power C, Kuh D, Morton S. From developmental origins of adult disease to life course research on adult disease and aging: insights from birth cohort studies. Annu Rev Public Health. 2013;34:7‐28. [DOI] [PubMed] [Google Scholar]
- 18. Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early‐life infection. J Neurosci. 2011;31:15511‐15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yin P, Li Z, Wang YY, et al. Neonatal immune challenge exacerbates seizure‐induced hippocampus‐dependent memory impairment in adult rats. Epilepsy Behav. 2013;27:9‐17. [DOI] [PubMed] [Google Scholar]
- 20. Tien LT, Kaizaki A, Pang Y, Cai Z, Bhatt AJ, Fan LW. Neonatal exposure to lipopolysaccharide enhances accumulation of alpha‐synuclein aggregation and dopamine transporter protein expression in the substantia nigra in responses to rotenone challenge in later life. Toxicology. 2013;308:96‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Debnath M, Venkatasubramanian G, Berk M. Fetal programming of schizophrenia: select mechanisms. Neurosci Biobehav Rev. 2015;49:90‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allswede DM, Buka SL, Yolken RH, Torrey EF, Cannon TD. Elevated maternal cytokine levels at birth and risk for psychosis in adult offspring. Schizophr Res. 2016;172:41‐45. [DOI] [PubMed] [Google Scholar]
- 23. Meyer U, Nyffeler M, Engler A, et al. The time of prenatal immune challenge determines the specificity of inflammation‐mediated brain and behavioral pathology. J Neurosci. 2006;26:4752‐4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder‐specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R‐33R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wischhof L, Irrsack E, Osorio C, Koch M. Prenatal LPS‐exposure–a neurodevelopmental rat model of schizophrenia–differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog Neuropsychopharmacol Biol Psychiatry. 2015;57:17‐30. [DOI] [PubMed] [Google Scholar]
- 26. Heinonen K, Eriksson JG, Lahti J, et al. Late preterm birth and neurocognitive performance in late adulthood: a birth cohort study. Pediatrics. 2015;135:e818‐e825. [DOI] [PubMed] [Google Scholar]
- 27. Lardenoije R, Iatrou A, Kenis G, et al. The epigenetics of aging and neurodegeneration. Prog Neurogibol. 2015;131:21‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2:607‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu X. DNA methylation and cognitive aging. Oncotarget. 2015;6:13922‐13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leenen FA, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenet. 2016;8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grova N, Schroeder H, Olivier JL, Turner JD. Epigenetic and neurological impairments associated with early life exposure to persistent organic pollutants. Int J Genomics. 2019;2019:2085496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Everson TM, Marsit CJ, Michael O'Shea T, et al. Epigenome‐wide analysis identifies genes and pathways linked to neurobehavioral variation in preterm infants. Sci Rep. 2019;9:6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith AK, Kilaru V, Klengel T, et al. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet B Neuropsychiatr Genet. 2015;168b:36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braun PR, Han S, Hing B, et al. Genome‐wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sparrow S, Manning JR, Cartier J, et al. Epigenomic profiling of preterm infants reveals DNA methylation differences at sites associated with neural function. Transl Psychiatry. 2016;6:e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cruickshank MN, Oshlack A, Theda C, et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan Q, Li S, Frost M, et al. Epigenetic signature of preterm birth in adult twins. Clin Epigenet. 2018;10:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akakpo L, Pierre WC, Jin C, Londono I, Pouliot P, Lodygensky GA. User‐independent diffusion tensor imaging analysis pipelines in a rat model presenting ventriculomegalia: a comparison study. NMR Biomed. 2017;30:e3793. [DOI] [PubMed] [Google Scholar]
- 39. Guevara E, Pierre WC, Tessier C, et al. Altered functional connectivity following an inflammatory white matter injury in the newborn rat: a high spatial and temporal resolution intrinsic optical imaging study. Front Neurosci. 2017;11:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lodygensky GA, Kunz N, Perroud E, et al. Definition and quantification of acute inflammatory white matter injury in the immature brain by MRI/MRS at high magnetic field. Pediatr Res. 2014;75:415‐423. [DOI] [PubMed] [Google Scholar]
- 41. Clancy B, Finlay BL, Darlington RB, Anand KJS. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931‐937. 10.1016/j.neuro.2007.1001.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramachandra R, Subramanian T. Atlas of the Neonatal Rat Brain. Boca Raton, FL: CRC Press; 2011. [Google Scholar]
- 43. Legault L‐M, Chan D, McGraw S. Rapid multiplexed reduced representation bisulfite sequencing library prep (rRRBS). Bio‐Protoc. 2019;9:e3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Magnus N, Garnier D, Meehan B, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci U S A. 2014;111:3544‐3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McGraw S, Zhang JX, Farag M, et al. Transient DNMT1 suppression reveals hidden heritable marks in the genome. Nucleic Acids Res. 2015;43:1485‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome‐scale DNA methylation profiling. Nat Protoc. 2011;6:468‐481. [DOI] [PubMed] [Google Scholar]
- 47. Boyle P, Clement K, Gu H, et al. Gel‐free multiplexed reduced representation bisulfite sequencing for large‐scale DNA methylation profiling. Genome Biol. 2012;13:R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009;10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biol. 2012;13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rahimi S, Martel J, Karahan G, et al. Moderate maternal folic acid supplementation ameliorates adverse embryonic and epigenetic outcomes associated with assisted reproduction in a mouse model. Hum Reprod. 2019;34:851‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Whidden L, Martel J, Rahimi S, Chaillet JR, Chan D, Trasler JM. Compromised oocyte quality and assisted reproduction contribute to sex‐specific effects on offspring outcomes and epigenetic patterning. Hum Mol Genet. 2016;25:4649‐4660. [DOI] [PubMed] [Google Scholar]
- 52. Smith ZD, Shi J, Gu H, et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017;549:543‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tripathi S, Pohl MO, Zhou Y, et al. Meta‐ and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18:723‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sandoval J, Heyn H, Moran S, et al. Validation of a DNA methylation microarray for 450 000 CpG sites in the human genome. Epigenetics. 2011;6:692‐702. [DOI] [PubMed] [Google Scholar]
- 56. Rhee SY, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nat Rev Genet. 2008;9:509‐515. [DOI] [PubMed] [Google Scholar]
- 57. Halder R, Hennion M, Vidal RO, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102‐110. [DOI] [PubMed] [Google Scholar]
- 58. Kim‐Ha J, Kim Y‐J. Age‐related epigenetic regulation in the brain and its role in neuronal diseases. BMB Rep. 2016;49:671‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neal M, Richardson JR. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim Biophys Acta. 2018;1864:432‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weng YL, Joseph J, An R, Song H, Ming GL. Epigenetic regulation of axonal regenerative capacity. Epigenomics. 2016;8:1429‐1442. [DOI] [PubMed] [Google Scholar]
- 61. Doherty TS, Forster A, Roth TL. Global and gene‐specific DNA methylation alterations in the adolescent amygdala and hippocampus in an animal model of caregiver maltreatment. Behav Brain Res. 2016;298:55‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McCoy CR, Rana S, Stringfellow SA, et al. Neonatal maternal separation stress elicits lasting DNA methylation changes in the hippocampus of stress‐reactive Wistar Kyoto rats. Eur J Neuorsci. 2016;44:2829‐2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lussier AA, Bodnar TS, Mingay M, et al. Prenatal alcohol exposure: profiling developmental DNA methylation patterns in central and peripheral tissues. Front Genet. 2018;9:610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schlesinger F, Smith AD, Gingeras TR, Hannon GJ, Hodges E. De novo DNA demethylation and noncoding transcription define active intergenic regulatory elements. Genome Res. 2013;23:1601‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rauscher GH, Kresovich JK, Poulin M, et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer. 2015;15:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karlsson L, Barbaro M, Ewing E, Gomez‐Cabrero D, Lajic S. Epigenetic alterations associated with early prenatal dexamethasone treatment. J Endocr Soc. 2019;3:250‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fan LW, Mitchell HJ, Tien LT, Rhodes PG, Cai Z. Interleukin‐1beta‐induced brain injury in the neonatal rat can be ameliorated by alpha‐phenyl‐n‐tert‐butyl‐nitrone. Exp Neurol. 2009;220:143‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lodygensky GA, Rademaker K, Zimine S, et al. Structural and functional brain development after hydrocortisone treatment for neonatal chronic lung disease. Pediatrics. 2005;116:1‐7. [DOI] [PubMed] [Google Scholar]
- 70. Thompson DK, Omizzolo C, Adamson C, et al. Longitudinal growth and morphology of the hippocampus through childhood: impact of prematurity and implications for memory and learning. Hum Brain Mapp. 2014;35:4129‐4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Monson BB, Anderson PJ, Matthews LG, et al. Examination of the pattern of growth of cerebral tissue volumes from hospital discharge to early childhood in very preterm infants. JAMA Pediatr. 2016;170:772‐779. [DOI] [PubMed] [Google Scholar]
- 72. Ianov L, Riva A, Kumar A, Foster TC. DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age‐related cognitive impairment. Front Aging Neurosci. 2017;9:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Frost PS, Barros‐Aragao F, da Silva RT, et al. Neonatal infection leads to increased susceptibility to Abeta oligomer‐induced brain inflammation, synapse loss and cognitive impairment in mice. Cell Death Dis. 2019;10:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bilbo SD, Biedenkapp JC, Der‐Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection‐induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase‐1 inhibition. J Neurosci. 2005;25:8000‐8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Knuesel I, Chicha L, Britschgi M, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. 2014;10:643‐660. [DOI] [PubMed] [Google Scholar]
- 78. Pierre WC, Smith PLP, Londono I, Chemtob S, Mallard C, Lodygensky GA. Neonatal microglia: the cornerstone of brain fate. Brain Behav Immun. 2017;59:333‐345. [DOI] [PubMed] [Google Scholar]
- 79. Degos V, Peineau S, Nijboer C, et al. G protein‐coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation‐induced sensitization to excitotoxic neurodegeneration. Ann Neurol. 2013;73:667‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tartaglione AM, Venerosi A, Calamandrei G. Early‐life toxic insults and onset of sporadic neurodegenerative diseases‐an overview of experimental studies. Curr Top Behav Neurosci. 2016;29:231‐264. [DOI] [PubMed] [Google Scholar]
- 81. Mottahedin A, Ardalan M, Chumak T, Riebe I, Ek J, Mallard C. Effect of neuroinflammation on synaptic organization and function in the developing brain: implications for neurodevelopmental and neurodegenerative disorders. Front Cell Neurosci. 2017;11:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71:882‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Numata S, Ye T, Hyde TM, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90:260‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lister R, Mukamel E. Turning over DNA methylation in the mind. Front Neurosci. 2015;9:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alisch RS, Barwick BG, Chopra P, et al. Age‐associated DNA methylation in pediatric populations. Genome Res. 2012;22:623‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gentilini D, Mari D, Castaldi D, et al. Role of epigenetics in human aging and longevity: genome‐wide DNA methylation profile in centenarians and centenarians’ offspring. Age. 2013;35:1961‐1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shinozaki G, Braun PR, Hing BWQ, et al. Epigenetics of delirium and aging: potential role of DNA methylation change on cytokine genes in glia and blood along with aging. Front Aging Neurosci. 2018;10:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Horvath S, Erhart W, Brosch M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci. 2014;111:15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre‐frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer's disease related cognitive functioning. Aging. 2015;7:1198‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hodgson K, Carless MA, Kulkarni H, et al. Epigenetic age acceleration assessed with human white‐matter images. J Neurosci. 2017;37:4735‐4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev. 2018;174:3‐17. [DOI] [PubMed] [Google Scholar]
- 93. Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD. Experience‐dependent epigenomic reorganization in the hippocampus. Learn Mem. 2017;24:278‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials