

Abstract
While (Ph2CN2)B(C6F5)3 is unstable, single electron transfer from Cp*2Co affords the isolation of stable products [Cp*2Co][Ph2CNNHB(C6F5)3] 1 and [Cp*Co(C5Me4CH2B(C6F5)3)] 2. The analogous combination of Ph2CN2 and BPh3 showed no evidence of adduct formation and yet single electron transfer from Cp*2Cr affords the species [Cp*2Cr][PhC(C6H4)NNBPh3] 3 and [Cp*2Cr][Ph2CNNHBPh3] 4. Computations showed both reactions proceed via transient radical anions of the diphenyldiazomethane–borane adducts to effect C−H bond activations.
Keywords: borane, DFT, diazomethane, one-electron reduction, radical anions
Single electron transfer to diphenyldiazomethane–borane adducts generates transient radical anions that effect C−H bond activations. Such reductions provide a strategy to stabilize weak and even undetectable donor–acceptor adducts.
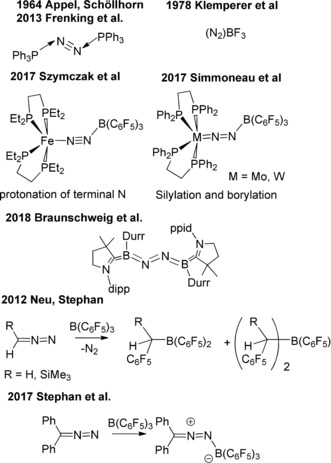
The activation of small molecules has been a major driver of organometallic chemistry over the last 60 years. Such efforts have spawned great interest and important developments. In recent years, such inquiries have begun to permeate main group chemistry. One avenue of main group chemistry exploited for the activation of small molecules has been frustrated Lewis pair (FLP) chemistry.1 While this initially emerged from the finding of the heterolytic activation of H2 by combinations of Lewis acids and bases,2 subsequent efforts demonstrated reactivity of FLPs with a wide range of small molecules.3 Noticeably absence from these investigations have been studies involving dinitrogen.
Organometallic chemists have studied metal–N2 systems since the seminal report of A. D. Allen4 who described the first transition metal–dinitrogen complex. Over the past 50 years numerous advances have emerged from the luminaries of organometallic chemistry including Schrock,5 Cummins,6 Peters,7 Fryzuk,8 Evans,9 Gambarotta,10 Nishibiashi,11 Holland,12 Chatt,13 and Liddle14 among others.15 Avenues to metal‐mediated N2 chemistry have typically involved stoichiometric reductants.8d More recently, in 2017 the Szymczak16 and Simonneau17 groups demonstrated the utility of a Lewis acidic borane in promoting reactivity of metal‐bound N2 fragments, effecting protonation, borylation and silylation of N2 bound between the metal and boron.
Main group interactions with N2 have drawn much less attention. A number of computational studies have addressed the interactions of N2 with Lewis acids, while the species (N2)BF3 was spectroscopically characterized upon generation by supersonic expansion at 600 torr and 170 K.18 The compound Ph3PNNPPh3 19 although not derived from N2, was controversially described as N2 stabilized by two phosphine donors.20 However, in a truly seminal finding, Braunschweig et al.21 described the first metal‐free capture of N2 using a cAAC‐stabilized borylene (cAAC: cyclic (alkyl)(amino) carbene).
In our own efforts towards main group‐N2 chemistry, we initiated studies of diazomethanes which liberate N2. Such systems may provide insight for the design of main group‐N2 systems.22 In 2012, we reported the insertion of diazomethanes into B−C bonds of electrophilic boranes with the liberation of N2 (Scheme 1).23 Such insertions were recently exploited in organic synthesis by Melen et al.24 In recent work,25 we showed that the sterically‐encumbered diazomethane, Ph2CN2, does not insert but rather forms a highly reactive, yet isolable borane‐adduct, (Ph2CN2)B(C6F5)3. Moreover, we also showed weak Lewis acid–base adducts were stabilized by stoichiometric reduction.26 This notion was also exploited by the Erker group in the isolation of Lewis acid stabilized radicals.27 Herein, we probe the impact of reduction on the reactivity of the unstable (Ph2CN2)B(C6F5)3, demonstrating that single electron transfer to diazomethane‐borane adducts stabilizes weak B⋅⋅⋅N interactions providing reactive transient radicals which effect C−H bond activation.
Scheme 1.
Interactions of main group systems with N2‐fragments.
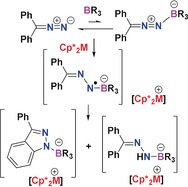
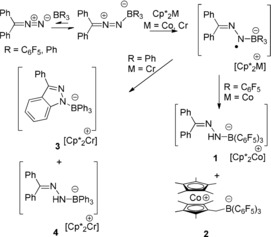
A 1:1 combination of diphenyldiazomethane (Ph2CN2) with B(C6F5)3 in chlorobenzene was stirred at −35 °C. Addition of an equal molar amount of Cp*2Co immediately gave a yellow solution. The crude 19F NMR spectrum showed two sets of resonances at −134.0, −163.9, and −167.3 ppm and −130.2, −161.9, −162.7 ppm, attributable to inequivalent C6F5 rings. The 11B NMR spectrum showed two resonances at −7.6 and −13.0 ppm attributable to two tetra‐coordinated boron species, 1 and 2, respectively. 1H NMR data showed resonances at 6.61 and 2.43 ppm attributable to NH and CH2 fragments. Fractional recrystallization permitted formulation of the two products 1 and 2 by X‐ray crystallographic analysis. Compound 1 was found to be the salt [Cp*2Co][Ph2CNNHB(C6F5)3] (Figure 1 a). While the cation was unexceptional, the diazoborate anion was derived from the interaction of the hydrazide bound to borane. The B−N(H) bond length in 1 is 1.539(7) Å, while the N−N and N−C bonds lengths is 1.342(5) and 1.303(7) Å, respectively. The B−N−N angle was determined to be 118.2(3)° while the C−N−N angle is 120.9(4)°. The second isolated product was confirmed to be [Cp*Co(C5Me4CH2B(C6F5)3)] 2 (Figure 1 b). In this species, one of the hydrogen atoms in one of the Cp* methyl groups has been replaced by borane, affording the zwitterionic CoIII‐borate 2, with a methylene–boron B−C bond length of 1.66(1) Å.
Figure 1.
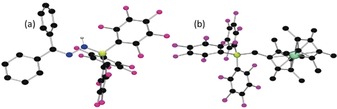
POV‐Ray depiction of the anion of a) 1 and b) 2. The cation and hydrogen atoms (except NH) are omitted for clarity. C: black, N: blue, F: pink, B: yellow‐green, Co: green, and H: grey.
Collectively, the identification of 1 and 2 is consistent with two possible reaction mechanisms involving single electron transfer from a CoII center to either B(C6F5)3 28 or the diazomethane adduct of the borane, (Ph2CN2)B(C6F5)3.25 It is noteworthy that although C−H activation by the radical [B(C6F5)3)].−,29 is expected to give the anion [HB(C6F5)3]−, independent combination of diazomethane with [HB(C6F5)3]− showed no reaction. This supports the view that compound 1 is formed through hydrogen atom abstraction from Cp*2Co by the transient diazomethane‐borane adduct radical anion [Ph2CN2B(C6F5)3].− (Scheme 2), consistent with the overall reaction ratio of diazomethane:Cp*2Co:B(C6F5)3 of 1:2:2.
Scheme 2.
Reactions of Ph2CN2 with B(C6F5)3 and Cp*2Co, and with BPh3 and Cp*2Cr.
Ph2CN2 was combined with BPh3 in chlorobenzene at −35 °C. Monitoring the solution by multinuclear NMR spectroscopy revealed no evidence of adduct formation. This is consistent with the poor Lewis basicity of the diazomethane and the weaker Lewis acidity of the BPh3 in comparison to B(C6F5)3, in line with the computed free energies (see Supporting Information). Addition of Cp*2Cr to a mixture of BPh3 and diazomethane at −35 °C generated an orange solution. The 11B NMR spectrum showed resonances at 23.0 and −3.5 ppm consistent with the formation of two products, 3 and 4 which were isolated by fractional crystallization. An X‐ray diffraction study revealed species 3 to be [Cp*2Cr][PhC(C6H4)NNBPh3] (Scheme 2, Figure 2 a). While the cation was typical, the anion of 3 was shown to be a borate with a substituent derived from the cyclization of the N2 fragment onto the ortho position of one of the aryl rings on the diazomethane carbon. The resulting five membered ring which is fused to the aryl ring is 1,3‐disubstituted with and phenyl ring on carbon and BPh3 bound to nitrogen. The resulting N−B bond is 1.566(6) Å, while the N−N and new N−C bond distances are determined to be 1.312(5) and 1.351(5) Å. The second product 4 was also characterized crystallographically revealing its formulation as [Cp*2Cr][Ph2CNNHBPh3] (Scheme 2, Figure 2 b). The B−N and N−N distances in the anion of 4 were determined to be 1.562(4) Å and 1.322(3) Å, respectively.
Figure 2.
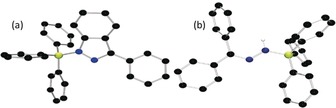
POV‐Ray depiction of the anion of a) 3 and b) 4. The cation and hydrogen atoms (except NH) are omitted for clarity. C: black, N: blue, and B: yellow‐green.
In contrast, the reaction of 9‐diazofluorene ((C12H8)CN2) with B(C6F5)3 did not form an adduct but led to loss of N2 (Scheme 3) and the formation of the carboboration product (C12H8)C(C6F5)(B(C6F5)2) as confirmed spectroscopically and crystallographically (see Supporting Information). This is consistent with observations seen for less sterically encumbered diazomethanes.23, 30 However, monitoring the reaction of (C12H8)CN2 with BPh3 and Cp*2Cr by 11B NMR spectroscopy revealed the generation of three products as evidenced by the resonances at 26.2, 2.4, and −1.7 ppm. The peaks at −1.7 and 2.4 ppm were unambiguously assigned to [Cp*2Cr][C12H8CNNHBPh3] 5 and [Cp*Cr(C5Me4CH2BPh3)] 7 by NMR and crystallographic methods (Figure 3, see Supporting Information). The remaining resonance at 26.2 ppm, was attributed to the species [Cp*2Cr][C13H7N2BPh3] 6 by analogy to 3. These results suggest that the electron transfer to the weak adducts gives a transient radical [(C12H8)CN2BPh3].−, which reacts further through competitive pathways involving either intramolecular cyclization or intermolecular H‐atom abstraction.
Scheme 3.
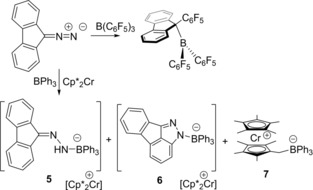
Reactions of (C12H8)CN2 with BPh3 and Cp*2Cr.
Figure 3.
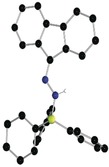
POV‐Ray depiction of the anion of 5. The cation and hydrogen atoms (except NH) are omitted for clarity. C: black, N: blue, and B: yellow‐green.
The mechanism of these reactions were probed by density functional theory (DFT) calculations at the PW6B95‐D3/def2‐QZVP + COSMO‐RS// TPSS‐D3/def2‐TZVP + COSMO level of theory in chlorobenzene solution.31 The reaction of Ph2CN2 with BPh3 and Cp*2Cr is initiated by single electron transfer from Cp*2Cr to the unstable Ph2CN2⋅BPh3 adduct (Figure 4) affording the radical anion [Ph2CNNHBPh3].− INT1 (spin on N next to B: 0.53e) is 4.3 kcal mol−1 endergonic. Alternative pathways involving electron transfers to separated BPh3 and Ph2CN2 species (14.8 and 8.9 kcal mol−1) are significantly less favorable. Similarly, further electron transfer to INT1 is unlikely (12.2 kcal mol−1 endergonic). The N‐centered radical INT1 may then add intramolecularly to the ortho position of a phenyl ring (via transition structure TS1) to give INT2 affording delocalization of the spin onto the ring. From here, a highly exergonic H‐transfer to another Ph2CN2 molecule (via TS2) gives the anion of 3 and the neutral N‐radical (Ph2CNNH). (spin on N next to H: 0.54e) with a moderate overall barrier of 23.5 kcal mol−1. The latter radical is readily reduced by Cp*2Cr through electron transfer and trapped by BPh3 giving the anion of 4 (−66.4 kcal mol−1).
Figure 4.
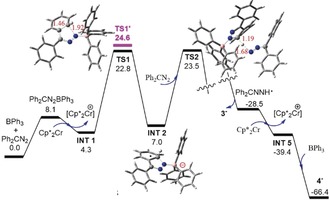
DFT‐computed free energy profile (kcal mol−1, at 298 K and 1 mol L−1 reference concentration) for the formation of anion of 3 and 4. Selected bond lengths are given in angstroms while selected C: grey, H: white, N: blue, and B: pink.
For the reaction of Ph2CN2 with the stronger Lewis acid B(C6F5)3 and the more reductive Cp*2Co,32 electron transfer from Cp*2Co to the reversible adduct (Ph2CNN)B(C6F5)3 is −15.3 kcal mol−1 exergonic affording the radical anion [Ph2CN2B(C6F5)3]⋅−. This intermediate is computed to effect H‐atom from one methyl group of Cp*2Co over a barrier of 16.6 kcal mol−1 to form the stable anion of 1 (see Supporting Information). The alternative intramolecular pathway, analogous to that above encounters a higher overall barrier of 23.3 kcal mol−1 (see Supporting Information).
To garner further support for the computed mechanism, efforts to observe the transient radical adducts in the reactions were undertaken, but were unsuccessful. However, monitoring the reaction of (C12H8)CN2, Cp*2Fe and Al(C6F5)3 in C6H5Cl at room temperature by EPR spectroscopy revealed a pentet resonance at g(iso)=2.0039, with (14N) hyperfine couplings of 3.70 G and 3.58 G. This signal was similar to the related N‐based radicals33 and was attributed to the radical species [Ph2CN2Al(C6F5)3].−. This signal slowly degrades at room temperature over 5 h, leaving a broad resonance attributed to an organic radical (see Supporting Information). Subsequent addition of Ph3SnH generated a mixture of products, from which [Cp*2Fe][(C12H8)CNNHAl(C6F5)3] 8 was identified by NMR spectroscopy while single crystals of [Cp*2Fe][(C12H8)CHAl(C6F5)3] 9 were obtained from the reaction mixture (Figure 5). Compound 8 was independently prepared and crystallographically characterized from the reaction of (C12H8)CNNH2, Cp*2Fe, and Al(C6F5)3. The formulations of 8 and 9 are consistent with the generation of the spectroscopically observed radical anions [(C12H8)CN2Al(C6F5)3].− and [(C12H8)CAl(C6F5)3].− (Scheme 4).
Figure 5.
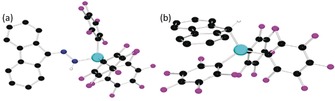
POV‐Ray depiction of the anion of a) 8 and b) 9. The cation and hydrogen atoms (except the NH and the Al‐bound CH) are omitted for clarity. C: black, N: blue, Al: Cyan, and F: pink.
Scheme 4.
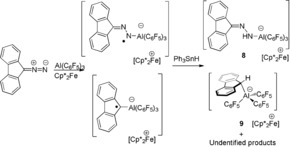
Reactions of (C12H8)CN2, Al(C6F5)3 and Cp*2Fe with Ph3SnH.
In conclusion, we have demonstrated that single electron transfer to unstable diazomethane‐borane adducts, accesses reactive radical anions that effect H‐atom abstraction from C−H bonds. The resulting anionic species are significantly more stable than the corresponding neutral adducts, suggesting that in situ reductions may be a useful strategy to infer the presence of weakly bound adducts. We suggest that this strategy could be exploited in developing main group‐N2 chemistry. At the same time, the potential utility of main group radical anions in C−H bond homolysis offers an interesting prospect for C−H functionalization.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
D.W.S. gratefully acknowledges the financial support from NSERC Canada and the award of Canada Research Chair. D.W.S. is also grateful for the award of an Einstein Visiting Fellowship at TU Berlin. L.L.C. is grateful for the award of NSERC scholarship. Z.‐W.Q. thanks the Deutsche Forschungsgemeinschaft (Leibniz Prize to Prof. Stefan Grimme) for funding.
L. L. Cao, J. Zhou, Z.-W. Qu, D. W. Stephan, Angew. Chem. Int. Ed. 2019, 58, 18487.
In memory of Nicholas C. Payne
The copyright line for this article was changed on 2 January 2020 after original online publication.
Contributor Information
Dr. Zheng‐Wang Qu, Email: qu@thch.uni-bonn.de.
Prof. Dr. Douglas W. Stephan, Email: dstephan@chem.utoronto.ca.
References
- 1. Stephan D. W., Science 2016, 354, aaf7229. [Google Scholar]
- 2. Welch G. C., Juan R. R. S., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. McCahill J. S. J., Welch G. C., Stephan D. W., Angew. Chem. Int. Ed. 2007, 46, 4968–4971; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5056–5059; [Google Scholar]
- 3b. Dureen M. A., Brown C. C., Stephan D. W., Organometallics 2010, 29, 6594–6607; [Google Scholar]
- 3c. Dureen M. A., Stephan D. W., J. Am. Chem. Soc. 2009, 131, 8396–8398; [DOI] [PubMed] [Google Scholar]
- 3d. Dureen M. A., Welch G. C., Gilbert T. M., Stephan D. W., Inorg. Chem. 2009, 48, 9910–9917; [DOI] [PubMed] [Google Scholar]
- 3e. Otten E., Neu R. C., Stephan D. W., J. Am. Chem. Soc. 2009, 131, 9918–9919; [DOI] [PubMed] [Google Scholar]
- 3f. Neu R. C., Otten E., Stephan D. W., Angew. Chem. Int. Ed. 2009, 48, 9709–9712; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9889–9892; [Google Scholar]
- 3g. Morton J. G. M., Dureen M. A., Stephan D. W., Chem. Commun. 2010, 46, 8947–8949; [DOI] [PubMed] [Google Scholar]
- 3h. Chi J. J., Johnstone T. C., Voicu D., Mehlmann P., Dielmann F., Kumacheva E., Stephan D. W., Chem. Sci. 2017, 8, 3270–3275; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3i. Chen J., Falivene L., Caporaso L., Cavallo L., Chen E. Y., J. Am. Chem. Soc. 2016, 138, 5321–5333; [DOI] [PubMed] [Google Scholar]
- 3j. Sajid M., Elmer L.-M., Rosorius C., Daniliuc C. G., Grimme S., Kehr G., Erker G., Angew. Chem. Int. Ed. 2013, 52, 2243–2246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2299–2302; [Google Scholar]
- 3k. Dobrovetsky R., Stephan D. W., J. Am. Chem. Soc. 2013, 135, 4974–4977; [DOI] [PubMed] [Google Scholar]
- 3l. Sajid M., Stute A., Cardenas A. J. P., Culotta B. J., Hepperle J. A. M., Warren T. H., Schirmer B., Grimme S., Studer A., Daniliuc C. G., Frohlich R., Petersen J. L., Kehr G., Erker G., J. Am. Chem. Soc. 2012, 134, 10156–10168; [DOI] [PubMed] [Google Scholar]
- 3m. Stephan D. W., Erker G., Chem. Sci. 2014, 5, 2625–2641; [Google Scholar]
- 3n. Ye K. Y., Bursch M., Qu Z. W., Daniliuc C. G., Grimme S., Kehr G., Erker G., Chem. Commun. 2017, 53, 633–635; [DOI] [PubMed] [Google Scholar]
- 3o. Longobardi L. E., Wolter V., Stephan D. W., Angew. Chem. Int. Ed. 2015, 54, 809–812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 823–826. [Google Scholar]
- 4. Allen A. D., Senoff C. V., J. Chem. Soc. Chem. Commun. 1965, 621–622. [Google Scholar]
- 5.
- 5a. Schrock R. R., Acc. Chem. Res. 2005, 38, 955–962; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Schrock R. R., Philos. Trans. R. Soc. London Ser. A 2005, 363, 959–969; [DOI] [PubMed] [Google Scholar]
- 5c. Schrock R. R., Angew. Chem. Int. Ed. 2008, 47, 5512–5522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5594–5605. [Google Scholar]
- 6. Cummins C. C., Chem. Commun. 1998, 1777–1786. [Google Scholar]
- 7. Anderson J. S., Rittle J., Peters J. C., Nature 2013, 501, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Fryzuk M. D., Johnson S. A., Coord. Chem. Rev. 2000, 200–202, 379–409; [Google Scholar]
- 8b. MacKay B. A., Fryzuk M. D., Chem. Rev. 2004, 104, 385–401; [DOI] [PubMed] [Google Scholar]
- 8c. MacLachlan E. A., Fryzuk M. D., Organometallics 2006, 25, 1530–1543; [Google Scholar]
- 8d. Burford R. J., Fryzuk M. D., Nat. Rev. Chem. 2017, 1, 0026. [Google Scholar]
- 9.
- 9a. Woen D. H., Chen G. P., Ziller J. W., Boyle T. J., Furche F., Evans W. J., J. Am. Chem. Soc. 2017, 139, 14861–14864; [DOI] [PubMed] [Google Scholar]
- 9b. Evans W. J., Lee D. S., Can. J. Chem. 2005, 83, 375–384. [Google Scholar]
- 10.
- 10a. Gambarotta S., J. Organomet. Chem. 1995, 500, 117–126; [Google Scholar]
- 10b. Gambarotta S., Inorg. Chem. Highlights 2002, 285–295; [Google Scholar]
- 10c. Gambarotta S., Scott J., Angew. Chem. Int. Ed. 2004, 43, 5298–5308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 5412–5422. [Google Scholar]
- 11. Tanaka H., Nishibayashi Y., Yoshizawa K., Acc. Chem. Res. 2016, 49, 987–995. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Holland P. L., Dalton Trans. 2010, 39, 5415–5425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. MacLeod K. C., Holland P. L., Nat. Chem. 2013, 5, 559–565; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. McWilliams S. F., Holland P. L., Acc. Chem. Res. 2015, 48, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chatt J., J. Organomet. Chem. 1975, 100, 17–28. [Google Scholar]
- 14.
- 14a. Doyle L. R., Wooles A. J., Jenkins L. C., Tuna F., McInnes E. J. L., Liddle S. T., Angew. Chem. Int. Ed. 2018, 57, 6314–6318; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6422–6426; [Google Scholar]
- 14b. Gardner B. M., Liddle S. T., Eur. J. Inorg. Chem. 2013, 3753–3770; [Google Scholar]
- 14c. Liddle S. T., Angew. Chem. Int. Ed. 2015, 54, 8604–8641; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8726–8764. [Google Scholar]
- 15. Kuganathan N., Green J. C., Himmel H.-J., New J. Chem. 2006, 30, 1253–1262. [Google Scholar]
- 16. Geri J. B., Shanahan J. P., Szymczak N. K., J. Am. Chem. Soc. 2017, 139, 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simonneau A., Turrel R., Vendier L., Etienne M., Angew. Chem. Int. Ed. 2017, 56, 12268–12272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12436–12440. [Google Scholar]
- 18. Janda K. C., Bernstein L. S., Steed J. M., Novick S. E., Klemperer W., J. Am. Chem. Soc. 1978, 100, 8074. [Google Scholar]
- 19. Appel R., Schöllhorn R., Angew. Chem. Int. Ed. Engl. 1964, 3, 805; [Google Scholar]; Angew. Chem. 1964, 76, 992. [Google Scholar]
- 20. Holzmann N., Dange D., Jones C., Frenking G., Angew. Chem. Int. Ed. 2013, 52, 3004–3008; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3078–3082. [Google Scholar]
- 21.
- 21a. Légaré M.-A., Bélanger-Chabot G., Dewhurst R. D., Welz E., Krummenacher I., Engels B., Braunschweig H., Science 2018, 359, 896–900; [DOI] [PubMed] [Google Scholar]
- 21b. Broere D. L., Holland P. L., Science 2018, 359, 871; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Légaré M.-A., Rang M., Bélanger-Chabot G., Schweizer J. I., Krummenacher I., Bertermann R., Arrowsmith M., Holthausen M. C., Braunschweig H., Science 2019, 363, 1329–1332. [DOI] [PubMed] [Google Scholar]
- 22. Itoh T., Nakata Y., Hirai K., Tomioka H., J. Am. Chem. Soc. 2006, 128, 957–967. [DOI] [PubMed] [Google Scholar]
- 23. Neu R. C., Jiang C., Stephan D. W., Dalton Trans. 2013, 42, 726–736. [DOI] [PubMed] [Google Scholar]
- 24. Santi M., Ould D. M. C., Wenz J., Soltani Y., Melen R. L., Wirth T., Angew. Chem. Int. Ed. 2019, 58, 7861–7865; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7943–7947. [Google Scholar]
- 25. Tang C. N., Liang Q. M., Jupp A. R., Johnstone T. C., Neu R. C., Song D. T., Grimme S., Stephan D. W., Angew. Chem. Int. Ed. 2017, 56, 16588–16592; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16815–16819. [Google Scholar]
- 26. Liu L., Cao L. L., Shao Y., Stephan D. W., J. Am. Chem. Soc. 2017, 139, 10062–10071. [DOI] [PubMed] [Google Scholar]
- 27. Tao X., Daniliuc C. G., Knitsch R., Hansen M. R., Eckert H., Lübbesmeyer M., Studer A., Kehr G., Erker G., Chem. Sci. 2018, 9, 8011–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kwaan R. J., Harlan C. J., Norton J. R., Organometallics 2001, 20, 3818–3820. [Google Scholar]
- 29. Harlan C. J., Hascall T., Fujita E., Norton J. R., J. Am. Chem. Soc. 1999, 121, 7274–7275. [Google Scholar]
- 30. Neu R. C., Stephan D. W., Organometallics 2012, 31, 46–49. [Google Scholar]
- 31.
- 31a.TURBOMOLE, version 7.3, 2018;
- 31b. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 2, 799; [Google Scholar]
- 31c. Eckert F., Klamt A., AIChE J. 2002, 48, 369–385; [Google Scholar]
- 31d.F. Eckert, A. Klamt, COSMOtherm, Version C3.0, Release 16.01, 2015;
- 31e. Eichkorn K., Weigend F., Treutler O., Ahlrichs R., Theor. Chem. Acc. 1997, 97, 119; [Google Scholar]
- 31f. Weigend F., Häser M., Patzelt H., Ahlrichs R., Chem. Phys. Lett. 1998, 143, 294; [Google Scholar]
- 31g. Weigend F., Furche F., Ahlrichs R., J. Chem. Phys. 2003, 119, 12753; [Google Scholar]
- 31h. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; [DOI] [PubMed] [Google Scholar]
- 31i. Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057–1065; [DOI] [PubMed] [Google Scholar]
- 31j. Tao J., Perdew J. P., Staroverov V. N., Scuseria G. E., Phys. Rev. Lett. 2003, 91, 146401; [DOI] [PubMed] [Google Scholar]
- 31k. Deglmann P., May K., Furche F., Ahlrichs R., Chem. Phys. Lett. 2004, 384, 103; [Google Scholar]
- 31l. Zhao Y., Truhlar D. G., J. Phys. Chem. A 2005, 109, 5656–5667; [DOI] [PubMed] [Google Scholar]
- 31m. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 31n. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 31o. Grimme S., Chem. Eur. J. 2012, 18, 9955. [DOI] [PubMed] [Google Scholar]
- 32. Robbins J. L., Edelstein N., Spencer B., Smart J. C., J. Am. Chem. Soc. 1982, 104, 1882–1893. [Google Scholar]
- 33.
- 33a. Berezin A. A., Constantinides C. P., Mirallai S. I., Manoli M., Cao L. L., Rawson J. M., Koutentis P. A., Org. Biomol. Chem. 2013, 11, 6780–6795; [DOI] [PubMed] [Google Scholar]
- 33b. Cowley H. J., Hayward J. J., Pratt D. R., Rawson J. M., Dalton Trans. 2014, 43, 1332–1337; [DOI] [PubMed] [Google Scholar]
- 33c. Waked A. E., Memar R. O., Stephan D. W., Angew. Chem. Int. Ed. 2018, 57, 11934–11938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12110–12114. [Google Scholar]
- 34.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201912338 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary