

Abstract
Previous analysis of next‐generation sequencing (NGS) hereditary pan‐cancer panel testing demonstrated that approximately 40% of TP53 pathogenic and likely pathogenic variants (PVs) detected have NGS allele frequencies between 10% and 30%, indicating that they likely are acquired somatically. These are seen more frequently in older adults, suggesting that most result from normal aging‐related clonal hematopoiesis. For this analysis, apparent heterozygous germline TP53 PV carriers (NGS allele frequency 30–70%) were offered follow‐up testing to confirm variant origin. Ninety‐eight probands had samples submitted for follow‐up family member testing, fibroblast testing, or both. The apparent heterozygous germline TP53 PV was not detected in 32.6% (15/46) of submitted fibroblast samples, indicating that it was acquired somatically, either through clonal hematopoiesis or via constitutional mosaicism. Notably, no individuals with confirmed germline or likely germline TP53 PVs met classic Li–Fraumeni syndrome (LFS) criteria, only 41% met Chompret LFS criteria, and 59% met neither criteria, based upon provider‐reported personal and family cancer history. Comprehensive reporting of TP53 PVs detected using NGS, combined with follow‐up analysis to confirm variant origin, is advised for clinical testing laboratories. These findings underscore the investment required to provide individuals and family members with clinically accurate genetic test results pertaining to their LFS risk.
Keywords: acquired somatically, hereditary pan‐cancer gene panel, Li–Fraumeni syndrome, next‐generation sequencing, pathogenic variant, TP53
In next‐generation sequencing (NGS) hereditary cancer panel testing, TP53 pathogenic variants whose NGS allele frequencies might appear consistent with those expected for germline variants, but the variant might be acquired somatically. Confirming TP53 variant origin can markedly impact medical management for tested individuals and their family members. The report highlights the need for a conservative approach to initial reporting of TP53‐positive findings, along with investment in follow‐up testing to deliver clinically accurate results.
1. INTRODUCTION
Individuals who have Li–Fraumeni syndrome (LFS; MIM# 151623) have a high risk of developing sarcomas, brain tumors, adrenocortical carcinomas, premenopausal breast cancer, leukemia, and other cancers early in life (Sorrell, Espenschied, Culver, & Weitzel, 2013). Though no studies have directly measured life expectancy in LFS, it is known to be severely reduced (Evans & Ingham, 2013). Germline pathogenic variants (PVs) in TP53 are the most common cause of LFS. Children who carry a germline PV in TP53 have an approximately 42% risk of developing a TP53‐related cancer before age 16 (Le Bihan, Moutou, Brugières, Feunteun, & Bonaïti‐Pellié, 1995), and adult men have an approximately 73% lifetime risk of cancer (Chompret et al., 2000). Women who have germline TP53 PVs have nearly a 100% chance of developing breast cancer during their lifetimes (Chompret et al., 2000). For these reasons, recent expert panel recommendations emphasize comprehensive, lifelong screening across numerous cancer types, beginning as soon as an individual has been confirmed to carry a TP53 PV or meets the “classic clinical definition” of LFS with no TP53 PV. This follows a modified version of the “Toronto protocol” (Kratz et al., 2017), which factors in the age and sex of the individual. In addition, National Comprehensive Cancer Network (NCCN) guidelines recommend that individuals with LFS receive aggressive cancer screening, including: annual breast MRI starting at age 20; colonoscopy every 2–5 years starting at age 25; whole‐body MRI; esophagogastroduodenoscopy; physical exam every 6–12 months; and a dermatologic exam starting at age 18 years (Daly et al., 2019). For women with LFS, risk‐reducing mastectomy also should be considered.
Due to the high cancer risks conferred by germline TP53 PVs, hereditary pan‐cancer gene panels typically include TP53. Recent work showed that apparent germline TP53 PVs were not uncommon among individuals who received hereditary cancer panel testing, accounting for 1% of germline PVs identified (Rosenthal, Bernhisel, Brown, Kidd, & Manley, 2017). Next‐generation sequencing (NGS) is used to accommodate the large number of genes often included in hereditary cancer panels; however, NGS is more sensitive than traditional sequencing methods and can detect variants at very low‐allele frequencies. Heterozygous germline variants will have NGS sequence reads with approximately a 1:1 ratio to those of the wild‐type allele, producing an NGS allele frequency of about 50%. Allele frequencies that differ significantly from 50% in blood‐derived samples suggest that the variant occurs in only a proportion of blood cells and therefore was acquired somatically. Somatic variants also can be detected in saliva‐derived samples, although the allele frequencies in saliva may be different than in blood, as saliva samples often contain a mix of epithelial and white blood cells.
A previous study showed that likely somatic PVs accounted for 0.71% of all PVs identified during hereditary cancer panel testing, with somatic PVs occurring most commonly in TP53 and among older individuals (Coffee et al., 2017). This was consistent with previous work, which has shown that clonal expansion of blood cell subpopulations carrying somatic variants during hematopoiesis, also termed clonal hematopoiesis of indeterminate potential (CHIP), has been observed for a number of genes (Genovese et al., 2014; Jaiswal et al., 2014; Mitchell et al., 2018; Xie et al., 2014) and occurs with normal aging (Steensma et al., 2015). However, it was notable that in one case, the NGS allele frequency of a TP53 PV increased from less than 30% (likely somatic range) to 45% (apparent germline range) when re‐tested after 3 months (Coffee et al., 2017). This led to the hypothesis that the NGS allele frequency range for somatically acquired TP53 PVs may overlap with that expected for heterozygous germline variant alleles, thus potentially confounding test result interpretation.
Unlike germline PVs, variants acquired somatically through CHIP do not carry hereditary cancer risk, though they are associated with other risks such as hematologic cancer, cardiovascular disease, and all‐cause mortality (Genovese et al., 2014; Gillis et al., 2017; Jaiswal et al., 2014; Jaiswal et al., 2017; Takahashi et al., 2017). Given the severe clinical interventions for individuals with LFS, it is critical that apparent germline PVs in TP53 be investigated further to determine whether they are truly in the germline. For this reason, our laboratory offers follow‐up testing to individuals with an apparent germline TP53 PV identified during the course of clinical hereditary cancer testing, to determine whether the PV was germline or acquired somatically. This report presents the results of follow‐up testing.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
Only deidentified data collected as part of clinical testing were used in this analysis. All individuals provided informed consent for clinical testing. The analysis set did not include individuals from states with laws that prevent the use of deidentified genetic data. The corresponding and senior authors confirm that they had full access to all data used in the analysis and take responsibility for data integrity and the accuracy of the data analysis. All authors are employees of Myriad Genetic Laboratories, Inc., and receive salary and stock options as compensation.
2.2. Cohort and genetic testing
This analysis assessed individuals who had hereditary pan‐cancer panel testing (Myriad Genetic Laboratories, Inc., Salt Lake City, UT) as part of normal clinical care between September 2013 and February 2018. Testing was performed using blood or saliva samples. Validation of the NGS panel has been described previously and included analysis of TP53 (Judkins et al., 2015). Sequence variants and large rearrangements (LRs) were analyzed relative to the wild‐type TP53 gene sequence (RefSeq NM_000546.5). For sequence variants, apparent germline PVs were defined as those identified with an NGS allele frequency between 30% and 70% and which received a laboratory classification of Pathogenic or Likely Pathogenic. This threshold was assigned to minimize false negative results and was based on our laboratory's experience and expertise in clinical genetic testing. The broad range captures potential germline allele frequency imbalances that may occur due to technical or biological reasons. For LR variants, apparent germline PVs were identified through NGS dosage analysis, which used normalized read counts from sequencing amplicons to determine gene copy number. While this approach did not allow assignment of precise allele frequencies for LRs, dosage increases or decreases relative to those expected for a heterozygous germline deletion or duplication could be evaluated. A germline LR typically presents as a 50% increase or decrease in dosage.
All sequence PVs were confirmed by Sanger sequencing. Pathogenic LRs were detected by NGS and confirmed by microarray, multiplex ligation‐dependent probe amplification, and/or repeat NGS. Individuals who were found to have an apparent heterozygous germline PV in TP53 were offered follow‐up testing at no additional cost to determine whether the variant was germline or somatic. Clinical information, including age, personal cancer history, and family cancer history, was obtained from provider‐completed test request forms and deidentified.
2.3. Follow‐up testing
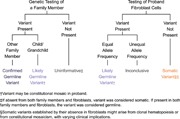
Follow‐up testing was performed either on blood or saliva samples from informative family members or on a cultured fibroblast sample from the proband (fibroblast cell culturing was performed by ARUP Laboratories, Salt Lake City, UT). DNA samples were extracted using standard methods. Follow‐up fibroblast and family testing consisted of Sanger single‐site analysis for the TP53 PV that was initially identified in the proband. Figure 1 depicts the interpretation of the PV identified in the proband based on the results of follow‐up, confirmatory testing. When family member testing was performed, the TP53 PV was “confirmed germline” if it also was identified in a relative who was not a child or grandchild. In this scenario, the PV was considered constitutionally present in all tissues, consistent with a molecular diagnosis of LFS. The PV was considered “likely germline” when it was present in a child or grandchild of the proband. In this scenario, it is likely that the PV is constitutional in the proband, yet it remains possible that it was acquired during early embryonic development and segregated to germ cells and other tissues, but not all tissues, rendering the proband constitutionally mosaic for the TP53 PV. Family member testing was considered “uninformative” when the TP53 PV was not detected in the tested family member.
Figure 1.
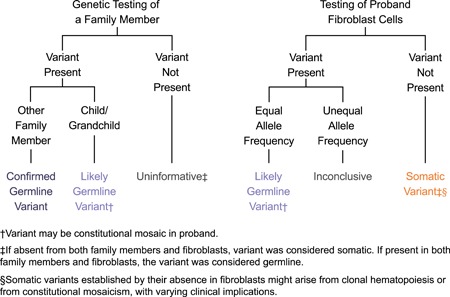
Follow‐up testing schema. The possible interpretations of follow‐up test results are provided for family testing and fibroblast testing in the proband
When fibroblast testing was performed, the TP53 PV was considered “somatic” if it was absent from the proband fibroblasts. This demonstrates that the PV is not present in all tissues; however, it remains undetermined whether the PV is restricted to blood (CHIP) or is present in some but not all tissues (constitutional mosaic). The PV was considered “likely germline” if it was present in the fibroblast sample from the proband. While identification of the PV in skin fibroblasts is strongly supportive of a constitutional variant, it does not definitively confirm the presence in the germline. Fibroblast testing results were “inconclusive” when initial NGS pan‐cancer panel testing showed an allele frequency consistent with a germline variant, but the ratio of wild‐type to PV allele in the fibroblast sample was significantly skewed from the expected 50:50 ratio. Such skewing might reflect the presence of more than one cell population in the skin biopsy, or it might result from technical or cell culture artifacts.
2.4. Analysis
Analyses were performed for all individuals with an apparent germline PV in TP53, were stratified by variant origin (confirmed germline, likely germline, somatic, uninformative, inconclusive, and unknown), and included age at testing and age at cancer diagnosis (if applicable). For the subset of individuals who had follow‐up testing, the results of follow‐up testing and NGS allele frequency were also assessed.
For all individuals who carried an apparent germline PV in TP53, clinical criteria for LFS were evaluated using classic LFS (Li et al., 1988) and revised Chompret LFS criteria (Bougeard et al., 2015). Classic LFS criteria were: proband diagnosed with sarcoma before 45 years of age, with a first‐degree relative diagnosed with any cancer before 45 years of age, and an additional first‐ or second‐degree relative diagnosed with any cancer before 45 years of age or a sarcoma at any age. Chompret LFS criteria were: (a) proband diagnosed with an LFS tumor (soft tissue sarcoma, osteosarcoma, brain tumor, adrenocortical carcinoma, leukemia, lung, or premenopausal breast [DCIS, invasive, or lobular invasive]) before 46 years of age, and at least one first‐ or second‐degree relative diagnosed with one LFS tumor before 56 years of age or with multiple primaries at any age; or (b) a proband with multiple tumors (except multiple breast tumors), the first of which occurred before age 46, and two of which belong to the LFS tumor spectrum; or (c) a proband with adrenocortical carcinoma, choroid plexus carcinoma, or rhabdomyosarcoma of embryonal anaplastic subtype, regardless of family history; or (d) breast cancer before 31 years of age.
3. RESULTS
A total of 221 individuals were identified as carrying an apparent TP53 PV (NGS allele frequency of 30–70%). The median age at testing was 41 years (range 15–88), and 28.5% (63/221) of individuals were older than 50 years (Figure 2). Overall, 79.6% (176/221) had a reported personal history of cancer at the time of testing. Of all individuals who had a reported personal history of cancer and age of diagnosis, 17.7% (29/174) were diagnosed after age 50. An additional 19.5% (43/221) of apparent germline TP53 PV carriers had no reported personal cancer history, and two individuals had no information reported about personal cancer history.
Figure 2.
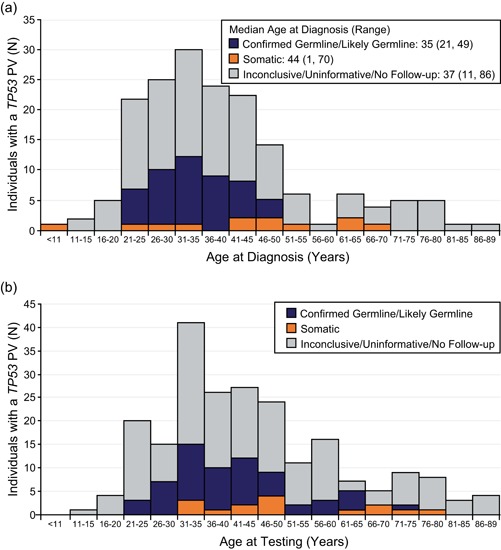
(a) Age at diagnosis and (b) age at testing for individuals with an apparent germline TP53 PV identified by hereditary cancer genetic testing. Results are stratified according to the results of follow‐up testing
3.1. Results of follow‐up testing
Ninety‐eight individuals had follow‐up testing to determine whether the TP53 PV was present in the germline. This included family member testing only (52/98, 53.1%), fibroblast testing only (25/98, 25.5%), or both (21/98, 21.4%; Table 1). When follow‐up included family member testing only, the proband's PV was present in a parent, sibling or relative other than a child in 28.8% (15/52) of cases. The confirmed germline status for the PV in these individuals is consistent with a molecular diagnosis of LFS. The PV was identified in a child for an additional 25.0% (13/52) of cases, demonstrating that the variant was present in germ cells and was likely constitutional in the proband. Importantly, this result does not rule out the alternative possibility that the proband was constitutionally mosaic for TP53, and therefore these PVs were considered likely, and not confirmed, germline. However, the child of the proband would inherit the TP53 PV in either scenario (germline PV or constitutional mosaic PV), which is consistent with a molecular diagnosis of LFS in the child. For 46.2% (24/52) of cases wherein the TP53 PV was absent from tested relative(s), the results were considered uninformative. It is important to note that absence of the PV in both parents could represent acquisition of a de novo TP53 PV in the proband, present in a proportion of a parent's germline cells (i.e., the parent is germline mosaic). It has been estimated that at least 14% of TP53 PVs are de novo mutations (Renaux‐Petel et al., 2017).
Table 1.
Summary of TP53 follow‐up testing and interpretation
| Follow‐up test result interpretation | |||||
|---|---|---|---|---|---|
| Follow‐up testing performed | Confirmed germline | Likely germline | Somatic | Inconclusive/uninformative* | Total |
| Family testing only | 15 (28.8%) | 13 (25.0%) | 0 | 24 (46.2%) | 52 |
| Fibroblast testing only | 0 | 14 (56.0%) | 9 (36.0%) | 2 (8.0%) | 25 |
| Family and fibroblast testing | 4 (19.0%) | 10 (47.6%) | 6 (28.6%) | 1 (4.8%) | 21 |
| Total | 19 (19.4%) | 37 (37.8%) | 15 (15.3%) | 27 (27.6%) | 98 |
Fibroblast testing results were inconclusive when initial testing showed an allele frequency consistent with a germline variant, but the ratio of wild‐type to PV allele in the fibroblast sample was significantly skewed from the expected 50:50 ratio. Family member testing was considered uninformative when the TP53 PV was not detected in the tested family member.
When follow‐up included fibroblast testing only, the TP53 PV was detected in fibroblasts in 56.0% (14/25) of cases and was designated as likely germline (Table 1). The PV was absent from fibroblasts and thus designated as somatic in 36.0% (9/25) of cases. In 8.0% (2/25) of cases, the variant status was determined to be inconclusive; while the variant appeared to be germline in blood, the ratio was significantly skewed in fibroblasts, indicating possible mosaicism in skin tissue.
When follow‐up included both family member and fibroblast testing, the PV was absent in both fibroblasts and tested relatives in 28.6% (6/21) of cases and was considered somatic. In an additional 66.7% (14/21) of cases, the PV was present in both fibroblasts and tested relatives and was designated as confirmed germline or likely germline depending on the relationship of the tested relatives. For one case (4.8%; 1/21), the PV ratio was significantly skewed in fibroblasts and determined to be inconclusive.
3.2. Confirmed and likely germline TP53 PV carriers
Overall, follow‐up testing confirmed germline status for the original TP53 PV identified during hereditary cancer testing in 19.4% (19/98) of individuals. In another 37.8% (37/98), the PV was considered likely germline (Table 1). The NGS allele frequencies for confirmed germline or likely germline sequence PVs in TP53 (87.5%; 49/56) ranged from 36% to 55% at the time of initial testing, with most falling between 45% and 55% (Figure 3). Allele frequencies around 50% are consistent with those typically seen for heterozygous germline variants observed in other genes on the panel (data not shown). LRs such as single‐ or multi‐exon deletions or duplications comprised 12.5% (7/56) of confirmed germline or likely germline PVs. Follow‐up family member testing showed that 57.1% (4/7) of LRs represented a confirmed germline variant in the proband, and 42.9% (3/7) were likely germline.
Figure 3.
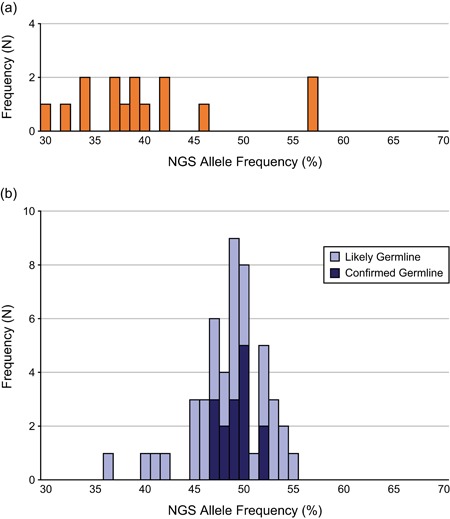
Allele frequencies for (a) somatic TP53 PVs (N = 15) and (b) confirmed germline (N = 19) and likely germline (N = 37) TP53 PVs. Only sequence variants are included. NGS, next‐generation sequencing; PV, pathogenic variant
Among the individuals who had a confirmed germline or likely germline PV in TP53, 78.6% (44/56) had a reported personal history of cancer. The median age at diagnosis among these individuals was 35 years (range 21–49 years), and 20.4% (9/44) were diagnosed after age 40. Based on personal and family cancer history information supplied by the provider on the test request form, 41.1% (23/56) of individuals with confirmed germline or likely germline TP53 PVs met clinical criteria for Chompret LFS (Table 2). None met the criteria for classic LFS.
Table 2.
Clinical Li–Fraumeni syndrome (LFS) classification, stratified by TP53 follow‐up testing result
| Follow‐up test result interpretation | ||||
|---|---|---|---|---|
| Confirmed germline/likely germline (N = 56) | Somatic (N = 15) | Inconclusive/uninformative (N = 27) | Unknown* (N = 123) | |
| Did not meet clinical criteria for LFS | 33 (58.9%) | 12 (80.0%) | 18 (66.7%) | 86 (69.9%) |
| Met Chompret criteria for LFS | 23 (41.1%) | 3 (20.0%) | 9 (33.3%) | 37 (30.1%) |
| Met classic criteria for LFS | 0 | 0 | 0 | 0 |
Includes individuals for whom no follow‐up testing was performed.
3.3. Somatic TP53 PV carriers
Among individuals with an apparent germline PV who had follow‐up fibroblast testing, TP53 PVs were found to be somatic in 32.6% (15/46) of cases (Table 1). While confirmed germline/likely germline PVs shared similar NGS allele frequency ranges, grouping around 50%, the somatic PV allele frequencies were dispersed more evenly (Figure 3).
Eighty percent (12/15) of individuals with a somatic TP53 PV had a reported personal history of cancer, with a median age at diagnosis of 44 years (Figure 2). For six somatic PV carriers, early‐onset breast cancer (ages 25, 28, 32, 33, 41, and 42) was reported. An additional six somatic PV carriers were not diagnosed with cancer until after age 45. There were no clear differences in the NGS allele frequencies of somatic PVs based on age at diagnosis. Allele frequencies for somatic PVs found in individuals diagnosed at an early age ranged from 30% to 46% compared with 34–57% for those diagnosed at a later age. The remaining three individuals with somatic PVs and no reported personal cancer history were in their late 40s at the time of testing (allele frequencies 37%, 40%, and 57%). No somatic TP53 PV carriers met clinical criteria for classic LFS; however, three individuals (20.0%) met criteria for Chompret LFS (Table 2).
4. DISCUSSION
This report describes the results of follow‐up testing to determine variant origin for individuals found to harbor apparent germline TP53 PVs as part of NGS hereditary cancer genetic testing. Follow‐up testing consisted of family testing and testing of fibroblasts from the proband. Family member testing can confirm that a PV is present in the germline (through the presence in the proband's sibling or parent) or likely germline (through the presence in the proband's child or grandchild). The result is uninformative when a parent appears to be negative for the PV detected in the proband. This is because three possible explanations exist. First, the PV might have been acquired during gametogenesis in the parent and is present only in a proportion of the parent's germ cells (i.e., parent is germline mosaic). It has been estimated that at least 14% of TP53 PVs represent de novo mutations (Renaux‐Petel et al., 2017). Second, the PV might have been acquired early in development and then segregated into some, but not all tissues (i.e., proband is constitutional mosaic). Third, the PV might have been acquired somatically in the proband's blood later in life, via CHIP.
On the other hand, fibroblast testing can determine that a PV is likely germline (present in fibroblasts) or that the PV was acquired somatically (absent in fibroblasts). Fibroblast testing, with or without additional family testing, showed that a substantial proportion (32.6%) of apparent germline TP53 PVs were acquired somatically. Although this proportion might reflect some self‐selection among individuals who elected to undergo follow‐up testing, it underscores the important fact that NGS allele frequency alone cannot determine the germline or somatic status of a TP53 PV. Therefore, while follow‐up fibroblast testing cannot definitively distinguish between CHIP and constitutional mosaicism to establish the origin of the TP53 PV, it can provide additional information that, when considered in context of clinical factors such as age and cancer history, informs screening and management approaches.
Most individuals who carried a somatic PV in TP53 reported a personal history of cancer. Furthermore, half of the somatic PV carriers were diagnosed when younger than 45 years of age. While only three individuals with a somatic PV met Chompret criteria for LFS, cancer at younger ages is consistent with the expected phenotype for LFS and meets NCCN criteria for TP53 testing (Daly et al., 2019). Overall, these data indicate that both genetic test results and the clinical presentation for individuals who carry somatically acquired TP53 PVs can resemble those of LFS or other hereditary cancer syndromes.
Similar to the somatic PV carriers, the majority of individuals with a confirmed germline PV in TP53 also reported a personal history of cancer. This included several individuals who were diagnosed after age 40, which is inconsistent with the expected clinical presentation of individuals with LFS. Overall, 58.9% (33/56) of confirmed germline or likely germline TP53 PV carriers did not meet current clinical criteria for LFS. Conflicting genetic findings and clinical phenotype may result in uncertainty regarding medical management. However, the findings presented here demonstrate that a large proportion of confirmed germline or likely germline PVs were identified among individuals whose reported personal and family cancer history on test requisition forms suggest that they do not meet the traditional criteria for LFS. Unlike previous studies that also suggest that the LFS phenotype is broader than is currently defined (Amadou, Waddington Achatz, & Hainaut, 2018; de Andrade et al., 2019, 2017; Rana et al., 2018), this analysis is not confounded by the presence of somatic carriers of TP53 PVs.
There is significant interest in understanding the origin of somatically acquired PVs in TP53. Individuals with somatic TP53 PVs who were diagnosed with cancer at young ages might be constitutionally mosaic for the PV. For the four women described here who were diagnosed with breast cancer in their late 20s and early 30s, detection of the PV in the relevant tissue type would support its contribution to the disease. The absence of TP53 PVs from the fibroblasts of three individuals in their late 40s with no personal cancer history could be consistent either with constitutional mosaicism or with CHIP. TP53 is one of the most common genes known to accrue somatic PVs in blood due to CHIP (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). CHIP has been shown to be present in about 10% of individuals older than 65 years, and its prevalence increases with age (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). Association of somatically acquired, low‐allele‐frequency (10–30%) TP53 PVs with aging has been seen previously in individuals tested using NGS hereditary cancer gene panels (Coffee et al., 2017). It follows that further expansion of a hematopoietic cell subclone carrying a TP53 PV could occur over time, reaching an NGS allele frequency that overlaps with those seen for heterozygous germline variants. The observation that subclones with TP53 PVs in mouse bone marrow chimeras expand over time, specifically after exposure to chemotherapy, further supports this hypothesis (Wong et al., 2015).
Although it is likely that CHIP underlies the acquisition of somatic TP53 PVs in this testing population, particularly among older individuals, it remains possible that some individuals are constitutionally mosaic for the variant. It is important to distinguish between these two mechanisms in tested individuals, as the implications for clinical management likely will differ. Constitutional mosaicism, wherein the TP53 PV is present in tissues vulnerable to LFS‐associated cancers, might be expected to cause early‐onset cancers. CHIP, as a normal byproduct of aging, might hold other implications for medical management. While the possible origins of somatic variants are not easily distinguishable, confirming whether a PV in TP53 within the 30–70% AF range is somatic versus germline can inform whether the extensive clinical interventions for germline PV carriers are appropriate. This distinction can both prevent unnecessary interventions for individuals who have nongermline PVs, as well as under‐treatment for individuals who harbor germline TP53 PVs but do not meet clinical LFS criteria.
A primary strength of the analysis is the large clinical data set that afforded the broad analysis and follow‐up testing in individuals tested for hereditary cancer risk. Personal and family cancer history entered on the test request form by the ordering provider further enhances data analysis and interpretation but comes with a limitation: the possibility that the information is less than complete. For some tested individuals, the family cancer history might be more extensive than was reported and could bring the family cancer history phenotype in closer alignment with LFS diagnostic criteria. Cytotoxic cancer therapy is known to be associated with CHIP (Coombs et al., 2017; Gillis et al., 2017; Slavin et al., 2019; Takahashi et al., 2017) and should be considered by ordering providers as a potential contributor to acquisition of somatic TP53 PVs. Unfortunately, cancer treatment information was not available in this clinical testing data set; this is an important area for investigation using study cohorts that are designed to capture such information.
In summary, this study demonstrates that TP53 PVs with NGS allele frequencies consistent with those expected for a germline PV can be either inherited or acquired somatically. Therefore, clinical reporting of TP53 PVs identified by NGS should include the possibility of either germline or somatic origin and the corresponding clinical implications. For individuals who carry germline TP53 PVs, confirmation of the test result is essential to providing appropriate medical management. Determining that a TP53 PV was acquired somatically may help prevent unnecessary medical intervention for the proband and undue anxiety for family members who otherwise may think that they are at risk of carrying the PV. These findings highlight the need for a conservative approach to the initial reporting of TP53‐positive NGS test results, combined with investment in follow‐up testing to deliver clinically accurate results.
CONFLICT OF INTERESTS
All authors are employed by Myriad Genetic Laboratories, Inc. and receive salaries as compensation.
ACKNOWLEDGMENTS
The authors thank Jennifer Logan, PhD, and Krystal Brown, PhD, both employees of Myriad Genetic Laboratories, Inc., for editorial assistance with the manuscript. This study was funded by Myriad Genetic Laboratories, Inc.
Coffee B, Cox HC, Bernhisel R, et al. A substantial proportion of apparently heterozygous TP53 pathogenic variants detected with a next‐generation sequencing hereditary pan‐cancer panel are acquired somatically. Human Mutation. 2020;41:203–211. 10.1002/humu.23910
References
REFERENCES
- Amadou, A. , Waddington Achatz, M. I. , & Hainaut, P. (2018). Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: Temporal phases of Li‐Fraumeni syndrome. Current Opinion in Oncology, 30(1), 23–29. 10.1097/cco.0000000000000423 [DOI] [PubMed] [Google Scholar]
- de Andrade, K. C. , Frone, M. N. , Wegman‐Ostrosky, T. , Khincha, P. P. , Kim, J. , Amadou, A. , … Achatz, M. I. (2019). Variable population prevalence estimates of germline TP53 variants: A gnomAD‐based analysis. Human Mutation, 40(1), 97–105. 10.1002/humu.23673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade, K. C. , Mirabello, L. , Stewart, D. R. , Karlins, E. , Koster, R. , Wang, M. , … Achatz, M. I. (2017). Higher‐than‐expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Human Mutation, 38(12), 1723–1730. 10.1002/humu.23320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan, C. , Moutou, C. , Brugières, L. , Feunteun, J. , & Bonaïti‐Pellié, C. (1995). ARCAD: A method for estimating age‐dependent disease risk associated with mutation carrier status from family data. Genetic Epidemiology, 12(1), 13–25. 10.1002/gepi.1370120103 [DOI] [PubMed] [Google Scholar]
- Bougeard, G. , Renaux‐Petel, M. , Flaman, J. M. , Charbonnier, C. , Fermey, P. , Belotti, M. , … Frebourg, T. (2015). Revisiting Li‐Fraumeni syndrome From TP53 mutation carriers. Journal of Clinical Oncology, 33(21), 2345–2352. 10.1200/jco.2014.59.5728 [DOI] [PubMed] [Google Scholar]
- Chompret, A. , Brugieres, L. , Ronsin, M. , Gardes, M. , Dessarps‐Freichey, F. , Abel, A. , & Feunteun, J. (2000). P53 germline mutations in childhood cancers and cancer risk for carrier individuals. British Journal of Cancer, 82(12), 1932–1937. 10.1054/bjoc.2000.1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee, B. , Cox, H. C. , Kidd, J. , Sizemore, S. , Brown, K. , Manley, S. , & Mancini‐DiNardo, D. (2017). Detection of somatic variants in peripheral blood lymphocytes using a next generation sequencing multigene pan cancer panel. Cancer Genetics, 211, 5–8. 10.1016/j.cancergen.2017.01.002 [DOI] [PubMed] [Google Scholar]
- Coombs, C. C. , Zehir, A. , Devlin, S. M. , Kishtagari, A. , Syed, A. , Jonsson, P. , … Berger, M. F. (2017). Therapy‐related clonal hematopoiesis in patients with non‐hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell, 21(3), 374–382. 10.1016/j.stem.2017.07.010. e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly, M.B. , Pilarski, R. , Berry, M. , Buys, S.S. , Friedman, S. , & Garber, J.E. (2019). Assessment: Breast and Ovarian (Version 3.2019). Retrieved from https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf [Google Scholar]
- Evans, G. , & Ingham, S. (2013). Reduced life expectancy seen in hereditary diseases which predispose to early‐onset tumors. The Application of Clinical Genetics, 6, 53–61. 10.2147/tacg.s35605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, G. , Kähler, A. K. , Handsaker, R. E. , Lindberg, J. , Rose, S. A. , Bakhoum, S. F. , … McCarroll, S. A. (2014). Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. New England Journal of Medicine, 371(26), 2477–2487. 10.1056/NEJMoa1409405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, N. K. , Ball, M. , Zhang, Q. , Ma, Z. , Zhao, Y. , Yoder, S. J. , … Padron, E. (2017). Clonal haemopoiesis and therapy‐related myeloid malignancies in elderly patients: A proof‐of‐concept, case‐control study. The Lancet Oncology, 18(1), 112–121. 10.1016/S1470-2045(16)30627-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, S. , Fontanillas, P. , Flannick, J. , Manning, A. , Grauman, P. V. , Mar, B. G. , … Ebert, B. L. (2014). Age‐related clonal hematopoiesis associated with adverse outcomes. New England Journal of Medicine, 371(26), 2488–2498. 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, S. , Natarajan, P. , Silver, A. J. , Gibson, C. J. , Bick, A. G. , Shvartz, E. , … Ebert, B. L. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine, 377(2), 111–121. 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judkins, T. , Leclair, B. , Bowles, K. , Gutin, N. , Trost, J. , McCulloch, J. , … Timms, K. (2015). Development and analytical validation of a 25‐gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer, 15(1), 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz, C. P. , Achatz, M. I. , Brugières, L. , Frebourg, T. , Garber, J. E. , Greer, M. L. C. , … Malkin, D. (2017). Cancer screening recommendations for individuals with Li‐Fraumeni syndrome. Clinical Cancer Research, 23(11), e38–e45. 10.1158/1078-0432.Ccr-17-0408 [DOI] [PubMed] [Google Scholar]
- Li, F. P. , Fraumeni JF, Jr, Jr. , Mulvihill, J. J. , Blattner, W. A. , Dreyfus, M. G. , Tucker, M. A. , & Miller, R. W. (1988). A cancer family syndrome in twenty‐four kindreds. Cancer Research, 48(18), 5358–5362. [PubMed] [Google Scholar]
- Mitchell, R. L. , Kosche, C. , Burgess, K. , Wadhwa, S. , Buckingham, L. , Ghai, R. , … Usha, L. (2018). Misdiagnosis of Li‐Fraumeni syndrome in a patient with clonal hematopoiesis and a somatic TP53 mutation. Journal of the National Comprehensive Cancer Network: JNCCN, 16(5), 461–466. 10.6004/jnccn.2017.7058 [DOI] [PubMed] [Google Scholar]
- Rana, H. Q. , Gelman, R. , LaDuca, H. , McFarland, R. , Dalton, E. , Thompson, J. , … Garber, J. E. (2018). Differences in TP53 mutation carrier phenotypes emerge from panel‐based testing. Journal of the National Cancer Institute, 110(8), 863–870. 10.1093/jnci/djy001 [DOI] [PubMed] [Google Scholar]
- Renaux‐Petel, M. , Charbonnier, F. , Théry, J. C. , Fermey, P. , Lienard, G. , Bou, J. , … Bougeard, G. (2017). Contribution of de novo and mosaic TP53 mutations to Li‐Fraumeni syndrome. Journal of Medical Genetics, 55(3), jmedgenet‐2017‐104976. 10.1136/jmedgenet-2017-104976 [DOI] [PubMed] [Google Scholar]
- Rosenthal, E. T. , Bernhisel, R. , Brown, K. , Kidd, J. , & Manley, S. (2017). Clinical testing with a panel of 25 genes associated with increased cancer risk results in a significant increase in clinically significant findings across a broad range of cancer histories. Cancer Genetics, 218‐219, 58–68. 10.1016/j.cancergen.2017.09.003 [DOI] [PubMed] [Google Scholar]
- Slavin, T. P. , Coffee, B. , Bernhisel, R. , Logan, J. , Cox, H. C. , Marcucci, G. , … Mancini‐DiNardo, D. (2019). Prevalence and characteristics of likely‐somatic variants in cancer susceptibility genes among individuals who had hereditary pan‐cancer panel testing. Cancer Genetics, 235‐236, 31–38. 10.1016/j.cancergen.2019.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell, A. D. , Espenschied, C. R. , Culver, J. O. , & Weitzel, J. N. (2013). Tumor protein p53 (TP53) testing and Li‐Fraumeni syndrome: Current status of clinical applications and future directions. Molecular Diagnosis & Therapy, 17(1), 31–47. 10.1007/s40291-013-0020-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steensma, D. P. , Bejar, R. , Jaiswal, S. , Lindsley, R. C. , Sekeres, M. A. , Hasserjian, R. P. , & Ebert, B. L. (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 126(1), 9–16. 10.1182/blood-2015-03-631747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K. , Wang, F. , Kantarjian, H. , Doss, D. , Khanna, K. , Thompson, E. , … Futreal, P. A. (2017). Preleukaemic clonal haemopoiesis and risk of therapy‐related myeloid neoplasms: A case‐control study. The Lancet Oncology, 18(1), 100–111. 10.1016/S1470-2045(16)30626-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, T. N. , Ramsingh, G. , Young, A. L. , Miller, C. A. , Touma, W. , Welch, J. S. , … Wilson, R. K. (2015). Role of TP53 mutations in the origin and evolution of therapy‐related acute myeloid leukaemia. Nature, 518(7540), 552–555. 10.1038/nature13968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, M. , Lu, C. , Wang, J. , McLellan, M. D. , Johnson, K. J. , Wendl, M. C. , … Ding, L. (2014). Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine (New York, NY, United States), 20(12), 1472–1478. 10.1038/nm.3733 [DOI] [PMC free article] [PubMed] [Google Scholar]