

Abstract
Maribavir is an investigational drug being evaluated in transplant recipients with cytomegalovirus infection. To understand potential drug‐drug interactions, we examined the effects of multiple doses of maribavir on cytochrome P450 (CYP) 2D6 and P‐glycoprotein (P‐gp) activity using probe substrates in healthy volunteers. During this phase 1 open‐label study (NCT02775240), participants received the probe substrates digoxin (0.5 mg) and dextromethorphan (30 mg) before and after maribavir (400 mg twice daily for 8 days). Serial plasma samples were analyzed for digoxin, dextromethorpha, dextrorphan, and maribavir concentrations. Pharmacokinetic parameters were calculated (noncompartmental analysis) and analyzed with a linear mixed‐effects model for treatment comparison to estimate geometric mean ratios (GMRs) and 90% confidence intervals (CIs). CYP2D6 polymorphisms were genotyped using polymerase chain reaction. Overall, 17 of 18 participants (94.4%) completed the study. All participants were genotyped as CYP2D6 intermediate/extensive metabolizers. GMR (90%CI) of digoxin Cmax, AUClast, and AUC0‐∞ with and without maribavir was 1.257 (1.139‐1.387), 1.187 (1.088‐1.296), and 1.217 (1.110‐1.335), respectively, outside the “no‐effect” window (0.8‐1.25). GMR (90%CI) of dextromethorphan AUClast and AUClast ratio of dextromethorphan/dextrorphan were 0.877 (0.692‐1.112) and 0.901 (0.717‐1.133), respectively, marginally outside the no‐effect window, although large variability was observed in these pharmacokinetic parameters. Pharmacokinetic parameters of dextrorphan were unaffected. Maribavir inhibited P‐gp activity but did not affect CYP2D6 activity. Maribavir's effect on the pharmacokinetics of P‐gp substrates should be evaluated individually, and caution should be exercised with P‐gp substrates with narrow therapeutic windows.
Keywords: maribavir, pharmacokinetics, digoxin, dextromethorphan, P‐glycoprotein, cytochrome P450 2D6
Cytomegalovirus (CMV) reactivation is a significant posttransplant complication associated with substantial morbidity and reduced long‐term survival following both hematopoietic stem cell transplant (HSCT) and solid‐organ transplantation (SOT).1, 2 CMV infection, including reactivation of latent infection, most commonly occurs during the first year after transplantation and represents a clinical challenge and significant risk to the vulnerable transplant recipient.3, 4 The primary strategy for anti‐CMV management in HSCT and SOT recipients is preemptive antiviral therapy and antiviral prophylaxis, respectively, with appropriate antivirals usually administered shortly after transplantation for ≥3 months according to the transplant type and patient immune status.4 In the United States, antiviral drugs, such as ganciclovir and valganciclovir, are approved for the prevention of CMV infection whereas letermovir is approved for CMV infection prophylaxis; other antivirals such as foscarnet and cidofovir are used off‐label.5, 6, 7, 8 Ganciclovir and valganciclovir are considered first‐line treatments for CMV management, whereas foscarnet and cidofovir are reserved for the second‐line treatment of resistant/refractory CMV.4 However, there are some limitations associated with the use of these agents including treatment‐limiting toxicities (neutropenia, anemia, and thrombocytopenia for ganciclovir and valganciclovir and renal impairment for foscarnet and cidofovir)5, 9, 10, 11, 12 and significant drug interactions leading to contraindication or requiring dose adjustment and careful drug monitoring (letermovir).13, 14 These limitations are often associated with failing to prevent CMV infection and disease and development of drug resistance.
Maribavir is an investigational drug for the treatment of CMV infection/disease in transplant recipients and was granted breakthrough therapy designation by the US Food and Drug Administration (FDA) in January 2018.9 Maribavir is a potent and selective orally bioavailable antiviral drug with a novel mechanism of action against CMV and a favorable safety profile without myelosuppression or nephrotoxicity.10, 11 Currently approved anti‐CMV agents inhibit DNA polymerase. However, maribavir, a benzimidazole riboside, inhibits the protein kinase UL97, which is important for CMV DNA replication and encapsidation and prevents the egress of viral capsids from the nuclei of infected cells.12, 13, 14 Maribavir has demonstrated in vitro activity against CMV wild‐type viruses as well as strains resistant to current anti‐CMV therapies.12, 15
As well as demonstrating in vitro activity, maribavir has been evaluated in phase 2 studies in SOT and HSCT recipients, including those with CMV resistant or who are refractory to ganciclovir/valganciclovir, foscarnet, or cidofovir.10, 11 Maribavir is currently being investigated for the treatment of CMV infection in 2 phase 3 studies, in treatment‐naive HSCT recipients (NCT02927067), and in SOT or HSCT with CMV resistant or refractory to (val)ganciclovir or foscarnet (NCT02931539).
Transplant recipients receive numerous comedications, including immunosuppressants to prevent graft rejection and therapies to manage comorbidities.16, 17 Most clinically relevant drug‐drug interactions (DDIs) result from interactions in metabolism and disposition mediated by cytochrome P450 (CYP) enzymes and/or manipulation of transporters such as the P‐glycoprotein (P‐gp) drug efflux transporter.18
In vitro data have demonstrated that maribavir is not an inhibitor of major CYP enzymes, uridine diphosphate glucuronosyltransferases, or transporters, except for weak inhibition of CYP1A2 (half‐maximal inhibitory concentration [IC50], 40 µM), CYP2C9 (IC50, 18 µM), CYP2C19 (IC50, 35 µM), P‐gp (IC50, 33.7 µM), and breast cancer resistance protein (BCRP) efflux transporter (IC50, 7 µM) — Shire data on file: reversible CYP inhibition, V9079M‐SHP620; time‐dependent inhibition kinetics, V8576M‐SHP620; BCRP inhibition [and substrate], V7317M‐SHP620 and Koszalka et al.19 However, data from a healthy volunteer study indicated that maribavir had no effect on the pharmacokinetics (PK) of voriconazole (a CYP2C19 substrate).20 Furthermore, findings from a study evaluating administration of a probe cocktail (midazolam, caffeine, warfarin, omeprazole, and dextromethorphan) and multiple doses of maribavir in healthy volunteers suggested that maribavir did not affect the activity of CYP1A2, CYP2C9, and CYP3A, but reduced the activity of CYP2D6;21 however, this effect on CYP2D6 may have been because of intraindividual variability. Also, the inhibition of CYP2D6 was based on the ratio of dextromethorphan to its active metabolite dextrorphan excreted in urine, which has been suggested as a less appropriate method of assessment than the plasma PK profile of dextromethorphan and dextrorphan.21, 22, 23, 24
The study reported here aimed to examine the effects of multiple oral doses of maribavir on CYP2D6 and P‐gp activity in healthy adults by assessing the plasma, PK profile of the probe drugs dextromethorphan and digoxin, respectively. The safety and tolerability of coadministered maribavir, digoxin, and dextromethorphan were also evaluated.
Methods
Study Design
The protocol and informed consent were reviewed and approved by the institutional review board (Integreview, Austin, Texas). All participants provided written informed consent. The study was performed in accordance with the ethical principles stated in the Declaration of Helsinki and the International Council for Harmonisation for Good Clinical Practice.
This phase 1 open‐label, 2‐period fixed‐sequence study in healthy volunteers was conducted at a single study center (Clinical Pharmacology Miami Inc., Miami, Florida) between July 21 and September 12, 2016. After a 28‐day screening period, there were 2 treatment periods: treatment 1 (days 1‐7) and treatment 2 (days 8‐15). A follow‐up telephone call occurred 7 ± 2 days after the last dose of maribavir received.
Participants
To be included in the study, participants had to be healthy (as determined by medical history, physical examination, vital signs, electrocardiogram [ECG], and clinical laboratory tests), nonsmoking, aged 18‐50 years, with a body mass index (BMI) between 18.5 and 30.0 kg/m2 inclusive, and a hemoglobin level ≥ 7.45 mmol/L (≥12.0 g/dL; normal ranges for women and men were 120‐160 and 130‐170 g/L, respectively).
Participants were excluded if they had a clinically significant history of any disorder/medical condition that could alter the metabolism of drugs or had hypersensitivity to the investigational products or related compounds. Other exclusion criteria included a history of alcohol/substance abuse, use of an investigational product, or use of any medication including over‐the‐counter, herbal, or homeopathic preparations such as St. John's wort and Ginkgo biloba (with the exception of hormonal replacement therapy and occasional use of ibuprofen and acetaminophen). Consumption of known CYP3A modulators including grapefruit or grapefruit juice, oranges, Seville oranges, apples or apple juice, vegetables from the mustard green family, charbroiled meats, or products containing these ingredients was also prohibited within 14 days prior to the first dose of maribavir. Women were excluded if they were nursing or pregnant.
Study Medication
Eligible participants were allocated a 4‐digit number and assigned to receive study medication orally as follows: on day 1, a single 0.5‐mg dose of digoxin and a single 30‐mg dose of dextromethorphan (treatment 1); on days 8 to 15, maribavir 400 mg (2 × 200 mg) twice daily approximately 12 hours apart; and on day 13, a 0.5‐mg single dose of digoxin and 30‐mg single dose of dextromethorphan (coadministered with maribavir; treatment 2).
Dextromethorphan and digoxin were administered at doses of 30 and 0.5 mg, respectively, in line with doses described previously. These are well‐established probes for the evaluation of the effect of pharmacologic agents on the activity of CYP2D6 and P‐gp, respectively.22, 25, 26 The 400‐mg twice‐daily dosing regimen for maribavir was used in this study, as this is the regimen being evaluated in the ongoing phase 3 trials for the treatment of CMV infection in transplant recipients (NCT02931539; NCT02927067).
On days 1 and 13, participants fasted for at least 10 hours prior and 4 hours after administration of digoxin and dextromethorphan or maribavir. There was a washout period of at least 7 days between the morning dose of digoxin and dextromethorphan on day 1 and the administration of the first dose of maribavir on day 8.
Pharmacokinetic Evaluation
Blood samples were collected at the following times for digoxin, dextromethorphan, and dextrorphan: predose and 0.25, 0.5, 1.0, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 48, and 72 hours after dosing on day 1 and day 13. For maribavir, samples were collected at predose and 0.25, 0.5, 1.0, 1.5, 2, 3, 4, 5, 6, 8, and 12 hours after dosing on day 13.
Plasma concentrations of maribavir, digoxin, dextromethorphan, and dextrorphan (total dextrorphan as the combined concentration of the free and glucuronide‐conjugated dextrorphan) were assessed using liquid chromatography with tandem mass spectrometry by Celerion (Lincoln, Nebraska).
The linear range for the maribavir assay was between 0.200 and 200 µg/mL, and the percentage bias was 2.0%, 0.6%, and –3.9% with quality control (QC) samples of 0.600, 5.00, and 75.0 µg/mL, respectively. The linear range of the assay for digoxin was between 10.0 and 3500 pg/mL, and the percentage bias was 4.0%, 2.2%, and ‐5.2% with QC samples at 30.0, 180, and 2500 pg/mL, respectively. The linear range of the dextromethorphan assay was 0.200 to 200 ng/mL, and the dextrorphan assay was linear between 2.50 and 2500 ng/mL. At 0.600, 6.40, and 150 ng/mL, the percentage bias for dextromethorphan was 2.2%, 3.0%, and 2.0%, respectively. At 7.50, 80.0, and 1880 ng/mL, the percentage bias for dextrorphan was 3.5%, 1.1%, and 0%, respectively.
All QC samples were stored in freezers at ‐20°C alongside study samples and were assayed with study samples against prepared calibration standards. The assays were validated, and QC and calibration standard data were designated as acceptable in accordance with FDA guidance and European Medicines Agency guidance.27, 28
PK parameters were calculated by noncompartmental analysis based on concentration‐time profiles and actual postdose times using Phoenix WinNonlin version 6.3 (Pharsight Corporation, Mountain View, California). PK parameters for maribavir included time of maximum observed concentration sampled during a dosing interval (Tmax), maximum observed concentration occurring at Tmax (Cmax), area under the curve from time 0 to the end of the dosing interval at steady state (AUCtau), the predose concentration, concentration at the end of the dosing interval (Ctau), apparent oral clearance at steady state equal to dose/AUCtau (CL/F), and terminal half‐life (t1/2). PK parameters for digoxin, dextromethorphan, and dextrorphan included Cmax, area under the curve from the time of dosing to the last measurable concentration (AUClast), and area under the curve extrapolated to infinity (AUC0‐∞). The ratio of AUClast for dextromethorphan over AUClast for dextrorphan (AUClast parent/metabolite ratio) was also calculated.
Pharmacogenomic Assessments and Analyses
Blood samples were collected on day 1 prior to dose administration and sent to Genelex Corporation (Seattle, Washington) for analysis to determine genetic variation of the polymorphism of CYP2D6 enzyme activity. Genomic DNA from ethylenediaminetetraacetic acid‐treated blood samples was extracted using a MagJET Whole Genomic DNA Extraction Kit (ThermoFisher, Waltham, Massachusetts) and/or a EZ1 DNA Blood Kit (Qiagen, Hilden, Germany). Analysis of CYP2D6 alleles *1,*12, *14, *15, *17, *19, *20, *29, *35, *36, and *41 and gene duplications was performed using a polymerase chain reaction test followed by a single‐base primer extension for variant detection using mass spectrometry (MassARRAY Analyzer 4 System, Agenda Bioscience, San Diego, California).
Participants were classified on the basis of their CYP2D6 genotype as poor metabolizers, intermediate metabolizers, extensive metabolizers, or ultrarapid metabolizers, as defined in Table 1.
Table 1.
Definitions of Metabolism Profiles Assigned to Study Participants on CYP2D6 Genotyping
| Metabolism Classification | Definitions |
|---|---|
| Poor metabolizers | Participants carrying 2 inactive CYP2D6 alleles, CYP2D6*3, *4, *5, *6, *7, *8, *11, *12, *14, *15, *19, *20, and *36 |
| Intermediate metabolizers | Participants carrying 1 inactive and 1 active CYP2D6 allele (CYP2D6*1, *2, *2A, and *35) or 2 partially active alleles (CYP2D6*9, *10, *17, *29, and *41) |
| Extensive metabolizers | Participants carrying 2 active alleles or 1 active and 1 partially active allele |
| Ultrarapid metabolizers | Participants carrying 3 or more active alleles |
CYP, cytochrome P450.
Safety Measurements
Safety was evaluated throughout the study by monitoring adverse events (AEs), physical examination results, clinical laboratory values, and vital sign measurements, including 12‐lead ECG readings. In addition, AEs were assessed through general questioning at screening, on every day of the treatment periods, and during follow‐up. An overall summary of the number of participants with treatment‐emergent adverse events (TEAEs) and the severity of TEAEs was recorded by treatment throughout the study. At screening and on days −1, 1, 4, 8, 13, and 16, physical examinations and ECGs were performed. Vital signs were recorded at screening and on day −1, days 1 to 4, and days 8 to 16.
Statistical Analyses
All statistical analyses were performed using SAS software, version 9.4 (SAS Institute, Inc., Cary, North Carolina). Unless otherwise specified, continuous variables were summarized by descriptive statistics, including the number of participants, mean, standard deviation (SD), median, minimum, and maximum. Categorical and continuous variables were summarized by the number and the percentage of participants in each category. Geometric means and the coefficients of variation (CV%) of the geometric means were also calculated for PK parameters.
Prior studies on digoxin and dextromethorphan have reported intrasubject variability of AUC0‐12 to be 25% and 32%, respectively.22, 29 Assuming an intrasubject variability of 32%, the required sample size of 14 participants was determined using nQuery 7.0 (Statistical Solutions Ltd, Cork, Ireland). The sample size calculation was based on the ability to show equivalence of the AUC between digoxin and dextromethorphan (with and without maribavir coadministration) in the absence of a significant DDI using a 90% confidence interval (CI) of 0.75 to 1.33 for the geometric least‐squares means ratio, with two 1‐sided tests, and a significance (α) of 0.05 with at least 80% power. To allow for dropouts, 18 participants were enrolled in the study.
For PK parameters, log‐transformed PK parameters (Cmax, AUC0‐∞, and AUClast) were compared in the presence and absence of maribavir using a linear mixed‐effects model with treatment regimen as the fixed effect and subject as the random effect. The ratio of geometric means in PK parameters and the corresponding 2‐sided 90%CIs were obtained by back‐transformation of the least‐square mean differences and the corresponding 2‐sided 90%CIs obtained on the log scale. It is noted that although the 0.75‐1.33 acceptance limits were planned a priori, the relatively conservative acceptance limits of 0.8‐1.25 were also used in the results section to interpret the study outcome.
Results
Study Population
A total of 18 participants were screened and enrolled in the study. All participants received at least 1 dose of maribavir and were included in both the safety and PK sets. Of these, 17 participants (94.4%) received a dose of maribavir and other test products (digoxin and dextromethorphan) and completed the study. Only 1 participant withdrew from the study during treatment 1.
The baseline characteristics of participants are summarized in Table 2. The mean ± SD age was 38.1 ± 8.7 years; the majority of participants were white (72.2%) and male (61.1%). The mean ± SD BMI was 27.4 ± 2.62 kg/m2; 14 participants (77.8%) were overweight (BMI, 25.0 to <30.0 kg/m2). All participants were identified as either extensive (n = 13, 72.2%) or intermediate (n = 5, 27.8%) metabolizers for the CYP2D6 isozyme.
Table 2.
Demographic Characteristics (Safety Analysis Set, n = 18)
| Characteristica | Total |
|---|---|
| Age,b years | |
| Mean (SD) | 38.1 (8.72) |
| Median (min‐max) | 40.5 (19‐47) |
| Sex | |
| Male | 11 (61.1) |
| Female | 7 (38.9) |
| Ethnicity | |
| Hispanic or Latino | 12 (66.7) |
| Non‐Hispanic or Latino | 6 (33.3) |
| Race | |
| White | 13 (72.2) |
| Black or African American | 5 (27.8) |
| Weight, kg | |
| Mean (SD) | 81.3 (11.6) |
| Median (min‐max) | 80.4 (61.4‐108.0) |
| Height, cm | |
| Mean (SD) | 172.1 (11.3) |
| Median (min‐max) | 175.5 (153‐190) |
| BMI,c kg/m2 | |
| Mean (SD) | 27.4 (2.62) |
| Median (min‐max) | 28.6 (22.0‐29.9) |
| CYP2D6 phenotypes | |
| Extensive metabolizer | 13 (72.2) |
| Intermediate metabolizer | 5 (27.8) |
BMI, body mass index; CYP, cytochrome P450; max, maximum; min, minimum; SD, standard deviation.
All data are presented as n (%) unless otherwise stated. All values are rounded to 3 significant figures; min and max values for age are reported as whole numbers.
aThe baseline value for a characteristic was the value from the point as specified in the statistical analysis plan.
bAge was calculated as the difference between date of birth and date on informed consent, truncated to years.
cBMI was calculated as (weight [kg]/height [m2]).
Pharmacokinetics
The PK parameters for maribavir are summarized in Table 3. Geometric mean was 91.5 µg·h/mL for AUCtau, 17.6 µg/mL for Cmax, 2.13 µg/mL for Ctau, 2.19 L/h for CL/F, and 4.0 hours for t1/2 for maribavir (400 mg twice daily for 7 days).
Table 3.
Summary of Pharmacokinetic Parameters for Maribavir 400 mg Twice Daily Calculated by Noncompartmental Analysis (n = 18)
| GM (95%CI); GM CV% | Median (Min‐Max) | |||||
|---|---|---|---|---|---|---|
| Cmax (µg/mL) | AUCtau (µg·h/mL) | Ctau (µg/mL) | C0 (µg/mL) | CL/F (L/h) | t½ (h) | Tmax (h) |
| 17.6 | 91.5 | 2.13 | 2.64 | 2.19 | 4.0 | 2.00 |
| (15.5‐19.9); | (79.7‐105); | (1.66‐2.73); | (1.92‐3.62); | (1.90‐2.51); | (3.7‐4.5); | (0.500‐3.00) |
| 25.1% | 27.4% | 51.4% | 68.2% | 27.4% | 19.3% | |
AUCtau, area under the curve from time 0 to the end of the dosing interval at steady state; C0, predose concentration; CI, confidence interval; CL/F, apparent oral clearance at steady state, equal to dose/AUCtau; Cmax, maximum observed concentration occurring at Tmax; Ctau, concentration at the end of the dosing interval; CV, coefficient of variation; GM, geometric mean; Max, maximum; Min, minimum; t½, terminal half‐life; Tmax, time of maximum observed concentration sampled during a dosing interval.
Mean plasma concentration‐time profiles of digoxin with and without maribavir are provided in Figure 1. The results showed that the 90%CI for AUClast for digoxin fell within the predefined no‐effect range of 0.75 to 1.33 (Table 4). Notably, the upper limit of the 90%CI for the geometric mean ratio of digoxin Cmax and AUC0‐∞ was above 1.33. Using this no‐effect range, maribavir did not significantly affect the AUClast of digoxin; however, Cmax and AUC0‐∞ of digoxin increased by 25.7% and 21.7%, respectively, when coadministered with maribavir. Using the no‐effect range of 0.8 to 1.25, the 90%CI of geometric mean ratio for AUClast for digoxin also fell outside the no‐effect range (Table 4); AUClast for digoxin increased by 18.7% when coadministered with maribavir (Table 4).
Figure 1.
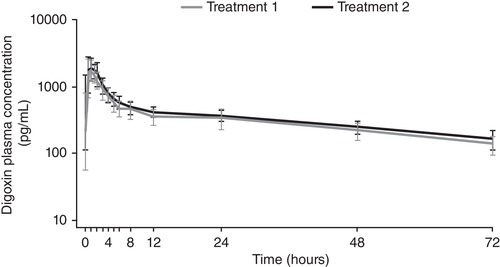
Mean plasma concentration‐time profile for digoxin by treatment period: log scale (pharmacokinetic set, n = 18). Treatment 1: digoxin 0.5 mg and dextromethorphan 30 mg administered on day 1. Treatment 2: maribavir 400 mg (2 × 200 mg) twice daily from days 8 to 15. On day 13, digoxin 0.5 mg and dextromethorphan 30 mg were administered with the morning dose of maribavir 400 mg (2 × 200 mg). Error bars represent the standard deviation.
Table 4.
Statistical Comparison of Digoxin Pharmacokinetic Parameters Between Treatment Periods (Participants Who Completed the Study, n = 17)
| Parameter | Treatment 1a GM | Treatment 2b GM | Ratio of GMs (90%CI) | Intrasubject Variability CV% |
|---|---|---|---|---|
| Cmax (ng/mL) | 1.87 | 2.35 | 1.257 (1.139‐1.387) | 16.6 |
| AUClast (ng·h/mL) | 22.5 | 26.7 | 1.187 (1.088‐1.296) | 14.7 |
| AUC0‐∞ (ng·h/mL) | 30.6 | 37.3 | 1.217 (1.110‐1.335) | 15.5 |
AUC0‐∞, area under the curve extrapolated to infinity, calculated using the observed value of the last nonzero concentration; AUClast, area under the curve from the time of dosing to the last measurable concentration; CI, confidence interval; Cmax, maximum observed concentration occurring at Tmax; CV, coefficient of variation; GM, geometric mean.
aDigoxin 0.5 mg and dextromethorphan 30 mg administered on day 1.
bMaribavir 400 mg (2 × 200 mg) twice daily from days 8 to 15. On day 13, digoxin 0.5 mg and dextromethorphan 30 mg were administered with the morning dose of maribavir 400 mg (2 × 200 mg).
Mean plasma concentration‐time profiles of dextromethorphan and dextrorphan during coadministration of maribavir are presented in Figure 2. The 90%CIs of the geometric mean ratios for Cmax of dextromethorphan and Cmax, AUClast, and AUC0‐∞ of dextrorphan were within the no‐effect range (0.75‐1.33); however, the lower limit of the 90%CIs of geometric mean ratios for the AUClast of dextromethorphan and AUClast ratio of dextromethorphan/dextrorphan were below the predefined lower limit of the no‐effect range (Table 5). Maribavir did not affect the Cmax of dextromethorphan or the Cmax, AUClast, or AUC0‐∞ of dextrorphan. However, the AUClast of dextromethorphan and AUClast ratio of dextromethorphan over dextrorphan were on average decreased by 12.3% and 9.9%, respectively, when coadministered with maribavir. Using the no‐effect range of 0.80‐1.25, the lower limit of the 90%CI of the geometric mean ratio for the Cmax of dextromethorphan was also below the no‐effect range, indicating a decrease of 6.1% when coadministered with maribavir (Table 5).
Figure 2.
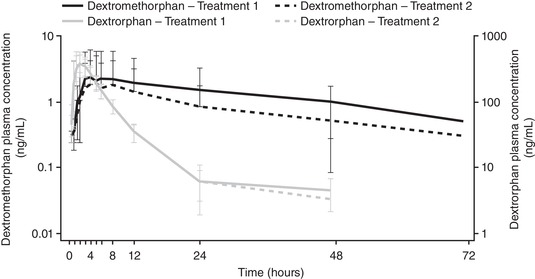
Mean plasma concentration‐time profile for dextromethorphan and dextrorphan by treatment, with log scale (pharmacokinetic set, n = 18).a Treatment 1: digoxin 0.5 mg and dextromethorphan 30 mg administered on day 1. Treatment 2: maribavir 400 mg (2 × 200 mg) twice daily from days 8 to 15. On day 13, digoxin 0.5 mg and dextromethorphan 30 mg was administered with the morning dose of maribavir 400 mg (2 × 200 mg). Error bars represent the standard deviation. aSamples with concentrations of less than the LLOQ (0.2 ng/mL for dextromethorphan and 2.5 ng/mL for dextrorphan) were treated as zero when calculating mean concentration.
Table 5.
Statistical Comparison of Pharmacokinetic Parameters Between Treatments for Dextromethorphan, Dextrorphan, and Dextromethorphan/Dextrorphan (Parent/Metabolite) Ratio (Participants Who Completed the Study, n = 17)
| Analyte | Parameter | Unit | Treatment 1a GM | Treatment 2b GM | Ratio of GMs (90%CI) | Intrasubject Variability CV% |
|---|---|---|---|---|---|---|
| Dextromethorphan | Cmax | ng/mL | 1.21 | 1.14 | 0.939 (0.774‐1.139) | 33.1 |
| AUClast | ng·h/mL | 7.72 | 6.77 | 0.877 (0.692‐1.112) | 41.1 | |
| Dextrorphan | Cmax | ng/mL | 424 | 401 | 0.944 (0.884‐1.009) | 11.1 |
| AUC0‐ ∞ | ng·h/mL | 2250 | 2180 | 0.972 (0.944‐1.001) | 4.68 | |
| AUClast | ng·h/mL | 2170 | 2110 | 0.974 (0.949‐0.999) | 4.27 | |
| Dextromethorphan/dextrorphan (parent/metabolite) ratio | AUClast ratio | 0.00360 | 0.00320 | 0.901 (0.717‐1.133) | 39.6 |
AUC0‐∞, area under the curve extrapolated to infinity, calculated using the observed value of the last nonzero concentration; AUClast, area under the curve from the time of dosing to the last measurable concentration; CI, confidence interval; Cmax, maximum observed concentration occurring at Tmax; CV, coefficient of variation; GM, geometric mean.
aDigoxin 0.5 mg and dextromethorphan 30 mg administered on day 1.
bMaribavir 400 mg (2 × 200 mg) twice daily from days 8 to 15. On day 13, digoxin 0.5 mg and dextromethorphan 30 mg were administered with the morning dose of maribavir 400 mg (2 × 200 mg).
Safety
All participants received digoxin and dextromethorphan on day 1 (treatment 1), and 17 participants received digoxin, dextromethorphan, and maribavir on day 13 (treatment 2). Mean ± SD duration of exposure to maribavir was 8.00 ± 0 days. Assessment of physical examinations, vital signs, ECG, and laboratory parameters revealed no clinically significant changes from baseline or between groups.
In total, 23 TEAEs were reported for 14 participants (77.8%); see Table 6. The most frequently reported TEAEs were headache (n = 8, 44.4%), dysgeusia (n = 7, 38.9%), blurred vision (n = 2, 11.1%), and flatulence (n = 2, 11.1%). The incidence of TEAEs was higher during treatment 2 (70.6%) than treatment 1 (22.2%). All TEAEs reported were considered by the investigator to be related to maribavir, digoxin, or dextromethorphan. No serious TEAEs or deaths were reported. There were no TEAEs leading to dose discontinuation or permanent discontinuation. Most TEAEs were mild in intensity, and no severe TEAEs were reported. One participant experienced 3 moderate TEAEs that occurred during treatment 2.
Table 6.
Summary of TEAEs by Treatment Period (n = 18)
| Category, n (%) | Treatment 1a (n = 18) | Treatment 2b (n = 17) | Overall (n = 18) |
|---|---|---|---|
| Any TEAE | 4 (22.2) | 12 (70.6) | 14 (77.8) |
| TEAEs related to maribavir, digoxin, and dextromethorphan | 4 (22.2) | 12 (70.6) | 14 (77.8) |
| Serious TEAEs related to maribavir, digoxin, and dextromethorphan | 0 (0) | 0 (0) | 0 (0) |
| TEAEs leading to discontinuation | 0 (0) | 0 (0) | 0 (0) |
| Most common TEAEs occurring in ≥5% | |||
| Dysgeusia | 0 (0) | 7 (41.2) | 7 (38.9) |
| Headache | 2 (11.1) | 6 (35.3) | 8 (44.4) |
| Blurred vision | 0 (0) | 2 (11.8) | 2 (11.1) |
| Flatulence | 2 (11.1) | 0 (0) | 2 (11.1) |
| Nausea | 0 (0) | 1 (5.9) | 1 (5.6) |
| Dry eye | 1 (5.6) | 0 (0) | 1 (5.6) |
TEAE, treatment‐emergent adverse event.
Percentages are based on the number of participants in the safety analysis set receiving the corresponding treatment. Participants were counted by the treatment most recently taken when the event occurred. Participants were counted once per category per treatment.
An adverse event (classified by preferred term) that occurred during the study was considered a TEAE if it had a start date on or after the first dose of maribavir/digoxin and dextromethorphan or if it had a start date before the date of the first dose of maribavir/drug probes but increased in severity on or after the date of the first dose of maribavir/digoxin and dextromethorphan.
aDigoxin 0.5 mg and dextromethorphan 30 mg administered on day 1.
bMaribavir 400 mg (2 × 200 mg) twice daily from days 8 to 15. On day 13, digoxin 0.5 mg and dextromethorphan 30 mg were administered with the morning dose of maribavir 400 mg (2 × 200 mg).
Discussion
In this phase 1 study evaluating the effects of multiple doses of maribavir on CYP2D6 and P‐gp activity, dextromethorphan and digoxin were used as probe drugs to assess CYP2D6 and P‐gp activity, respectively, and were administered at the same time. The FDA recommends dextromethorphan as an in vitro and clinical substrate of CYP2D6‐mediated metabolism and digoxin as an in vitro substrate of P‐gp.25 Although digoxin is frequently administered alone, both probes have previously been administered together, and evaluation of the current literature did not reveal any interactions between the 2 probes.22, 25, 26 As such, it was assumed that no interaction would occur between digoxin and dextromethorphan/dextrorphan, although this has not been proven. Coadministration of digoxin and dextromethorphan was not expected to affect the PK of maribavir, which is primarily metabolized through oxidation catalyzed by CYP3A (primary, 70%‐85%) and CYP1A2 (secondary, 15%‐30%).30 Although digoxin has previously been described as an in vitro substrate of organic anion transporter polypeptides (OATP)1B3 (also known as OATP8), other investigations have concluded that it is not a substrate for human OATP1A2, OATP1B1, OATP1B3, and OATP2B1.31, 32 Nonetheless, maribavir was shown not to be a clinically relevant inhibitor of OATP1B3.30
Maribavir 400 mg administered twice daily increased digoxin Cmax and AUC0‐∞ by 25.7% and 21.7%, respectively, whereas AUClast was increased by 18.7% (a result outside the 0.80‐1.25 no‐effect range). This increase in digoxin exposure is consistent with the data from in vitro studies that have identified maribavir as a weak inhibitor of P‐gp with an IC50 of 33.7 µM (Shire data on file: P‐gp inhibition [and substrate]: V9052M‐SHP681). The observed in vivo inhibition of P‐gp activity is likely driven by maribavir's concentration inside the enterocyte, not plasma drug concentration, as free Cmax in plasma is estimated much lower at 0.70 uM based on the free fraction in plasma of 1.5%. Although maribavir had a mild inhibitory effect on P‐gp activity, the ability of maribavir to alter the concentrations of drugs that are substrates of P‐gp and with wide therapeutic windows is unlikely to be clinically meaningful. However, caution should be given to immunosuppressants frequently used in transplant recipients that are substrates for CYP3A, such as cyclosporin, tacrolimus, and sirolimus. Given the high substrate overlap between CYP3A and P‐gp, these immunosuppressants are likely to also be substrates of P‐gp. A phase 1 clinical probe‐cocktail study in healthy subjects demonstrated that maribavir 400 mg twice daily for 10 days did not affect the PK of oral midazolam, a probe substrate of CYP3A21; however, a drug‐interaction study conducted in kidney transplant recipients showed that maribavir 400 mg twice daily increased tacrolimus AUC and Cmax by 51% and 38%, respectively.33 The mechanism of the greater than expected increase in tacrolimus exposure following maribavir coadministration in kidney transplant recipients is currently unknown, but may be related to study design, the high variability in tacrolimus PK variability,34 differences in CYP3A/P‐pg activity between healthy subjects and transplant recipients,35, 36 variability in CYP3A/P‐gp activity over time posttransplant,37 and involvement of other transporters that may be affected by maribavir such as BCRP. Given the significant increase in tacrolimus exposure by maribavir, close therapeutic drug monitoring of immunosuppressants on initiation and discontinuation of maribavir treatment is required in ongoing phase 3 clinical trials with maribavir for the treatment of CMV infections in transplant recipients.
Maribavir had no effect on dextrorphan AUClast and Cmax. It was found to decrease dextromethorphan AUClast, and dextromethorphan/dextrorphan AUClast ratio (and dextromethorphan Cmax, when the 0.80‐.25 no‐effect range was used) by approximately 10% on average. Examination of individual data for dextromethorphan AUC, dextromethorphan Cmax, and dextromethorphan/dextrorphan AUClast ratio and treatment ratios revealed high PK variability (intrasubject variability was estimated at 41.4%, 33.1%, and 39.6%, respectively, higher than expected) and no consistent trend for dextromethorphan exposure decrease among subjects. Given the relatively small sample size of the study, the higher than expected PK variability of dextromethorphan and the small magnitude in the decrease in the dextromethorphan PK parameter, we believe maribavir does not affect CYP2D6 activity in vivo. This is in line with in vitro data demonstrating that maribavir has no effect on CYP2D6 activity (Shire data on file: reversible CYP inhibition: V9079M‐SHP620; time‐dependent inhibition kinetics: V8576M‐SHP620). A previous evaluation of CYP2D6 enzyme activity after multiple doses of maribavir in healthy volunteers demonstrated that maribavir increased the ratio of dextromethorphan and dextrorphan excreted in the urine by an average of 18%.21 However, based on the plasma PK results observed in the current study, it is possible that this increase of the urinary parent/metabolite ratio was likely because of the change of urinary clearance of dextrorphan by other mechanisms and not because of the inhibition of CYP2D6. Overall, based on accumulated in vitro and in vivo data, the effect of maribavir on the CYP2D6 enzyme activity is minimal and the potential for DDI mediated by CYP2D6 is low. It is unlikely that maribavir will affect the PK of drugs that are CYP2D6 substrates. Although all participants in this study were intermediate/extensive CYP metabolizers, the effect of maribavir on CYP2D6 activity is expected to be lower in poor metabolizers versus the intermediate/extensive metabolizers observed here. As such, we hypothesize that maribavir would also have no effect on CYP2D6 activity in CYP2D6 poor metabolizers.
There are a few limitations related to study design that present the potential for bias in study results. In this study, a fixed treatment sequence was used. However, the most desirable design for these studies would be to employ randomized treatment sequences; therefore, despite including a sufficiently long washout period, the treatment effect may have been confounded by a sequence effect. The fixed treatment sequence, small sample size, and higher than previously reported PK variability of dextromethorphan may have impacted the true treatment effect and limited the external validity of the study findings. In addition, the assumption that no interaction would occur between digoxin and dextromethorphan/dextrorphan is recognized as a possible study limitation, as a validation study has not been performed. In addition to the limitations above, this study also did not genotype participants for P‐gp; therefore, any treatment effect by P‐gp polymorphisms cannot be assessed. Finally, the current study was conducted in healthy volunteers, and extrapolation of study results to transplant recipients should be made with caution.
No new safety signals were identified during the administration of maribavir 400 mg twice daily for 7 days. The most frequently reported TEAEs during maribavir treatment were dysgeusia and headache, and this is consistent with the safety profile reported in previous studies with maribavir.38, 39, 40, 41, 42, 43 Dysgeusia has previously been described during maribavir administration and typically presents as a metallic taste that is generally mild and tolerable for most participants.38, 43 No clinically meaningful changes were observed in vital sign measurements or ECGs with maribavir, consistent with previous reports.43
Conclusions
As transplant recipients commonly use a number of therapies simultaneously and often take immunosuppressant agents with narrow therapeutic windows, a thorough review of all concurrent medications and assessment of potential DDI risk should be performed.44 Overall, in this phase 1 study, maribavir (400 mg twice daily) did not impact CYP2D6 activity and inhibited P‐gp activity. The clinical significance and effect of maribavir on the PK of drug substrates of P‐gp should be evaluated on a case‐by‐case basis and caution should be given to P‐gp substrates of narrow therapeutic window. No new safety signals were observed with maribavir (400 mg twice daily for 7 days), with headache and dysgeusia the most commonly reported TEAEs. These results are consistent with findings of previous in vitro and in vivo studies.
Conflicts of Interest
Ivy H. Song, Katarina Ilic, Joseph Murphy, and Patrick Martin are employees of Shire, a Takeda company, and Takeda stock owners. Kenneth Lasseter is an employee of Clinical Pharmacology of Miami Inc., which was funded by Shire Development LLC, Lexington, Massahusetts, a Takeda company, to perform the study. Under the direction of the authors, Amy Horne, MSc, and Yelena Lyustikman, MSc, of Caudex Health, Oxford, UK, provided writing assistance for this publication, funded by Shire International GmbH, a Takeda company. I.H.S., K.I., J.M., and P.M. have received salary and ownership of stocks from Takeda. K.L. has received study funding from Takeda. I.H.S., K.I., J.M., P.M., and K.L. have signed authorship agreements with Shire, a Takeda company. These authorship agreements are in accordance with ICMJE and GPP3 guidelines.
Acknowledgments
The authors acknowledge Jeff Wang (at Shire at the time of study) for his statistical contribution to the study. The authors also acknowledge Jingyang Wu and Prasant Mohanty for additional statistical support with the manuscript. Under the direction of the authors, Amy Horne, MSc, and Yelena Lyustikman, MSc, of Caudex Health, Oxford, UK, provided writing assistance for this publication, editorial assistance in formatting, proofreading, copyediting, fact checking, and coordination and collation of comments was also provided by Caudex Health, funded by Shire International GmbH, a Takeda company. Although employees of Shire were involved in the design, collection, analysis, interpretation, and fact checking of information, the content of this article, the interpretation of the data, and the decision to submit the article for publication was made by the authors independently.
Funding
This study was funded by Shire Development LLC, Lexington, Massachusetts, a Takeda company. Shire International GmbH, Zug, Switzerland, a Takeda company, provided funding to Caudex Health, Oxford, UK, for support in writing and editing this article.
Data‐Sharing Statement
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data behind the results reported in this article, will be available 3 months after the submission of a request to researchers who provide a methodologically sound proposal. The data will be provided after its deidentification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data requests should follow the process outlined in the Data‐Sharing section on Shire's website (http://www.shiretrials.com/en/our-commitment-to-transparency/data-sharing-with-researchers) and should be directed to clinicaltrialdata@shire.com.
Author Contributions
All authors contributed substantially to the scientific process leading to the writing of the article.
Conception and design of the study: Ivy H. Song, Joseph Murphy, and Patrick Martin.
Acquisition and analysis of data: Ivy H. Song, Katarina Ilic, Joseph Murphy, and Patrick Martin.
Drafting the manuscript or figures: Ivy H. Song, Katarina Ilic, Joseph Murphy, Kenneth Lasseter, and Patrick Martin.
All authors take final responsibility for the decision to submit for publication. All authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. The authors are entirely responsible for the scientific content of the article.
None of the authors of this manuscript are fellows of the American College of Clinical Pharmacology (FCP)
Prior presentation: A version of these data were presented at the annual Transplantation and Cellular Therapy (TCT) Meetings 2019, Houston, Texas, February 20‐24, 2019
References
- 1. Teira P, Battiwalla M, Ramanathan M, et al. Early cytomegalovirus reactivation remains associated with increased transplant‐related mortality in the current era: a CIBMTR analysis. Blood. 2016;127(20):2427‐2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hakimi Z, Aballea S, Ferchichi S, et al. Burden of cytomegalovirus disease in solid organ transplant recipients: a national matched cohort study in an inpatient setting. Transpl Infect Dis. 2017;19(5). [DOI] [PubMed] [Google Scholar]
- 3. Azevedo LS, Pierrotti LC, Abdala E, et al. Cytomegalovirus infection in transplant recipients. Clinics (Sao Paulo). 2015;70(7):515‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meesing A, Razonable RR. New developments in the management of cytomegalovirus infection after transplantation. Drugs. 2018;78(11):1085‐1103. [DOI] [PubMed] [Google Scholar]
- 5. Accessdata . Ganciclovir. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209347lbl.pdf. Accessed April 4, 2019.
- 6. Accessdata . Valcyte. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021304s012,022257s007lbl.pdf. Accessed March 15, 2019.
- 7. Accessdata . Letermovir. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209939orig1s000,209940orig1s000lbl.pdf. Accessed March 15, 2019.
- 8. Food and Drug Administration . Cytomegalovirus in transplantation: developing drugs to treat or prevent disease guidance for industry. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM608059.pdf. Accessed April 4, 2019.
- 9. Globenewswire . Shire PLC: Shire receives FDA breakthrough therapy designation for maribavir, an investigational treatment for cytomegalovirus (CMV) infection in transplant patients. https://www.globenewswire.com/news-release/2018/01/04/1283243/0/en/Shire-plc-Shire-Receives-FDA-Breakthrough-Therapy-Designation-for-Maribavir-an-Investigational-Treatment-for-Cytomegalovirus-CMV-Infection-in-Transplant-Patients.html. Accessed March 15, 2019.
- 10. Maertens J, Cordonnier C, Jaksch P, et al. Maribavir versus valganciclovir for preemptive treatment of cytomegalovirus (CMV) viremia: a randomized, dose‐ranging, phase 2 study among hematopoietic stem cell transplant (SCT) and solid organ transplant (SOT) recipients. Open Forum Infect Dis. 2016;3(suppl 1):2287. [Google Scholar]
- 11. Papanicolaou GA, Silveira FP, Langston AA, et al. Maribavir for refractory or resistant cytomegalovirus infections in hematopoietic‐cell or solid‐organ transplant recipients: a randomized, dose‐ranging, double‐blind, phase 2 study. Clin Infect Dis. 2018;68(8):1256‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biron KK, Harvey RJ, Chamberlain SC, et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L‐riboside with a unique mode of action. Antimicrob Agents Chemother. 2002;46(8):2365‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamirally S, Kamil JP, Ndassa‐Colday YM, et al. Viral mimicry of Cdc2/cyclin‐dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009;5(1):e1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krosky PM, Baek MC, Coen DM. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol. 2003;77(2):905‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drew WL, Miner RC, Marousek GI, Chou S. Maribavir sensitivity of cytomegalovirus isolates resistant to ganciclovir, cidofovir or foscarnet. J Clin Virol. 2006;37(2):124‐127. [DOI] [PubMed] [Google Scholar]
- 16. Duncan MD, Wilkes DS. Transplant‐related immunosuppression: a review of immunosuppression and pulmonary infections. Proc Am Thorac Soc. 2005;2(5):449‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cimino FM, Snyder KA. Primary care of the solid organ transplant recipient. Am Fam Physician. 2016;93(3):203‐210. [PubMed] [Google Scholar]
- 18. Glotzbecker B, Duncan C, Alyea E 3rd, Campbell B, Soiffer R. Important drug interactions in hematopoietic stem cell transplantation: what every physician should know. Biol Blood Marrow Transplant. 2012;18(7):989‐1006. [DOI] [PubMed] [Google Scholar]
- 19. Koszalka GW, Johnson NW, Good SS, et al. Preclinical and toxicology studies of 1263W94, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob Agents Chemother. 2002;46(8):2373‐2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Downey R, Johnson J, Gelone S, Broom C, Villano S. Lack of a pharmacokinetic (PK) interaction between oral Maribavir, a novel anti‐cytomegalovirus agent and the CYP 2C19 substrate Voriconazole, in healthy volunteers. Clin Pharmacol Ther. 2009;85:PIII‐74. [Google Scholar]
- 21. Ma JD, Nafziger AN, Villano SA, Gaedigk A, Bertino JS Jr. Maribavir pharmacokinetics and the effects of multiple‐dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N‐acetyltransferase‐2, and xanthine oxidase activities in healthy adults. Antimicrob Agents Chemother. 2006;50(4):1130‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wyen C, Fuhr U, Frank D, et al. Effect of an antiretroviral regimen containing ritonavir boosted lopinavir on intestinal and hepatic CYP3A, CYP2D6 and P‐glycoprotein in HIV‐infected patients. Clin Pharmacol Ther. 2008;84(1):75‐82. [DOI] [PubMed] [Google Scholar]
- 23. Frank D, Jaehde U, Fuhr U. Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol. 2007;63(4):321‐333. [DOI] [PubMed] [Google Scholar]
- 24. Borges S, Li L, Hamman MA, Jones DR, Hall SD, Gorski JC. Dextromethorphan to dextrorphan urinary metabolic ratio does not reflect dextromethorphan oral clearance. Drug Metab Dispos. 2005;33(7):1052‐1055. [DOI] [PubMed] [Google Scholar]
- 25. Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin Pharmacol Ther. 2007;81(2):270‐283. [DOI] [PubMed] [Google Scholar]
- 26. Jetter A, Fatkenheuer G, Frank D, et al. Do activities of cytochrome P450 (CYP)3A, CYP2D6 and P‐glycoprotein differ between healthy volunteers and HIV‐infected patients? Antivir Ther. 2010;15(7):975‐983. [DOI] [PubMed] [Google Scholar]
- 27. Department of Health and Human Services (DHHS) FaDA , Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM) (US). Guidance for industry: Bioanalytical method of validation. https://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf. Accessed May 23, 2018. [Google Scholar]
- 28. European Medicines Agency . Guideline on bioanalytical method validation. https://www.ema.europa.eu/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed April 4, 2019. [DOI] [PubMed]
- 29. Kakuda TN, Van Solingen‐Ristea RM, Onkelinx J, et al. The effect of single‐ and multiple‐dose etravirine on a drug cocktail of representative cytochrome P450 probes and digoxin in healthy subjects. J Clin Pharmacol. 2014;54(4):422‐431. [DOI] [PubMed] [Google Scholar]
- 30. Sun K, Rong H, Song I, Welty D. In vitro metabolic and transporter profiling for maribavir (SHP620) [abstract]. 22nd North American ISSX Meeting. 2018. [Google Scholar]
- 31. Kullak‐Ublick GA, Ismair MG, Stieger B, et al. Organic anion‐transporting polypeptide B (OATP‐B) and its functional comparison with three other OATPs of human liver. Gastroenterology. 2001;120(2):525‐533. [DOI] [PubMed] [Google Scholar]
- 32. Taub ME, Mease K, Sane RS, et al. Digoxin is not a substrate for organic anion‐transporting polypeptide transporters OATP1A2, OATP1B1, OATP1B3, and OATP2B1 but is a substrate for a sodium‐dependent transporter expressed in HEK293 cells. Drug Metab Dispos. 2011;39(11):2093‐2102. [DOI] [PubMed] [Google Scholar]
- 33. Pescovitz MD, Bloom R, Pirsch J, Johnson J, Gelone S, Villano SA. A randomized, double‐blind, pharmacokinetic study of oral maribavir with tacrolimus in stable renal transplant recipients. Am J Transplant. 2009;9(10):2324‐2330. [DOI] [PubMed] [Google Scholar]
- 34. Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet. 2004;43(10):623‐653. [DOI] [PubMed] [Google Scholar]
- 35. Murakami T IM, Oda K. Modulation of P‐glycoprotein Expression and Function under Disease States in Rats and Humans. Int J Clin Pharmacol Pharmacother. 2017;2:130. [Google Scholar]
- 36. Lemahieu W, Maes B, Verbeke K, Rutgeerts P, Geboes K, Vanrenterghem Y. Cytochrome P450 3A4 and P‐glycoprotein activity and assimilation of tacrolimus in transplant patients with persistent diarrhea. Am J Transplant. 2005;5(6):1383‐1391. [DOI] [PubMed] [Google Scholar]
- 37. Storset E, Hole K, Midtvedt K, Bergan S, Molden E, Asberg A. The CYP3A biomarker 4beta‐hydroxycholesterol does not improve tacrolimus dose predictions early after kidney transplantation. Br J Clin Pharmacol. 2017;83(7):1457‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winston DJ, Young JA, Pullarkat V, et al. Maribavir prophylaxis for prevention of cytomegalovirus infection in allogeneic stem cell transplant recipients: a multicenter, randomized, double‐blind, placebo‐controlled, dose‐ranging study. Blood. 2008;111(11):5403‐5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goldwater DR, Dougherty C, Schumacher M, Villano SA. Effect of ketoconazole on the pharmacokinetics of maribavir in healthy adults. Antimicrob Agents Chemother. 2008;52(5):1794‐1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winston DJ, Saliba F, Blumberg E, et al. Efficacy and safety of maribavir dosed at 100 mg orally twice daily for the prevention of cytomegalovirus disease in liver transplant recipients: a randomized, double‐blind, multicenter controlled trial. Am J Transplant. 2012;12(11):3021‐3030. [DOI] [PubMed] [Google Scholar]
- 41. Lalezari JP, Aberg JA, Wang LH, et al. Phase I dose escalation trial evaluating the pharmacokinetics, anti‐human cytomegalovirus (HCMV) activity, and safety of 1263W94 in human immunodeficiency virus‐infected men with asymptomatic HCMV shedding. Antimicrob Agents Chemother. 2002;46(9):2969‐2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marty FM, Ljungman P, Papanicolaou GA, et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem‐cell transplants: a phase 3, double‐blind, placebo‐controlled, randomised trial. Lancet Infect Dis. 2011;11(4):284‐292; Erratum: 2011;2011:2343. [DOI] [PubMed] [Google Scholar]
- 43. Wang LH, Peck RW, Yin Y, Allanson J, Wiggs R, Wire MB. Phase I safety and pharmacokinetic trials of 1263W94, a novel oral anti‐human cytomegalovirus agent, in healthy and human immunodeficiency virus‐infected subjects. Antimicrob Agents Chemother. 2003;47(4):1334‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohammadpour N, Elyasi S, Vahdati N, Mohammadpour AH, Shamsara J. A review on therapeutic drug monitoring of immunosuppressant drugs. Iran J Basic Med Sci. 2011;14(6):485‐498. [PMC free article] [PubMed] [Google Scholar]