

Abstract
Succinate dehydrogenase (SDH) mutations lead to the accumulation of succinate, which acts as an oncometabolite. Germline SDHx mutations predispose to paraganglioma (PGL) and pheochromocytoma (PCC), as well as to renal cell carcinoma and gastro‐intestinal stromal tumors. The SDHx genes were the first tumor suppressor genes discovered which encode for a mitochondrial enzyme, thereby supporting Otto Warburg's hypothesis in 1926 that a direct link existed between mitochondrial dysfunction and cancer. Accumulation of succinate is the hallmark of tumorigenesis in PGL and PCC. Succinate accumulation inhibits several α‐ketoglutarate dioxygenases, thereby inducing the pseudohypoxia pathway and causing epigenetic changes. Moreover, SDH loss as a consequence of SDHx mutations can lead to reprogramming of cell metabolism. Metabolomics can be used as a diagnostic tool, as succinate and other metabolites can be measured in tumor tissue, plasma and urine with different techniques. Furthermore, these pathophysiological characteristics provide insight into therapeutic targets for metastatic disease. This review provides an overview of the pathophysiology and clinical implications of oncometabolite succinate in SDHx mutations.
Keywords: oncometabolites, paraganglioma, pheochromocytoma, SDH mutation, succinate
1. INTRODUCTION
Mutations of genes encoding for the succinate dehydrogenase (SDH) complex, associated with familial paraganglioma (PGL) and pheochromocytoma (PCC), lead to accumulation of succinate, which disturbs the metabolic regulation of the cell. Nowadays metabolic dysregulation is recognized as one of the eight hallmarks of cancer.1
Although germline mutations in SDHx genes are predominantly linked to PGL and PCC, these mutations also predispose to renal cell carcinoma (RCC), gastrointestinal stromal tumors (GISTs) and, possibly, pituitary adenomas. PCC, PGL and head and neck PGL (HNPGL) are rare neuroendocrine tumors arising from chromaffin cells that can synthesize and release catecholamines. Sympathetic PGLs are derived from sympathetic paraganglia in the chest, abdomen or pelvis. PCC are PGLs located in the adrenal medulla.2 HNPGLs are derived from parasympathetic paraganglia. Common locations for HNPGLs include the carotid body and the middle ear, as well as the vagus nerve and internal jugular vein. While parasympathetic PGLs are most often non‐functional tumors, PCC and sympathetic PGL release catecholamines into the circulation and can lead to severe (lethal) cardiovascular and cerebrovascular complications. Approximately, 40% of these tumors carry a germline mutation in one of more than 20 susceptibility genes, of which the SDHx genes are the most prevalent.3
In terms of genomic features, tumors related to SDHx mutations are classified as cluster I, along with Von Hippel Lindau (VHL), fumarate hydratase (FH), malate dehydrogenase 2 (MDH2), hypoxia induced factor (HIF2α) and isocitrate dehydrogenase (IDH)‐mutations and the recently identified SLC25A11.4 Cluster I germline mutations predispose to tumors characterized by a pseudohypoxic signature, in contrast to cluster II germline mutations, which are associated with abnormal kinase signaling pathways and include mutations in the genes of rearranged during transfection (RET), neurofibromatosis (NF1), transmembrane protein 127 (TMEM127), kinesin family member 1B (KIF1B), and MYC‐associated factor X (MAX). Cluster III is associated with the Wnt‐signaling pathway; it includes somatic mutations of cold shock domain‐containing E1 (CSDE1) and mastermind‐like transcriptional coactivator 3 (MAML3) fusion genes.5, 6
SDHx genes were the first to be recognized as tumor suppressor genes encoding a mitochondrial enzyme. This resulted in an upsurge of interest in the concept of aerobic glycolysis or the “Warburg effect,” reported by Otto Warburg in 1926, which is characterized by high glucose consumption and lactate production of cancer cells, even in the presence of oxygen.7 This metabolic dysregulation is in fact recognized as one of the eight hallmarks of cancer.
Defective SDH function triggers the accumulation of succinate, an intermediate metabolite of the tricarboxylic acid (TCA) cycle, which plays a crucial role in the generation of adenosine triphosphate (ATP) in mitochondria. Accumulation of succinate, along with other intermediate metabolites of the TCA cycle, can give rise to the development and progression of cancer. FH mutations lead to the accumulation of fumarate, and IDH mutations result in an accumulation of (R)‐2‐hydroxyglutarate. These oncometabolites modulate the activity of α‐ketoglutarate‐dependent dioxygenases, which are involved in the induction of the pseudohypoxia pathway and inhibit histones and DNA demethylases, resulting in a hypermethylator phenotype (also known as CpG island methylator phenotype [CIMP]). The SLC25A11 gene encodes for a mitochondrial carrier protein that is part of the malate‐asparate shuttle (this shuttle regenerates NADH to allow complex I to function), mediating the transport of α‐ketoglutarate from the mitochondrial matrix to the cytoplasm in exchange with malate. Preliminary results show that in SLC25A11‐mutated cells aspartate and glutamate concentration is increased inducing the pseudohypoxic pathway and hypermethylation.4
Recognition of these pathophysiological characteristics provides unique opportunities for diagnostic and therapeutic strategies. Over the past years, several excellent reviews, such as those by Kucklova et al, Morin et al and Vicha et al, have discussed the pathophysiology of SDHx‐related tumors.8, 9, 10 In the current review, we first present a short summary of the SDH protein and the clinical features of SDHx‐mutation carriers. We then focus on the oncometabolite succinate and its pivotal role in tumorigenesis in SDHx‐related tumors, as well as on the implications for clinical practice, especially diagnostics and therapeutic options related to metastatic disease.
2. SUCCINATE DEHYDROGENASE
SDH is a hetero‐tetrameric mitochondrial enzyme that plays a role in the TCA cycle and in the mitochondrial electron transport chain as complex II (Figure 1). SDH catalyzes the oxidation of succinate to fumarate in the TCA cycle and transfers electrons to the ubiquinone (coenzyme Q) pool in the respiratory chain. SDH subunit A (SDHA) and subunit B (SDHB) comprise the catalytic subunits in the hydrophilic head that protrudes into the mitochondrial matrix. SDH subunit C (SDHC) and subunit D (SDHD) are the ubiquinone‐binding and membrane‐anchoring subunits. SDH assembly factor (SDHAF) is required for the flavination of SDHA, which is essential for the formation of the SDH complex. The SDHA gene is located on chromosome 5p15.33 and contains 16 exons.11 SDHA is the major catalytic subunit, converting succinate to fumarate. It contains the binding site for succinate. The gene encoding for SDHB is located on chromosome 1p35‐36.1 and has eight exons12; the SDHB protein contains three Fe‐S centers and mediates electron transfer to the ubiquinone pool. The gene encoding SDHC is located at 1q21 and has six exons,13 and the SDHD gene is located on chromosome 11q23 and has four exons.14 SDHC and SDHD bind ubiquinone, generating protons eventually leading to the production of ATP.
Figure 1.
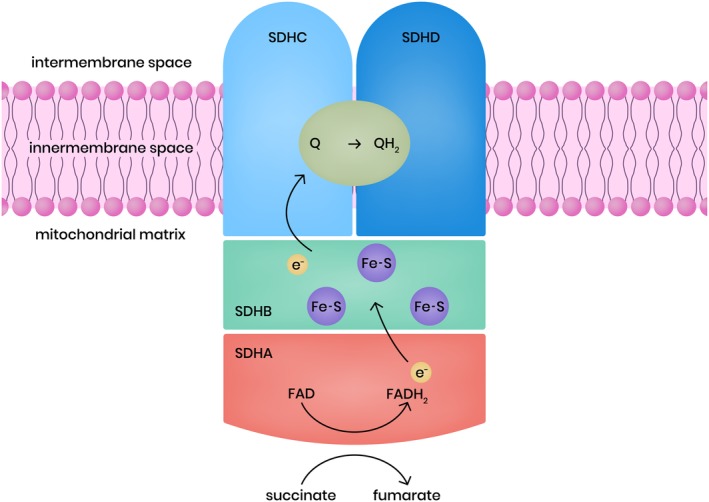
Succinate dehydrogenase (SDH) complex (simplified). The catalytic subunits SDH subunit A contains the flavin cofactor (FAD) which accepts electrons from succinate and passes them to Fe‐S center in the SDH subunit B subunit. The electrons are then passed the ubiquinone pool embedded in SDHC and SDHD subunits. Reduced Q (QH2 = ubiquinol) transfers electrons within the mitochondrial inner membrane space to complex III [Colour figure can be viewed at http://wileyonlinelibrary.com]
3. PHENOTYPE OF SDHX MUTATION CARRIERS
Although different SDHx mutations occur in genes encoding for a single enzyme, the clinical picture for each subunit differs with regard to penetrance, manifestations and rate of malignancy. International guidelines advice to screen all germline mutation carriers, however with different screenings strategies for different SDHx mutation carriers.15 Screenings advices do not only differ between the different mutations, but also over time, because studies on penetrance differ over time regarding the population studied (index included or not), the imaging methods used and the duration of follow‐up. Adherence to screening, leads to the detection of smaller PCC/PGL and might even lead to an improved survival for patients who develop metastases, although this is based on only few patients.16
Until now, a clear explanation for the difference of the clinical picture between different SDHx mutations is lacking, except for the hypothesis that the extent of SDH deficiency or loss depends on the subunit. Apart from the differences, all SDHx mutations are characterized by the (potential) presence of PGL/PCC. SDHx‐associated tumors harbor germline and somatic mutations, consistent with Knudson's second‐hit hypothesis.12 This hypothesis states that the combination of an inactivating germline mutation as a first hit, and somatic loss of function of the wild type allele as a second hit, is essential for tumor development.17 This second hit usually is an inactivation of the normal allele, that is, loss of 1p as was shown in a large genomic study.18
Germline SDHx mutations have been associated with neoplasms other than PGL/ PCC, such as RCC,19, 20, 21 GISTs and possibly pituitary adenoma.22, 23, 24 In addition, somatic SDHx mutations have been described in T‐cell leukemia.25 Because the discovery of SDHx genes is relatively recent, the full clinical phenotype of these carriers remains to be sufficiently clarified. The following paragraphs describe the currently known phenotype of each SDHx subunit (Table 1). The question of why SDHx mutations predispose to tumors in a select subset of tissues remains elusive.
Table 1.
Phenotypic features of SDHx mutation carriers
| Prevalence (%) | Penetrance | Mode of inheritance | PCC | sPGL | HNPGL | Multifocality | Metastasis | Other tumors | |
|---|---|---|---|---|---|---|---|---|---|
| SDHA | 1–7 | Low | AD | + | + | ++ | Rare | Yes | GIST, PA, NB |
| SDHB | 8–10 | Medium | AD | + | ++ | + | Rare | Frequent | GIST, RCC, PA |
| SDHC | 1–2 | Low | AD | + | + | ++ | Frequent | Rare | GIST, RCC, PA |
| SDHD | 5–9 | High | Paternal transmissiona | + | + | ++ | Frequent | Rare | GIST, PA, RCC |
| SDHAF2 | <1 | Unknown | Paternal transmissiona | − | − | ++ | Frequent | Unknown | PA |
Abbreviations: AD, autosomal dominant; GIST, gastrointestinal stromal tumor; HNPGL, head and neck paraganglioma; NB, neuroblastoma; PA, pituitary adenoma; PCC, pheochromocytoma; RCC, renal cell carcinoma; SDH, succinate dehydrogenase; SDHAF2, SDH assembly factor; SDHA, SDH subunit A; SDHB, SDH subunit B; SDHC, SDH subunit C; SDHD, SDH subunit D; sPGL, sympathetic paraganglioma; −, manifestation (to our knowledge) not described in these mutation carriers.+, manifestation present in these mutation carriers; ++, most common manifestation of these mutation carriers.
SDHD and SDHAF2 autosomal dominant with maternal imprinting.
3.1. SDHA mutations
Mutations in the SDHA gene were originally described as a cause of autosomal recessive juvenile encephalopathy, also known as Leigh syndrome.26 Later on, in 2010, a 32 year old woman with an abdominal PGL was reported to have a heterozygous SDHA germline mutation.27 Mutations in the SDHA gene remain a rare cause of PGL and account for 1% to 7% of all PGL cases.28, 29 About half of SDHA mutation carriers present with HNPGL, although sympathetic PGL and PCC are also reported.30, 31, 32 Recently, van der Tuin et al calculated the penetrance of SDHA mutation in a cohort comprising 86 patients (30 index and 56 non‐index patients). The penetrance for all manifestations was only 10% at age 70 in non‐index patients, but 50% at age 70 when both index and non‐index patients were included in the analysis.32
Metastatic disease was reported in 0% to 33% of PGL patients with SDHA mutations, although these reports included few patients (n = 4‐34).31, 32, 33, 34, 35, 36 GISTs and pituitary adenomas were reported in a small subset of patients.24, 31, 32, 37, 38 In a large pediatric GIST study of Boikos et al, a SDHA mutation, germline or somatic, was the most common molecular subtype39 Neuroblastoma was reported in one SDHA mutation carrier where it was possible to confirm loss of heterozygosity (LoH) in tumor tissue.38
3.2. SDHB mutations
Mutations in the SDHB gene are those most frequently found in PGL and account for about 10% of all cases of PGL.28 Most common manifestations are sympathetic PGLs (50%), whereas PCC and HNPGL occur less often (20%‐25% and 20%‐30%, respectively).15 Bilateral PCCs appear to be rare in SDHB mutation carriers. Penetrance of different manifestations decreases over time as more asymptomatic carriers are identified. Earlier studies of penetrance included mostly index patients, thereby overestimating the penetrance. A recent study by Andrews et al calculated the cumulative tumor risk in a large cohort of 673 SDHB‐ mutation carriers and corrected for such ascertainment bias by calculating not only the penetrance for only index patients, but also for a combination of index and non‐index patients.40 Their Kaplan‐Meier analysis showed an estimated risk for the combined manifestation of PCC, sympathetic PGL and HNPGL in non‐index patients to be 22% at age 60.40 In their retrospective cohort analysis (index and non‐index patients) the penetrance was 24% at age 60 years.40 Males seem to be slightly more at risk than females of developing a PGL.40, 41
SDHB‐related PGL/PCC are associated with a high risk of metastasis and poor prognosis. Earlier studies report a higher metastatic rate (31%‐97%)42, 43, 44, 45 than more recent studies.40 In a meta‐analysis including 12 studies comprising both asymptomatic SDHB carriers and carriers with manifest non‐metastatic disease, van Hulsteijn et al reported a metastatic rate of 17%.46 The risk of metastasis in HNPGL in SDHB mutation carriers appears to be lower compared to PGL developing at other sites.15 In a recent meta‐analysis of the outcomes of metastatic PGL and PCC, Hamidi et al found that the overall mortality in SDHB mutation carriers ranged from 35% to 55% (n = 96) compared to an overall mortality of 53% (95% confidence interval 43%‐63%) in the whole group of PGL/PCC.47 In the past, several studies have shown an association between SDHB‐related metastatic PGL/PCC and a shorter survival in patients compared to sporadic metastatic PGL/PCC.48, 49 In a recent analysis of Hescot et al, not the SDHB mutation status but hypersecretion of metanephrines and chromogranin A was found to be a significant prognostic factor for worst overall survival.50
Other SDHB‐associated tumors include RCC, although the risk for this manifestation seems low, varying between 4.7% and 8%.21, 40 GISTs are reported to occur in approximately 2% of SDHB carriers.51 Pituitary adenoma have been reported in nine cases, but only three had proven LoH (loss of heterozygosity) and abnormal SDHB expression, thus confirming involvement of SDHB mutation.24 Tufton et al reported a case of a SDHB mutation carrier with pituitary carcinoma.52
3.3. SDHC mutations
Mutations in the SDHC gene account for 1% to 2% of PGL/PCC cases.28 SDHC typically manifest as benign, non‐functional HNPGL, although sympathetic PGL and PCC are also reported.53, 54 Multiple HNPGL are common.54 Penetrance for all PGL/PCC manifestations in a cohort of 43 non‐index SDHC carriers was 25% at age 60.40
Although metastatic disease seems to be rare in SDHC mutation carriers, it has in a few cases been reported.40, 55, 56, 57 Eight RCC and multiple GISTs have been reported in SDHC carriers.19, 58, 59 Two cases of pituitary adenoma have been described, although for LoH studies no tissue was available to prove pathogenicity.24
3.4. SDHD mutations
A mutation in the SDHD gene accounts for approximately 5% to 9% of all cases of PGL/PCC.28, 29 This gene follows an autosomal dominant inheritance, modified by maternal imprinting. The predominant clinical feature of SDHD carriers is the development of (multiple) HNPGLs, as 85% of carriers develop tumors at this site.51 PCC and sympathetic PGL occur less frequently in 10% to 25% and 20% to 25% of carriers, respectively. Penetrance for 160 non‐index SDHD mutation carriers was 43% at age 60.40
Metastatic risk in SDHD carriers is low and occurs in 7% to 8% of cases.15, 60 Other associated tumors include RCC and GIST, although the lifetime risk for this manifestation is very low (<1%).40, 61, 62 Pituitary adenomas are reported in five SDHD mutation carriers; in two of these, both macroprolactinomas, the presence of LoH was proven.22, 23
3.5. SDHAF2 mutations
The SDHAF2 gene, like SDHD, is affected by maternal imprinting; therefore, only those carriers who inherit the mutation via the paternal line will develop the disease. Only a few cases of PGL/PCC associated with SDHAF2 mutations have been described, and these account for <1% of all cases of PGL.29 Germline pathogenic variants in SDHAF2 have been seen only in association with HNPGLs.31, 63, 64, 65, 66, 67 Kunst et al describe a large family of 16 patients, 11 with a HNPGL, primarily at carotid body and vagal locations. Within this family, the presence of multiple HNPGLs was common, and no cases of metastatic disease were found.65
4. CONSEQUENCES OF SDH DEFICIENCY OR LOSS
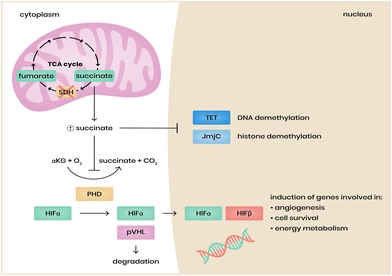
In SDHx germline mutation carriers affected by a second hit, SDH loss of function leads to the accumulation of succinate in the tumor cells,68, 69, 70, 71, 72 which is the hallmark of tumorigenesis of these tumors This accumulation inhibits several α‐ketoglutarate dioxygenases, which are involved in the induction of the pseudohypoxia pathway and in epigenetic DNA modifications. Moreover, SDH deficiency or loss may lead to overproduction of reactive oxygen species (ROS) and to a “rewiring” of the cell's metabolism.
4.1. Accumulation of succinate induces the pseudohypoxia pathway
Tumors harboring a SDHx mutation have a strong hypoxic signature. PGL/PCCs have historically been closely associated with hypoxia, because these highly vascularized tumors arise either in tissues known to be susceptible to low oxygen levels (adrenal medulla, organ of Zuckerkandl), or in cells known to serve as oxygen sensors (carotid body).
The major regulator of hypoxia response is the transcription factor HIF. HIF activity is regulated by TCA cycle metabolites. HIF is a heterodimer and consists of two subunits, one α subunit and one β subunit. There are three different α‐subunits: HIF1α, HIF2α and HIF3α, and two different β subunits: HIF1ß (aryl hydrocarbon receptor nuclear translocator [ARNT1]) and ARNT2. Whereas the β subunits are constitutively expressed, the active α subunits HIF1α and HIF2α are degraded in the presence of oxygen and therefore function as gatekeepers in response to low oxygen. Under normoxic conditions, HIFα is continuously synthesized, and propyl hydroxylase domain (PHD) marks it for degradation, involving the activity of the VHL ubiquitination complex (pVHL). The hydroxylation reaction performed by the PHD enzymes requires oxygen and α‐ketoglutarate as substrates, as well as iron and ascorbate as cofactors.73 Thus, during hypoxia PHD becomes inactive, and as a result HIFα escapes pVHL recognition and degradation. The unmodified HIFα molecule translocates to the nucleus, where it forms a transcriptionally active HIFα heterodimer with a stable HIFβ subunit. This active transcription factor induces a wide variety of target genes involved in cellular adaptation to hypoxia as in angiogenesis, energy metabolism, and cell survival.
In a SDH deficient condition, the excess of accumulated succinate is shuttled from the mitochondrial matrix to the cytoplasm, where it competes with α‐ketoglutarate in binding to PHD and inhibiting PHD. This consequently leads to the stabilization of HIFα even in the presence of oxygen, a condition known as pseudohypoxia.3, 68, 74, 75, 76
HIFα regulates the transcription of several genes known to be involved in tumorigenesis, angiogenesis, extracellular matrix elements and energy metabolism. HIF1α and HIF2α share the target genes vascular endothelial growth factor (VEGF), glucose transporters 1 and 3 (GLUT1 and GLUT3) and hexokinase 2. HIF1α stimulates the expression of various glycolytic enzymes and HIF2α stimulates the expression of platelet‐derived growth factor (PDGF) and erythropoietin (EPO).9 Pollard et al showed the overexpression of HIF1α in SDHx tumors compared to tumors with other germline mutations,71, 77 while others studies showed overexpression of HIF2α in SDHx related tumors compared to sporadic PGL/PCC.78, 79, 80 The role of HIF3α in relation to tumorigenesis remains to be elucidated, although, previous studies have indicated that HIF3α may suppress the expression of genes induced by HIF1α and HIF2α (for review see Reference 81).
Heat shock proteins (HSPs) are molecular chaperones that are important for protein assembly, folding and stability and play a central role in cell proliferation, survival and tumor progression. HSP90 is involved in the stability of HIF1α.9 HSP90 has been shown to be overexpressed in metastatic PGL/PCC compared with benign PGL/PCC.82 Inhibition of HSP90 leads to downregulation of HIF1α and is a potential target for therapy in metastatic PGL/PCC.83
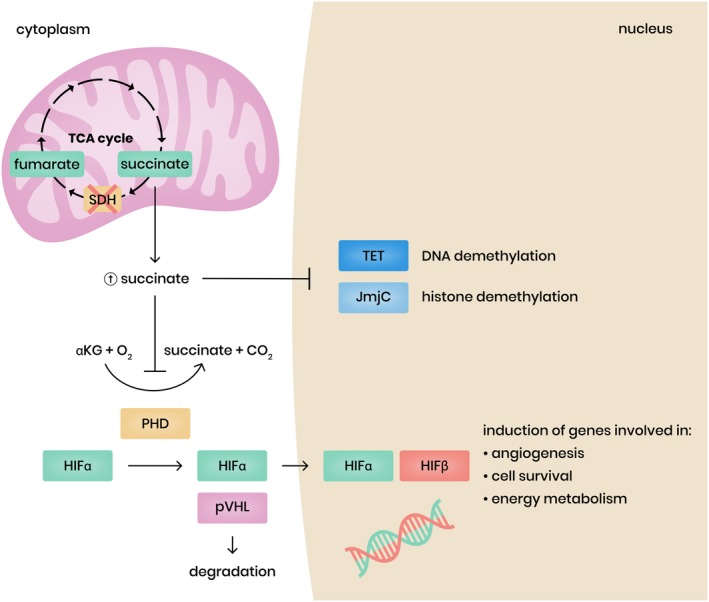
4.2. Accumulation of succinate leads to a hypermethylator phenotype
Recent studies have observed a hypermethylator phenotype in SDH deficient PGL/PCC.84, 85, 86 Next to PHD, accumulation of succinate competitively inhibits other α‐ketoglutarate‐dependent dioxygenases, including jumonji‐domain histone demethylases (JmjC) and the ten‐eleven translocation (TET) family of DNA methylase (Figure 2). Inhibition of these dioxygenases leads to hypermethylation of promotor regions (CpG islands) of several genes (also known as CpG island methylator phenotype [CIMP]). Because methylation triggers gene transcription deregulation, hypermethylation of tumor‐suppressor gene promotors plays an important role in tumorigenesis.
Figure 2.
Consequences of succinate dehydrogenase (SDH) loss. SDH loss leads to the accumulation of succinate which inhibits a‐ketoglutarate dependent dioxygenases including prolyl‐hydroxylases (PHD), ten–eleven translocation (TET) and jumonji C‐domain‐containing proteins (JmjC) [Colour figure can be viewed at http://wileyonlinelibrary.com]
Letouzé et al determined the DNA methylation profiles of a large PGL/PCC cohort. They identified 191 genes showing significant hypermethylation, due to an inhibition of DNA demethylation, and downregulation in SDHx‐related PGL/PCC.84 The most significant epigenetically silenced genes were those encoding phenyl‐ethanolamine‐N‐methyltransferase (PNMT) and keratine 19 (KRT19), which are involved in neuroendocrine differentiation and in epithelial‐to‐mesenchymal transition (EMT).84
PNMT catalyzes the conversion of norepinephrine to epinephrine. Next to the PNMT gene, four other genes that we found to be hypermethylated, are involved in the catecholamine secretion: SULT1A1, DRD2, NPY, and SLC6A2.84 Reduced expression of these genes leads to an immature catecholamine secretory profile with predominant excretion of norepinephrine and dopamine. In SDHB mutated tumors the level of hypermethylation seems to be significantly higher compared to other SDHx mutated tumors, and the expression of these target genes significantly lower. The authors hypothesize that SDH inactivation may be more complete in SDHB mutated tumors compared to tumors harboring a mutation in the other subunits, leading to a higher succinate accumulation and hence a stronger inhibition of α‐ketoglutarate‐dependent demethylation.84 This could be an explanation for the higher metastatic risk in SDHB‐related tumors.
The study of Richter et al confirmed that tumor succinate:fumarate ratios were higher in tumors of patients with SDHB mutations compared to tumors of patients with an SDHC/D mutation.87 EMT is a process by which epithelial cells lose their polarity and cell‐to‐cell adhesion, thereby gaining migratory and invasive properties to become mesenchymal stem cells. This process, normally occurring during embryonic development, can be reactivated in cancer cells and is involved in metastatic dispersion.88 Several genes and signaling pathways have been identified as involved in different parts of the induction of EMT. KRT19 encodes an intermediate filament required for the formation of desmosomes (structure specialized for cell‐to‐cell adhesion) and shown to be downregulated in SDHB metastatic PGL tissue samples unlike non‐SDHB metastatic PGL tissue samples.89 EMT is the first pathway identified that may be responsible for the specific metastatic properties of SDHB‐related PGL and PCC.
Kiss et al showed that the tumor suppressor gene P16 was hypermethylated in SDHB mutated tumor tissue samples as opposed to RET‐, VHL‐ or NF‐related PGL/PCC.90 P16 is an inhibitor of cyclin‐dependent kinases and plays an important role in cell cycle regulation by decelerating the cells progression from G1 phase to S phase, and acts therefore as a tumor suppressor. The authors showed that hypermethylation of P16 was associated with short disease‐related survival.90
4.3. SDH loss leads to overproduction of reactive oxygen species
Reactive oxygen species (ROS) are damaging molecules containing oxygen with an unpaired free electron, such as superoxide and hydrogen peroxide. Although ROS are critical for normal cell function, they also lead to oxidative damage of DNA, which leading to genomic instability and finally to apoptosis. Mitochondria are the major source of ROS, especially complexes I and III, although complex II can also produce a significant number.91, 92 Accumulation of succinate results in an over‐reduced ubiquinone pool resulting in a reverse electron transfer to complex I, where electrons escape as ROS.93 Excessive ROS levels have been shown to stabilize HIFα and induce the pseudohypoxia pathway in SDHx‐mutated PGL/PCC.94 In addition to the stabilization of HIFα, SDHx‐mutation‐induced increases in ROS have been shown to cause genomic instability that may contribute to tumorigenesis.95, 96 Nevertheless, experimental evidence for ROS in various models of SDH dysfunction is not consistent, as some suggests that ROS are increased or normal, a finding which is extensively reviewed by Kluckova and coworkers.8
4.4. SDH loss leads to changes in the cell's metabolism pathways
SDH deficiency or loss can lead to reprogramming of cancer‐related cell metabolism such as enhanced glycolysis (Warburg effect), as well as changes in anaplerotic pathways and in oxidative phosphorylation (Figure 3 ).
Figure 3.
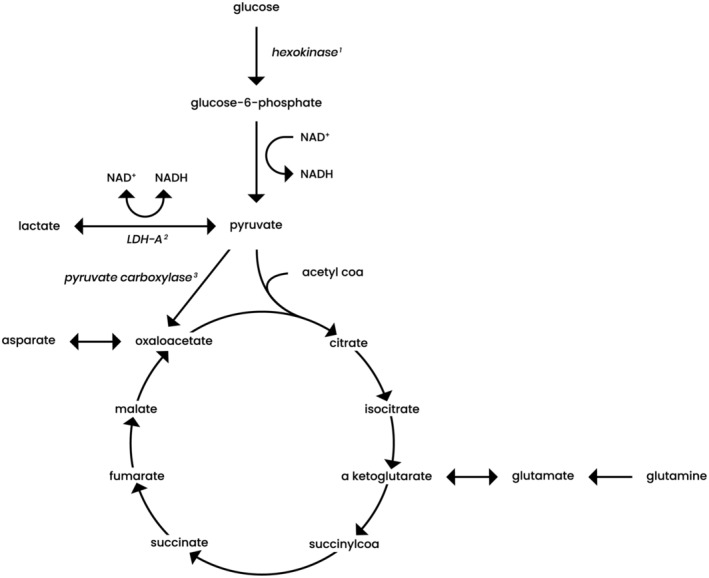
Metabolic pathways in which succinate dehydrogenase (SDH) loss is involved, including glycolysis, tricarboxylic acid cycle and anaplerotic reactions. The first step in glycolysis is the phosphorylation of glucose to glucose‐6‐phosphate by hexokinase1. Lactate dehydrogenase A (LDH‐A)2 catalyzes the conversion of pyruvate and lactate with concomitant conversion of nicotinamide adenine dinucleotide (NADH) and NAD+. Pyruvate carboxylase3 catalyzes the conversion of pyruvate to oxaloacetate. Proposed metabolic changes in SDH loss are enhanced glycolysis, via activation of LDH‐A and hexokinase. Furthermore, pyruvate carboxylase may be upregulated in SDH loss and there may be an increased glutamine metabolism. A more detailed explanation is described in the text
4.4.1. Warburg effect
As stated above, SDHx‐related tumors display the Warburg effect. The main driver of the Warburg effect is HIFα which induces expression of GLUT1 and GLUT3, hexokinase 2, pyruvate kinase variant M2 (PKM2) and lactate dehydrogenase A (LDH‐A), thereby enhancing the glycolytic pathway.97 PKM2 interacts with HIF1α in the nucleus, where it functions as a coactivator to increase the expression of HIF1α target genes that stimulate the shift from oxidative phosphorylation to glycolysis.98, 99 Favier et al and Fliedner et al found an overexpression of LDH‐A in SDHx‐related tumors.100, 101 LDH‐A converts pyruvate to lactate, thereby recovering the NAD+ needed to maintain glycolysis, critical for tumor proliferation in vivo.100 The generated lactate leads to an acid tumor microenvironment, which in turn may facilitate tumor invasion and migration and is correlated with a poor prognosis.102
4.4.2. Anaplerotic pathways
Pyruvate carboxylase, catalyzing pyruvate to oxaloacetate, an important anaplerotic reaction, may be upregulated in SDHx tumors. Cardaci et al showed that pyruvate carboxylase is upregulated in SDHB null cells. Silencing of the pyruvate carboxylase gene both significantly reduced the proliferation of SDH cells in vitro and delayed the onset of tumor in vivo, compared to SDH proficient cells/mice. By identifying pyruvate carboxylase as an essential gene for SDH‐deficient cells but dispensable for normal cells, this study unveils a metabolic vulnerability for potential treatment of SDHx‐associated tumors.103
Lussey‐Lepoutre et al showed that in SDHx‐mutated tumor cells the increased synthesis of oxaloacetate is essential in order to produce aspartate (as well as to continue a truncated oxidative TCA cycle). Aspartate is an important precursor for protein and nucleotide biosynthesis for anabolic purposes. In SDH deficient cells, as compared to wild type cells, knockdown of pyruvate carboxylase results in complete ablation of proliferation. The authors also showed the use of glutamine and glutamate to provide intermediates that are lacking due to TCA disturbance.104 Tannahill et al and Imperiale et al also showed an increased import and metabolism of glutamine in SDHx‐related tumors.105, 106
4.4.3. Oxidative phosphorylation
Disruption of complex II leads to changes in the TCA cycle, but also to changes in oxidative phosphorylation in the form of upregulation of complex I. Pang et al showed that in tumor tissue and in an SDHB‐ knockdown mouse cell line, complex I components and activity are upregulated.107 Consequently, the quantity of NAD+ in tumor tissue was 2.7‐fold higher in cluster I than in cluster II tumors. NAD+ is a cofactor that supports the poly (ADP‐ribose) polymerase (PARP) DNA repair way. PARP is an enzyme which produces ADP‐ribose‐conjugated PARP, involved in repair and stabilization of DNA. As an enhanced NAD+/PARP pathway was linked to chemoresistance in SDHB mutation carriers,107 inhibition of PARP could be a potential target to support chemotherapy, as further explained below.
5. APPLICATIONS FOR DIAGNOSTICS OF PGL/PCC
5.1. SDH and immunohistochemistry
In the vast majority of SDHx‐associated tumors, loss of SDHB protein expression can be detected by immunohistochemical staining with a high sensitivity and specificity (100% and 84%, respectively).33, 108 SDHB immunohistochemistry can therefore discriminate between SDHx‐related and non‐SDHx‐related PGL/PCC. Loss of both SDHB and SDHA immunoreactivity is shown only in the context of a SDHA mutation.3, 23, 33, 61, 108 SDHB and/or SDHA immunohistochemical expression could precedegenetic testing,33 or be used to classify variants of unknown significance.
The presence of an SDHB mutation is a predictor of metastasis in PGL/PCC. The current definition of a metastatic PGL or PCC according to the World Health Organization includes the presence of metastasis in non‐chromaffin tissue.2 In spite of attempts to develop an effective system for predicting the metastatic potential of PGL/PCC, none has yet resulted in a reliable classification. Recently, a grading system for PCC and PGL (GAPP) was developed.109 This score combines pathological features with biochemical phenotypes but does not include the SDHB mutational status of the tumor. Therefore, a combination of the GAPP and SDHB immunohistochemistry (modified‐GAPP or M‐GAPP) has been suggested as a valuable tool for predicting metastatic disease.109 Koh et al validated the M‐GAPP score in a retrospective cohort of 72 PGL/PCC patients with a mean follow‐up of 44 months. The M‐GAPP score was significantly higher in the 12 patients who developed metastatic disease.110
5.2. Metabolomics: measuring succinate levels in plasma, urine and tumor tissue
Succinate can be measured in plasma, urine and tumor tissue. Hobert et al measured succinate concentrations using gas chromatography‐mass spectrometry in plasma and urine of patients with germline SDHB, SDHD, PTEN mutations and patients with sporadic PGL/PCC. In three out of six SDHx mutation carriers (without PGL) elevated plasma succinate was recorded, while it was not elevated in any of patients with sporadic PGL/PCC.111
Tumor tissue can be used to measure the succinate:fumarate ratio using liquid chromatography‐mass spectrometry (LC‐MS).112, 113 An elevated succinate:fumarate ratio provides a diagnostic sensitivity of 93% and sensitivity of 97% to identify SDHx mutated PGL/PCC.87 Richter et al used 50 frozen specimens from 49 patients as a training set and 184 tumor samples as a validation set. In their study, succinate:fumarate ratios were higher in SDHB‐related PGL/PCC compared to SDHC/D tumors,87 thereby supporting Letouzé's suggestions84 that in a more complete inactivation of the SDH protein is present in SDHB‐mutation carriers. Measuring the succinate:fumarate ratio in tumor tissue can help to identify the underlying germline or somatic pathogenic mutation, especially when genetic mutation is inconclusive. Whether it may also have a prognostic value to predict metastatic disease still needs to be determined.
The studies of Lendvai et al and Imperiale et al confirm the findings that succinate:fumarate ratios are higher in SDHB‐ and SDHD‐related PGL/PCC than in apparently sporadic and non‐SDHx‐mutated PGL/PCC (n = 8).72, 106 Imperiale et al also found significantly lower levels of glutamate in SDHx‐related tumors.106 In an additional study, Richter et al, used LC‐MS to screen 395 PGL/PCC tissues for TCA cycle metabolites to indicate TCA cycle aberrations. SDHx‐mutated tumors were characterized by high succinate levels and low levels of all other TCA cycle metabolites including glutamate and aspartate.112
High resolution magic angle spinning (HR‐MAS) nuclear magnetic resonance (NMR) spectroscopy is a new technique that can be used to analyze catecholamine and succinate levels both in vivo and ex vivo. The HR‐MAS NMR technique was used by the group of Taïeb to investigate the metabolic profile of SDHx‐mutated tumor tissue and to compare this profile to the metabolic profile of apparently sporadic and VHL tumor tissue.114 SDHx‐related tumors had increased levels of succinate and significantly decreased levels of glutamate compared to apparently sporadic tumors and VHL‐related tumors.106 The same group also explored the possibility of quantification of oncometabolites in tissue when the tumor is still inside the patient, and shown in eight patients that 1H‐MRS (1high magnetic resonance spectroscopy) adequately detected succinate resonance peaks in four patients with an SDHx‐related tumor.115 In addition, Lussey‐Lepoutre et al used 1H‐MRS to detect succinate levels in both mice and patients with PGL in vivo. Five patients had a SDHx gene mutation and in these patients a succinate peak could be detected.116 This offers unique opportunities for better characterizing these tumors at a metabolic level.
5.3. Altered cell metabolism pathways: The use of 18F‐fluorodeoxyglucose positron emission tomography
According to Endocrine Society PGL/PCC guidelines, 18F‐fluorodeoxyglucose (FDG) positron emission tomography (PET)/computed tomography (CT) is the preferred imaging modality in SDHB‐mutated PGL/PCC.117 Recent studies have shown that SDHx‐related PGL/PCC might be better visualized by [68Ga]‐DOTA(0)‐Tyr(3)‐octreotate ([67GA]‐DOTATATE) PET/CT than 18F‐FDG PET/CT, especially those located in the head and neck region as well as metastatic PGL/PCC.118, 119, 120 The sensitivity of FDG‐PET for SDHx related tumors varies between 83% and 100%.118, 121, 122, 123
Like glucose, 18F‐FDG is taken up by tumor cells mostly via GLUT. After cell entry, 18F‐FDG is phosphorylated by hexokinase into 18F‐FDG‐6‐P, which, unlike glucose‐6‐P, cannot be further metabolized along the glycolytic pathway. Because the cell membrane is impermeable to 18F‐FDG‐6‐P, it accumulates within cells in a manner directly proportionate to their metabolic activity. An increased glucose uptake and consumption due to an increase in glycolysis leads to a high uptake of 18F‐FDG.124 18F‐FDG uptake in any cell is determined by expression of GLUTs and activity of hexokinase. Van Berkel et al studied the expression of GLUT and hexokinase in 27 tumor tissues from patients with hereditary tumors, using immunohistochemical staining and analyzed preoperative 18F‐FDG PET scans. The expression of hexokinase‐2 and hexokinase‐3 was significantly higher in SDHx‐mutated PGL/PCC than in sporadic tumors, and the mean standardized uptake values of the 18F‐FDG PET scans correlated with the expression of hexokinase‐2 and ‐3.124
Increased levels of succinate may also play a role in the high uptake of 18F‐FDG by SHDx‐related tumors. Garrigue et al showed that intratumoral injection of succinate significantly increased 18F‐FDG uptake in vivo and in vitro.125 Moreover, laser‐doppler did not show succinate induced 18F‐FDG uptake to be because of increased blood flow or increased capillary permeability.125
6. IMPLICATIONS FOR TREATMENT OF METASTATIC PGL/PCC
The cornerstone treatment for patients with benign SDHx related PGL/PCC is surgery.117 As described above, SDHB‐mutation carriers are those especially at risk of metastatic disease. Even for patients with metastatic PGL/PCC, resection of the primary tumor seems to be associated with a better overall survival.126 Metastases frequently occur in lymph nodes (distant and regional), bones, liver and lungs. Until now, there is no curative therapy for patients with metastatic disease. The main focus of treatment is on controlling the secretion of catecholamines, thereby alleviating symptoms and controlling tumor‐related complaints. Systemic treatment options include radionuclide therapy using 131I Metaiodobenzylguanidine (MIBG), peptide receptor radionuclide therapy (PRRT) and chemotherapy. As described above, insight in the pathways leading to tumor formation and potential metastatic disease in patients with SDHx mutations, may lead to a better response to existing therapies and provide us with a unique opportunity to develop novel targeted therapies.
6.1. Targeting the pseudohypoxia pathway
6.1.1. Restoration of PHD activity
Succinate competes with α‐ketoglutarate in binding to PHD, thereby inhibiting PHD activity; therefore excess of α‐ketoglutarate could restore PHD.11, 127, 128, 129 Increasing levels of intracellular α‐ketoglutarate have been shown to affect the levels of HIF1α in vitro.127 As succinate and hypoxia act synergistically in inhibiting PHD activity, not only administering α‐ketoglutarate but also inducing hyperoxia might restore PHD activity.130, 131 Increasing the α‐ketoglutarate levels in the cell, is challenging. In a recent mouse model study of breast cancer, the α‐ketoglutarate dehydrogenase (KGDH) inhibitor (AA6) was able to cause intracellular α‐ketoglutarate accumulation.132
6.1.2. HIF2α inhibition
In the HIF2α structure is a specific cavity which can be targeted.133, 134 Two compounds, PT2385 and PT2399, have been developed to serve as an HIF2α inhibitor. Both compounds, studied in vitro and in vivo, efficiently reduced the growth of clear cell RCCs.135, 136 A recent publication describes a phase I trial with PT2385 in patients with progressive clear cell RCCs. All 25 patients included in the expansion phase had locally advanced disease or disease that had progressed during a median of four prior regimens. Respectively, 2%, 12%, and 52% of patients had complete response, partial response and stable disease, results which are very promising.137 Although at present no intervention studies are being undertaken in patients with metastatic SDHx‐ related PGL/PCC, probably in the near future a phase II trial will start to evaluate HIF2α inhibitors for patients with metastatic PGL/PCC.133, 138
6.1.3. Tyrosine kinase inhibitors
Treatment with Tyrosine Kinase Inhibitors (TKI) targets the downstream pathway of HIF. Several TKI's that been described in case reports, series or phase II trials, such as sunitinib, cabozantinib, lenvatinib, pazopanib, and axitinib. An excellent overview of existing data and forthcoming trials was recently published by Toledo and Jiminez.139 All TKIs inhibit angiogenesis, by inhibiting the activation of the VEGF receptor (VEGFR).
Until now, most available data are for sunitinib. Besides inhibiting the activation of the VEGFR, sunitinib also inhibits the activation of the PDGF receptor and the receptor of RET, c‐KIT and Fms‐like tyrosine kinase (FLT). Canu et al reviewed the efficacy of sunitinib in 35 patients, of whom 13 were carriers of an SDHB germline mutation. Outcome did not differ between patients with or without an SDHB mutation.140 In a retrospective analysis of 17 patients with progressive disease, who received sunitinib, 47% had a partial response or stable disease. Positive responses were noted in carriers of SDHB mutations as well as in patients with apparently sporadic tumors. Progression‐free survival was only 4.1 months.141 Currently two phase II studies are being conducted; the Study Of Sunitinib In Patients With Recurrent Paraganglioma/Pheochromocytoma SNIPP (closed) and the First International Randomized Study in Malignant Progressive Pheochromocytoma and Paraganglioma (FIRSTMAPP).
Pazopanib, similar to sunitinib, also inhibits the action of VEGFR, PDGFR and the RET receptor, but additionally inhibits the fibroblast growth factor receptor (FGFR). Pazopanib was studied in a phase II trial, terminated due to poor accrual after including only seven patients.142 Six patients were evaluated, as one withdrew informed consent. Of the six only one patient had a partial response, lasting 2.4 years.
Preliminary results of axitinib were presented at the ASCO meeting in 2015. Axitinib only blocks VEGFR, and led to a partial response in three out of nine patients with metastatic PGL/PCC; moreover toxicity led to a high rate of dose reduction.143
Cabozantinib seems a promising TKI for patients with metastatic PGL/PCC, especially for patients with bone metastases. Cabozantinib also inhibits the c‐Met receptor pathways and may therefore delay the development of resistance, as this pathway is upregulated by VEGFR inhibition. Currently there is a phase II trial ongoing, with promising preliminary results.144
Another phase II trial is aiming to evaluate the response rate of lenvatinib in a group of 25 patients with metastatic PGL/PCC. Lenvatinib, like pazopanib, also inhibits FGFR.
6.1.4. Heat shock protein 90 inhibitors
Inhibition of HSP90 leads to downregulation of HIF1α and is a potential target for therapy in metastatic PGL/PCC. Giubellino et al showed potent inhibition of proliferation in PCC cell lines by tanespimycin (17‐AAG) and ganetespib. Furthermore, they showed the efficacy of 17‐AAG and ganetespib in reducing metastatic burden and increasing survival in a metastatic model of PCC.83 Chae et al suggested that HSP90 could be especially effective in SDHB‐mutated tumors. Genetic inactivation of SDHB leads to a recruitment of HSP90 to the mitochondria, to help compensate for the impaired oxidative phosphorylation. As HSP90 promotes the stability of HIFα, its inhibition can lead to the death of these cells.145
6.2. Targeting the hypermethylator phenotype of SDHx related PGL/PCC
Chemotherapy is, in contrast to therapies mentioned above, widely available for the treatment of metastatic PGL/PCC. The combination of cyclophosphamide, vincristine and dacarbazine (CVD) is the most studied and is currently first line chemotherapy in patients with a metastatic PGL/PCC. However, in the absence of prospective studies, the evidence is only based on small retrospective studies.141, 146, 147, 148, 149, 150, 151 In 2014, a meta‐analysis was performed suggesting a partial response of 37%.152
Some reports however, suggest a higher response rate to temozolomide, an oral alternative to dacarbazine, in patients with SDHB mutations. Temozolomide is a DNA alkylating agent, leading to methylation of the O6‐position of guanine, resulting in DNA adduction. These DNA adducts result in apoptosis of the malignant cell. The O(6)‐methylguanine‐DNA‐methyltransferase (MGMT) enzyme is capable of repairing the DNA adducts. Therefore, the efficacy of treatment with temozolomide is associated with the expression of MGMT in the tumor cells. In a study by Hadoux et al, 11 out of 14 patients with progressive metastatic disease, had a SDHB mutation.153 Thirty‐six percent had partial response, 55% stable disease and 9% progressive disease. The authors observed a longer progression‐free survival in patients with an SDHB mutation compared to patients without an SDHB mutation (19.7 vs 2.9 months). The higher response rate in patients with SDHB mutations could be caused by hypermethylation of the MGMT promotor region and consequently lower MGMT expression.
Recently two patients with a SDHB metastatic PGL/PCC showed a clinical benefit from temozolomide even after disease progression on CVD. Both patients showed hypermethylation of the MGMT promotor region, suggesting that monotherapy of temozolomide may benefit patients with metastatic SDHB‐related PGL/PCC.154 Very recently Jawed et al studied 12 patients with a metastatic PGL/PCC, all with SDHB mutation, who received CVD; a marked efficacy was noted.155 Two out of 12 patients had a complete remission and eight patients a partial response. The median duration of response was 478 days, with a median progression‐free survival of 930 days.
Decitabine, registered for the treatment of acute myeloid leukemia, is a cytidine deoxynucleoside‐analog. It inhibits DNA‐methyltransferase and therefore acts as a hypomethylating agent. In two cell models decitabine was able to induce cell death of SDH −/− cells.84, 156
6.3. Preventing ROS damage
Ascorbic acid, α‐tocopherol (vitamin E) and N‐acetylcysteine all act as antioxidants preventing ROS damage, thereby diminishing tumorigenesis primarily through decreasing DNA damage and mutations. There is, however, limited evidence for this efficacy in SDHx mutated PGL/PCC.157, 158
6.4. Targeting the altered cell's metabolism
6.4.1. Inhibiting glycolysis
As discussed above, SDHx‐related PGL/PCC are “glucose addicts.” Interventions aiming to inhibit glycolysis could therefore be an interesting and several potential options exists. WZB117 and STF‐31 are inhibitors of GLUT1, downregulating glycolysis and inhibiting cancer cell growth in vitro and vivo.159 Dichloroacetate (DCA) downregulates pyruvate dehydrogenase kinase. (Normally this upregulates pyruvate dehydrogenase involved in the glycolysis). This shifts glycolysis to oxidative phosphorylation and induces apoptosis in cancer cells. Pyruvate carboxylase was identified as an essential gene for SDH‐deficient cells (although dispensable for normal cells), a metabolic vulnerability offering a potential target for treatment of SDHx‐associated tumors.103
6.4.2. Inhibiting the effects of upregulation of complex I
As noted above another way to become resistant to chemotherapy is via the NAD+/PARP‐pathway. In the study of Pang et al the combination of temozolomide with a PARP inhibitor led to increased mouse survival in a metastatic PGL/PCC allograft model (52 days compared with 42 days).107 Notably, one of the postulated pathways through which metformin exerts an anti‐tumor effect is also through inhibition of complex I, implying that metformin could also act as a potential chemosensitizer in patients with SDHx‐related metastatic PGL/PCC.
7. CONCLUSION
Recent years, we have seen an increase in knowledge regarding the consequence of loss of the SDH enzyme in the pathogenesis of (metastatic) PGL/PCC in patients harboring an SDHx mutation. The accumulation of succinate and the impairment of the complex II function of oxidative phosphorylation leads, via the pseudohypoxic pathway, induction of ROS, and rewiring of the cell's metabolism to tumor formation. The advantages of new insight into these pathophysiological characteristics provide new directions for diagnostics and therapeutic options in metastatic SDHx‐related PGL and PCC.
CONFLICT OF INTEREST
All authors hereby declare that they are no conflicts of interest. Data sharing is not applicable to this article as no new data were created or analyzed in this study.
ACKNOWLEDGEMENTS
We gratefully acknowledge the contribution of M. Robledo from the Spanish National Cancer Research Center (CNIO), Madrid, Spain, for reading the manuscript and giving valuable comments.
Eijkelenkamp K, Osinga TE, Links TP, van der Horst‐Schrivers ANA. Clinical implications of the oncometabolite succinate in SDHx‐mutation carriers. Clin Genet. 2020;97:39–53. 10.1111/cge.13553
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/cge.13553/
REFERENCES
- 1. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lam AK. Update on adrenal tumours in 2017 world health organization (WHO) of endocrine tumours. Endocr Pathol. 2017;28(3):213‐227. [DOI] [PubMed] [Google Scholar]
- 3. Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108‐119. [DOI] [PubMed] [Google Scholar]
- 4. Buffet A, Morin A, Castro‐Vega LJ, et al. Germline mutations in the mitochondrial 2‐oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 2018;78(8):1914‐1922. [DOI] [PubMed] [Google Scholar]
- 5. Buffet A, Burnichon N, Amar L, Gimenez‐Roqueplo AP. Pheochromocytoma: when to search a germline defect? Presse Med. 2018;47(7–8 Pt 2):e109‐e118. [DOI] [PubMed] [Google Scholar]
- 6. Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309‐314. [DOI] [PubMed] [Google Scholar]
- 8. Kluckova K, Tennant DA. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018;372(2):367‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vicha A, Taieb D, Pacak K. Current views on cell metabolism in SDHx‐related pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21(3):R261‐R277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morin A, Letouze E, Gimenez‐Roqueplo AP, Favier J. Oncometabolites‐driven tumorigenesis: from genetics to targeted therapy. Int J Cancer. 2014;135(10):2237‐2248. [DOI] [PubMed] [Google Scholar]
- 11. Her YF, Maher LJ 3rd. Succinate dehydrogenase loss in familial paraganglioma: biochemistry, genetics, and epigenetics. Int J Endocrinol. 2015;2015:296167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69(1):49‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26(3):268‐270. [DOI] [PubMed] [Google Scholar]
- 14. Baysal BE, Ferrell RE, Willett‐Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848‐851. [DOI] [PubMed] [Google Scholar]
- 15. Tufton N, Sahdev A, Drake WM, Akker SA. Can subunit specific phenotypes guide surveillance imaging decisions in asymptomatic SDH mutation carriers? Clin Endocrinol (Oxf). 2019;90:31‐46. [DOI] [PubMed] [Google Scholar]
- 16. Buffet A, Ben Aim L, Leboulleux S, et al. Positive impact of genetic test on the management and outcome of patients with paraganglioma and/or pheochromocytoma. J Clin Endocrinol Metab. 2019;104(4):1109‐1118. [DOI] [PubMed] [Google Scholar]
- 17. Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157‐162. [DOI] [PubMed] [Google Scholar]
- 18. Castro‐Vega LJ, Letouze E, Burnichon N, et al. Multi‐omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanharanta S, Buchta M, McWhinney SR, et al. Early‐onset renal cell carcinoma as a novel extraparaganglial component of SDHB‐associated heritable paraganglioma. Am J Hum Genet. 2004;74(1):153‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100(17):1260‐1262. [DOI] [PubMed] [Google Scholar]
- 21. Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype‐phenotype‐proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31(1):41‐51. [DOI] [PubMed] [Google Scholar]
- 22. Xekouki P, Szarek E, Bullova P, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J Clin Endocrinol Metab. 2015;100(5):E710‐E719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papathomas TG, Gaal J, Corssmit EP, et al. Non‐pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase‐related PCC‐PGL syndromes: a clinicopathological and molecular analysis. Eur J Endocrinol. 2013;170(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 24. Denes J, Swords F, Rattenberry E, et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab. 2015;100(3):E531‐E541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baysal BE. A recurrent stop‐codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T‐cell acute leukemia. PLoS One. 2007;2(5):e436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourgeron T, Rustin P, Chretien D, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11(2):144‐149. [DOI] [PubMed] [Google Scholar]
- 27. Burnichon N, Brière JJ, Libé R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19(15):3011‐3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. NGS in PPGL (NGSnPPGL) Study Group , Toledo RA, Burnichon N, et al. Consensus statement on next‐generation‐sequencing‐based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2017;13(4):233‐247. [DOI] [PubMed] [Google Scholar]
- 29. Favier J, Amar L, Gimenez‐Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101‐111. [DOI] [PubMed] [Google Scholar]
- 30. Maniam P, Zhou K, Lonergan M, Berg JN, Goudie DR, Newey PJ. Pathogenicity and penetrance of germline SDHA variants in pheochromocytoma and paraganglioma (PPGL). J Endocr Soc. 2018;2(7):806‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bausch B, Schiavi F, Ni Y, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene‐informed prevention. JAMA Oncol. 2017;3:1204‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Tuin K, Mensenkamp AR, Tops CMJ, et al. Clinical aspects of SDHA‐related pheochromocytoma and paraganglioma: a nationwide study. J Clin Endocrinol Metab. 2018;103(2):438‐445. [DOI] [PubMed] [Google Scholar]
- 33. Papathomas TG, Oudijk L, Persu A, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a multinational study of the european network for the study of adrenal tumors (ENS@T). Mod Pathol. 2015;28(6):807‐821. [DOI] [PubMed] [Google Scholar]
- 34. Tufton N, Ghelani R, Srirangalingam U, et al. SDHA mutated paragangliomas may be at high risk of metastasis. Endocr Relat Cancer. 2017;24(7):L43‐L49. [DOI] [PubMed] [Google Scholar]
- 35. Casey RT, Challis BG, Marker A, et al. A case of a metastatic SDHA mutated paraganglioma re‐presenting twenty‐three years after initial surgery. Endocr Relat Cancer. 2017;24(8):L69‐L71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96(9):E1472‐E1476. [DOI] [PubMed] [Google Scholar]
- 37. Casey RT, Ascher DB, Rattenberry E, et al. SDHA related tumorigenesis: a new case series and literature review for variant interpretation and pathogenicity. Mol Genet Genomic Med. 2017;5(3):237‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dubard Gault M, Mandelker D, DeLair D, et al. GermlineSDHAmutations in children and adults with cancer. Cold Spring Harb Mol Case Stud. 2018;4(4):a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild‐type gastrointestinal stromal tumors: a report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016;2(7):922‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andrews KA, Ascher DB, Pires DEV, et al. Tumour risks and genotype‐phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet. 2018;55(6):384‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jochmanova I, Wolf KI, King KS, et al. SDHB‐related pheochromocytoma and paraganglioma penetrance and genotype‐phenotype correlations. J Cancer Res Clin Oncol. 2017;143(8):1421‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812‐8818. [DOI] [PubMed] [Google Scholar]
- 43. Benn DE, Gimenez‐Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827‐836. [DOI] [PubMed] [Google Scholar]
- 44. Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943‐951. [DOI] [PubMed] [Google Scholar]
- 45. Srirangalingam U, Walker L, Khoo B, et al. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH‐B) gene mutation carriers. Clin Endocrinol (Oxf). 2008;69(4):587‐596. [DOI] [PubMed] [Google Scholar]
- 46. van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EPM. Risk of malignant paraganglioma in SDHB‐mutation and SDHD‐mutation carriers: a systematic review and meta‐analysis. J Med Genet. 2012;49(12):768‐776. [DOI] [PubMed] [Google Scholar]
- 47. Hamidi O, Young WF Jr, Gruber L, et al. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: a systematic review and meta‐analysis. Clin Endocrinol (Oxf). 2017;87(5):440‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822‐3828. [DOI] [PubMed] [Google Scholar]
- 49. Assadipour Y, Sadowski SM, Alimchandani M, et al. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery. 2017;161(1):230‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hescot S, Curras‐Freixes M, Deutschbein T, et al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP‐prono study): an ENS@T retrospective study. J Clin Endocrinol Metab. 2019;2:213‐627 [DOI] [PubMed] [Google Scholar]
- 51. Benn DE, Robinson BG, Clifton‐Bligh RJ. 15 YEARS OF PARAGANGLIOMA: clinical manifestations of paraganglioma syndromes types 1‐5. Endocr Relat Cancer. 2015;22(4):T91‐T103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tufton N, Roncaroli F, Hadjidemetriou I, et al. Pituitary carcinoma in a patient with an SDHB mutation. Endocr Pathol. 2017;28(4):320‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schiavi F, Boedeker CC, Bausch B, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294(16):2057‐2063. [DOI] [PubMed] [Google Scholar]
- 54. Else T, Marvin ML, Everett JN, et al. The clinical phenotype of SDHC‐associated hereditary paraganglioma syndrome (PGL3). J Clin Endocrinol Metab. 2014;99(8):E1482‐E1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bickmann JK, Sollfrank S, Schad A, et al. Phenotypic variability and risk of malignancy in SDHC‐linked paragangliomas: lessons from three unrelated cases with an identical germline mutation (p.Arg133*). J Clin Endocrinol Metab. 2014;99(3):E489‐E496. [DOI] [PubMed] [Google Scholar]
- 56. Ong RKS, Flores SK, Reddick RL, Dahia PLM, Shawa H. A unique case of metastatic, functional, hereditary paraganglioma associated with an SDHC germline mutation. J Clin Endocrinol Metab. 2018;103(8):2802‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bourdeau I, Grunenwald S, Burnichon N, et al. A SDHC founder mutation causes paragangliomas (PGLs) in the french canadians: new insights on the SDHC‐related PGL. J Clin Endocrinol Metab. 2016;101(12):4710‐4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108(1):314‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gill AJ, Lipton L, Taylor J, et al. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology. 2013;45(7):689‐691. [DOI] [PubMed] [Google Scholar]
- 60. Timmers HJLM, Pacak K, Bertherat J, et al. Mutations associated with succinate dehydrogenase D‐related malignant paragangliomas. Clin Endocrinol (Oxf). 2008;68(4):561‐566. [DOI] [PubMed] [Google Scholar]
- 61. Gill AJ, Toon CW, Clarkson A, et al. Succinate dehydrogenase deficiency is rare in pituitary adenomas. Am J Surg Pathol. 2014;38(4):560‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Casey RT, Warren AY, Martin JE, et al. Clinical and molecular features of renal and pheochromocytoma/paraganglioma tumor association syndrome (RAPTAS): case series and literature review. J Clin Endocrinol Metab. 2017;102(11):4013‐4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325(5944):1139‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bayley JP, Kunst HPM, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11(4):366‐372. [DOI] [PubMed] [Google Scholar]
- 65. Kunst HPM, Rutten MH, de Monnink JP, et al. SDHAF2 (PGL2‐SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17(2):247‐254. [DOI] [PubMed] [Google Scholar]
- 66. Piccini V, Rapizzi E, Bacca A, et al. Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer. 2012;19(2):149‐155. [DOI] [PubMed] [Google Scholar]
- 67. Zhu WD, Wang ZY, Chai YC, Wang XW, Chen DY, Wu H. Germline mutations and genotype‐phenotype associations in head and neck paraganglioma patients with negative family history in China. Eur J Med Genet. 2015;58(9):433‐438. [DOI] [PubMed] [Google Scholar]
- 68. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77‐85. [DOI] [PubMed] [Google Scholar]
- 69. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25(34):4675‐4682. [DOI] [PubMed] [Google Scholar]
- 70. Lehtonen HJ, Makinen MJ, Kiuru M, et al. Increased HIF1 alpha in SDH and FH deficient tumors does not cause microsatellite instability. Int J Cancer. 2007;121(6):1386‐1389. [DOI] [PubMed] [Google Scholar]
- 71. Pollard PJ, Briere JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over‐expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14(15):2231‐2239. [DOI] [PubMed] [Google Scholar]
- 72. Lendvai N, Pawlosky R, Bullova P, et al. Succinate‐to‐fumarate ratio as a new metabolic marker to detect the presence of SDHB/D‐related paraganglioma: initial experimental and ex vivo findings. Endocrinology. 2014;155(1):27‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ploumakis A, Coleman ML. OH, the places you'll go! Hydroxylation, gene expression, and cancer. Mol Cell. 2015;58(5):729‐741. [DOI] [PubMed] [Google Scholar]
- 74. Briere JJ, Favier J, El Ghouzzi V, et al. Succinate dehydrogenase deficiency in human. Cell Mol Life Sci. 2005;62(19–20):2317‐2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gottlieb E, Tomlinson IPM. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5(11):857‐866. [DOI] [PubMed] [Google Scholar]
- 76. Briere JJ, Favier J, Benit P, et al. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA‐mutant fibroblasts under normoxic conditions. Hum Mol Genet. 2005;14(21):3263‐3269. [DOI] [PubMed] [Google Scholar]
- 77. Pollard PJ, El‐Bahrawy M, Poulsom R, et al. Expression of HIF‐1alpha, HIF‐2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91(11):4593‐4598. [DOI] [PubMed] [Google Scholar]
- 78. Gimenez‐Roqueplo AP, Favier J, Rustin P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002;87(10):4771‐4774. [DOI] [PubMed] [Google Scholar]
- 79. Gimenez‐Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69(6):1186‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lopez‐Jimenez E, Gomez‐Lopez G, Leandro‐Garcia LJ, et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL‐related pheochromocytomas. Mol Endocrinol. 2010;24(12):2382‐2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang SL, Wu C, Xiong ZF, Fang X. Progress on hypoxia‐inducible factor‐3: its structure, gene regulation and biological function (review). Mol Med Rep. 2015;12(2):2411‐2416. [DOI] [PubMed] [Google Scholar]
- 82. Xu Y, Zhang C, Chen D, et al. Effect of HSP90 inhibitor in pheochromocytoma PC12 cells: an experimental investigation. Tumour Biol. 2013;34(6):4065‐4071. [DOI] [PubMed] [Google Scholar]
- 83. Giubellino A, Sourbier C, Lee MJ, et al. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PLoS One. 2013;8(2):e56083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739‐752. [DOI] [PubMed] [Google Scholar]
- 85. Xiao M, Yang H, Xu W, et al. Inhibition of alpha‐KG‐dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Richter S, Peitzsch M, Rapizzi E, et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab. 2014;99(10):3903‐3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kuriyama S, Mayor R. Molecular analysis of neural crest migration. Philos Trans R Soc Lond B Biol Sci. 2008;363(1495):1349‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Loriot C, Burnichon N, Gadessaud N, et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J Clin Endocrinol Metab. 2012;97(6):E954‐E962. [DOI] [PubMed] [Google Scholar]
- 90. Kiss NB, Muth A, Andreasson A, et al. Acquired hypermethylation of the P16INK4A promoter in abdominal paraganglioma: relation to adverse tumor phenotype and predisposing mutation. Endocr Relat Cancer. 2013;20(1):65‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kluckova K, Sticha M, Cerny J, et al. Ubiquinone‐binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015;6:e1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287(32):27255‐27264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165‐181. [DOI] [PubMed] [Google Scholar]
- 94. Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia‐induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401‐408. [DOI] [PubMed] [Google Scholar]
- 95. Owens KM, Aykin‐Burns N, Dayal D, Coleman MC, Domann FE, Spitz DR. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2(−*) and H2O2. Free Radic Biol Med. 2012;52(1):160‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Slane BG, Aykin‐Burns N, Smith BJ, et al. Mutation of succinate dehydrogenase subunit C results in increased O2.‐, oxidative stress, and genomic instability. Cancer Res. 2006;66(15):7615‐7620. [DOI] [PubMed] [Google Scholar]
- 97. Ochoa‐Ruiz E, Diaz‐Ruiz R. Anaplerosis in cancer: another step beyond the warburg effect. AJMB. 2012;2(4):291‐303. [Google Scholar]
- 98. Semenza GL. Hypoxia‐inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia‐inducible factor 1 in cancer cells. Oncotarget. 2011;2(7):551‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fliedner SMJ, Kaludercic N, Jiang XS, et al. Warburg effect's manifestation in aggressive pheochromocytomas and paragangliomas: insights from a mouse cell model applied to human tumor tissue. PLoS One. 2012;7(7):e40949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Favier J, Briere JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4(9):e7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF‐kappaB/IL‐8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550‐2560. [DOI] [PubMed] [Google Scholar]
- 103. Cardaci S, Zheng L, MacKay G, et al. Pyruvate carboxylation enables growth of SDH‐deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. 2015;17(10):1317‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lussey‐Lepoutre C, Hollinshead KER, Ludwig C, et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun. 2015;6:8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature. 2013;496(7444):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Imperiale A, Moussallieh FM, Sebag F, et al. A new specific succinate‐glutamate metabolomic hallmark in SDHx‐related paragangliomas. PLoS One. 2013;8(11):e80539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pang Y, Lu Y, Caisova V, et al. Targeting NAD(+)/PARP DNA repair pathway as a novel therapeutic approach to SDHB‐mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res. 2018;24(14):3423‐3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10(8):764‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21(3):405‐414. [DOI] [PubMed] [Google Scholar]
- 110. Koh JM, Ahn SH, Kim H, et al. Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma. PLoS One. 2017;12(11):e0187398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hobert JA, Mester JL, Moline J, Eng C. Elevated plasma succinate in PTEN, SDHB, and SDHD mutation‐positive individuals. Genet Med. 2012;14(6):616‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Richter S, Gieldon L, Pang Y, et al. Metabolome‐guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med. 2019;21:705‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim E, Wright MJ, Sioson L, et al. Utility of the succinate: fumarate ratio for assessing SDH dysfunction in different tumor types. Mol Genet Metab Rep. 2016;10:45‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Imperiale A, Moussallieh FM, Roche P, et al. Metabolome profiling by HRMAS NMR spectroscopy of pheochromocytomas and paragangliomas detects SDH deficiency: clinical and pathophysiological implications. Neoplasia. 2015;17(1):55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Varoquaux A, le Fur Y, Imperiale A, et al. Magnetic resonance spectroscopy of paragangliomas: new insights into in vivo metabolomics. Endocr Relat Cancer. 2015;22(4):M1‐M8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lussey‐Lepoutre C, Bellucci A, Morin A, et al. In vivo detection of succinate by magnetic resonance spectroscopy as a hallmark of SDHx mutations in paraganglioma. Clin Cancer Res. 2016;22(5):1120‐1129. [DOI] [PubMed] [Google Scholar]
- 117. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915‐1942. [DOI] [PubMed] [Google Scholar]
- 118. Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]‐DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB‐associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015;21(17):3888‐3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Archier A, Varoquaux A, Garrigue P, et al. Prospective comparison of (68)ga‐DOTATATE and (18)F‐FDOPA PET/CT in patients with various pheochromocytomas and paragangliomas with emphasis on sporadic cases. Eur J Nucl Med Mol Imaging. 2016;43(7):1248‐1257. [DOI] [PubMed] [Google Scholar]
- 120. Kroiss A, Putzer D, Frech A, et al. A retrospective comparison between 68Ga‐DOTA‐TOC PET/CT and 18F‐DOPA PET/CT in patients with extra‐adrenal paraganglioma. Eur J Nucl Med Mol Imaging. 2013;40(12):1800‐1808. [DOI] [PubMed] [Google Scholar]
- 121. Kornaczewski ER, Pointon OP, Burgess JR. Utility of FDG‐PET imaging in screening for succinate dehydrogenase B and D mutation‐related lesions. Clin Endocrinol (Oxf). 2016;85(2):172‐179. [DOI] [PubMed] [Google Scholar]
- 122. Timmers HJLM, Chen CC, Carrasquillo JA, et al. Comparison of 18F‐fluoro‐L‐DOPA, 18F‐fluoro‐deoxyglucose, and 18F‐fluorodopamine PET and 123I‐MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94(12):4757‐4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Timmers HJLM, Kozupa A, Chen CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB‐associated pheochromocytoma and paraganglioma. J Clin Oncol. 2007;25(16):2262‐2269. [DOI] [PubMed] [Google Scholar]
- 124. van Berkel A, Rao JU, Kusters B, et al. Correlation between in vivo 18F‐FDG PET and immunohistochemical markers of glucose uptake and metabolism in pheochromocytoma and paraganglioma. J Nucl Med. 2014;55(8):1253‐1259. [DOI] [PubMed] [Google Scholar]
- 125. Garrigue P, Bodin‐Hullin A, Balasse L, et al. The evolving role of succinate in tumor metabolism: an (18)F‐FDG‐based study. J Nucl Med. 2017;58(11):1749‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Roman‐Gonzalez A, Zhou S, Ayala‐Ramirez M, et al. Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann Surg. 2018;268(1):172‐178. [DOI] [PubMed] [Google Scholar]
- 127. MacKenzie ED, Selak MA, Tennant DA, et al. Cell‐permeating alpha‐ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase‐deficient cells. Mol Cell Biol. 2007;27(9):3282‐3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Smith EH, Janknecht R, Maher LJ III. Succinate inhibition of alpha‐ketoglutarate‐dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16(24):3136‐3148. [DOI] [PubMed] [Google Scholar]
- 129. Jokilehto T, Jaakkola PM. The role of HIF prolyl hydroxylases in tumour growth. J Cell Mol Med. 2010;14(4):758‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Peters JP, Her YF, Maher LJ 3rd. Modeling dioxygenase enzyme kinetics in familial paraganglioma. Biol Open. 2015;4(10):1281‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Her YF, Nelson‐Holte M, Maher LJ 3rd. Oxygen concentration controls epigenetic effects in models of familial paraganglioma. PLoS One. 2015;10(5):e0127471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Atlante S, Visintin A, Marini E, et al. Alpha‐ketoglutarate dehydrogenase inhibition counteracts breast cancer‐associated lung metastasis. Cell Death Dis. 2018;9(7):756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Toledo RA. New HIF2α inhibitors: potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2017;24(9):C9‐C19. [DOI] [PubMed] [Google Scholar]
- 134. Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2alpha PAS‐B domain of the HIF2 transcription factor. Proc Natl Acad Sci USA. 2009;106(2):450‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF‐2 antagonist. Nature. 2016;539(7627):112‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Cho H, Kaelin WG. Targeting HIF2 in clear cell renal cell carcinoma. Cold Spring Harb Symp Quant Biol. 2016;81:113‐121. [DOI] [PubMed] [Google Scholar]
- 137. Courtney KD, Infante JR, Lam ET, et al. Phase I dose‐escalation trial of PT2385, a first‐in‐class hypoxia‐inducible factor‐2alpha antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. 2018;36(9):867‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tella SH, Taieb D, Pacak K. HIF‐2alpha: Achilles' heel of pseudohypoxic subtype paraganglioma and other related conditions. Eur J Cancer. 2017;86:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Toledo R, Jimenez C. Recent advances in the management of malignant pheochromocytoma and paraganglioma: focus on tyrosine kinase and hypoxia‐inducible factor inhibitors. F1000Res. 2018;7:F1000 10.12688/f1000research.13995.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Canu L, Pradella S, Rapizzi E, et al. Sunitinib in the therapy of malignant paragangliomas: report on the efficacy in a SDHB mutation carrier and review of the literature. Arch Endocrinol Metab. 2017;61(1):90‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ayala‐Ramirez M, Feng L, Habra MA, et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra‐adrenal paragangliomas: insights from the largest single‐institutional experience. Cancer. 2012;118(11):2804‐2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jasim S, Suman VJ, Jimenez C, et al. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine. 2017;57(2):220‐225. [DOI] [PubMed] [Google Scholar]
- 143. Burotto‐Pichun ME, Edgerly M, Velarde M, et al. Phase II clinical trial of axitinib in metastatic pheochromocytomas and paragangliomas (P/PG): preliminary results. Presented at ASCO Annual Meeting 2015 chicago, IL, USA. J Clin Oncol. 2015;33(suppl 7, abstr):457. [Google Scholar]
- 144. Jimenez C. Treatment for patients with malignant pheochromocytomas and paragangliomas: a perspective from the hallmarks of cancer. Front Endocrinol (Lausanne). 2018;9:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chae YC, Angelin A, Lisanti S, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun. 2013;4:2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer. 2013;4(2):103‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Patel SR, Winchester DJ, Benjamin RS. A 15‐year experience with chemotherapy of patients with paraganglioma. Cancer. 1995;76(8):1476‐1480. [DOI] [PubMed] [Google Scholar]
- 148. Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22‐year follow‐up of 18 patients. Cancer. 2008;113(8):2020‐2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Deutschbein T, Fassnacht M, Weismann D, Reincke M, Mann K, Petersenn S. Treatment of malignant phaeochromocytoma with a combination of cyclophosphamide, vincristine and dacarbazine: own experience and overview of the contemporary literature. Clin Endocrinol (Oxf). 2015;82(1):84‐90. [DOI] [PubMed] [Google Scholar]
- 150. Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267‐273. [DOI] [PubMed] [Google Scholar]
- 151. Asai S, Katabami T, Tsuiki M, Tanaka Y, Naruse M. Controlling tumor progression with cyclophosphamide, vincristine, and dacarbazine treatment improves survival in patients with metastatic and unresectable malignant pheochromocytomas/paragangliomas. Horm Cancer. 2017;8(2):108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EPM. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta‐analysis. Clin Endocrinol (Oxf). 2014;81(5):642‐651. [DOI] [PubMed] [Google Scholar]
- 153. Hadoux J, Favier J, Scoazec JY, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. 2014;135(11):2711‐2720. [DOI] [PubMed] [Google Scholar]
- 154. Tena I, Gupta G, Tajahuerce M, et al. Successful second‐line metronomic temozolomide in metastatic paraganglioma: case reports and review of the literature. Clin Med Insights Oncol. 2018;12:1179554918763367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Jawed I, Velarde M, Darr R, et al. Continued tumor reduction of metastatic pheochromocytoma/paraganglioma harboring succinate dehydrogenase subunit B mutations with cyclical chemotherapy. Cell Mol Neurobiol. 2018;38(5):1099‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Loriot C, Domingues M, Berger A, et al. Deciphering the molecular basis of invasiveness in sdhb‐deficient cells. Oncotarget. 2015;6(32):32955‐32965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ni Y, Eng C. Vitamin E protects against lipid peroxidation and rescues tumorigenic phenotypes in cowden/cowden‐like patient‐derived lymphoblast cells with germline SDHx variants. Clin Cancer Res. 2012;18(18):4954‐4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Gao P, Zhang H, Dinavahi R, et al. HIF‐dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]