

Abstract
Key points
Outer hair cells (OHCs) enhance the sensitivity and the frequency tuning of the mammalian cochlea.
Similar to the primary sensory receptor, the inner hair cells (IHCs), the mature functional characteristics of OHCs are acquired before hearing onset.
We found that OHCs, like IHCs, fire spontaneous Ca2+‐induced action potentials (APs) during immature stages of development, which are driven by CaV1.3 Ca2+ channels.
We also showed that the development of low‐ and high‐frequency hair cells is differentially regulated during pre‐hearing stages, with the former cells being more strongly dependent on experience‐independent Ca2+ action potential activity.
Abstract
Sound amplification within the mammalian cochlea depends upon specialized hair cells, the outer hair cells (OHCs), which possess both sensory and motile capabilities. In various altricial rodents, OHCs become functionally competent from around postnatal day 7 (P7), before the primary sensory inner hair cells (IHCs), which become competent at about the onset of hearing (P12). The mechanisms responsible for the maturation of OHCs and their synaptic specialization remain poorly understood. We report that spontaneous Ca2+ activity in the immature cochlea, which is generated by CaV1.3 Ca2+ channels, differentially regulates the maturation of hair cells along the cochlea. Under near‐physiological recording conditions we found that, similar to IHCs, immature OHCs elicited spontaneous Ca2+ action potentials (APs), but only during the first few postnatal days. Genetic ablation of these APs in vivo, using CaV1.3−/− mice, prevented the normal developmental acquisition of mature‐like basolateral membrane currents in low‐frequency (apical) hair cells, such as I K,n (carried by KCNQ4 channels), I SK2 and I ACh (α9α10nAChRs) in OHCs and I K,n and I K,f (BK channels) in IHCs. Electromotility and prestin expression in OHCs were normal in CaV1.3−/− mice. The maturation of high‐frequency (basal) hair cells was also affected in CaV1.3−/− mice, but to a much lesser extent than apical cells. However, a characteristic feature in CaV1.3−/− mice was the reduced hair cell size irrespective of their cochlear location. We conclude that the development of low‐ and high‐frequency hair cells is differentially regulated during development, with apical cells being more strongly dependent on experience‐independent Ca2+ APs.
Keywords: action potentials, auditory, calcium signals, cochlea, development, hair cells
Key points
Outer hair cells (OHCs) enhance the sensitivity and the frequency tuning of the mammalian cochlea.
Similar to the primary sensory receptor, the inner hair cells (IHCs), the mature functional characteristics of OHCs are acquired before hearing onset.
We found that OHCs, like IHCs, fire spontaneous Ca2+‐induced action potentials (APs) during immature stages of development, which are driven by CaV1.3 Ca2+ channels.
We also showed that the development of low‐ and high‐frequency hair cells is differentially regulated during pre‐hearing stages, with the former cells being more strongly dependent on experience‐independent Ca2+ action potential activity.
Introduction
The correct perception of acoustic stimuli in the mammalian cochlea depends on the sensory receptors, the inner and outer hair cells (IHCs and OHCs), and their respective afferent and efferent innervation (Goutman et al. 2015). While IHCs are the primary sensory receptors that relay sound information to type I afferent fibres, OHCs possess a unique combination of sensory and motor properties, which enhance the sensitivity and the frequency tuning within the cochlear partition (Dallos, 1992). Mature mouse OHCs express basolateral K+ channels with an unusually hyperpolarized activation (KCNQ4) (Kubisch et al. 1999; Marcotti & Kros, 1999). The current carried by these channels, I K,n, together with the resting depolarizing current flowing through the mechano‐electrical transducer channels, sets their resting membrane potential to near −40 mV (Johnson et al. 2011a). This relatively depolarized membrane potential ensures optimal activation of the motor protein prestin (Zheng et al. 2000; Liberman et al. 2002), which drives the somatic motility feedback mechanism characteristic of mature OHCs (Brownell et al. 1985; Ashmore, 1987). Another key feature of mature OHCs is that they are the primary target of the medial olivocochlear (MOC) efferent fibres (Liberman, 1980; Guinan et al. 1983; Simmons et al. 1996; Maison et al. 2003). Efferent fibres release the neurotransmitter acetylcholine (ACh), the role of which is to inhibit OHC electromotility and hence reduce the mechanical amplification of the cochlear partition (Guinan, 1996). OHC inhibition is achieved because ACh, by promoting Ca2+ influx through α9α10 nicotinic acetylcholine receptors (α9α10nAChRs), leads to the opening of the hyperpolarizing small conductance Ca2+‐activated K+ channels (SK2: Oliver et al. 2000; Katz et al. 2004; Lioudyno et al. 2004; Marcotti et al. 2004a).
OHCs from altricial rodents are very immature at birth and only begin to acquire the above biophysical and morphological characteristics of mature cells towards the end of the first postnatal week (He et al. 1994; Simmons et al. 1996; Oliver et al. 1997; Marcotti & Kros, 1999; Abe et al. 2007). On the other hand, IHCs start to acquire their mature basolateral current profile a few days later than OHCs, which corresponds to the onset of hearing at P12 (Kros et al. 1998; Marcotti et al. 2003a). Currently, it is not known whether the functional maturation of OHCs is solely dependent on an intrinsic genetic programme or can also be influenced by experience‐independent electrical activity similar to other sensory systems (e.g. Katz & Shatz, 1996; Blankenship & Feller 2010). Repetitive action potential activity has been shown to occur in the IHCs of the immature mouse cochlea (e.g. Kros et al. 1998; Tritsch et al. 2007; Johnson et al. 2011b, 2017; Sendin et al. 2014; Corns et al. 2018; Eckrich et al. 2018), which is used to influence the refinement of the exocytotic machinery (Johnson et al. 2013) and basolateral membrane currents (Brandt et al. 2003) in IHCs. OHCs appear less capable of firing spontaneous and repetitive AP activity in both mice and rats (Oliver et al. 1997; Marcotti & Kros, 1999; Weisz et al. 2012), although a previous study showed AP activity, but by using elevated extracellular Ca2+ and high intracellular EGTA (Beurg et al. 2008), and another has reported spontaneous Ca2+ signals during early postnatal stages (Ceriani et al. 2019).
Here we found that OHCs, under physiological recording conditions, are capable of firing spontaneous APs during early stages of development, just before the onset of functional maturation at ∼P7. Similar AP activity was previously described in IHCs, although this activity persists until near the onset of hearing at P12 (Johnson et al. 2011b, 2012). These APs are driven by CaV1.3 Ca2+ channels. Using CaV1.3 Ca2+ channel knockout mice (CaV1.3−/−), we found that pre‐hearing Ca2+‐dependent APs are required for the normal acquisition of morphological and biophysical features that are characteristic of fully functional mature OHCs and IHCs. However, low‐frequency (apical coil) hair cells were more susceptible to the absence of pre‐hearing Ca2+ APs than high‐frequency (basal coil) cells, indicating a differential regulation of hair cell development along the cochlea. The expression of the motor protein prestin and the associated OHC electromotility were normal in mature CaV1.3−/− mice, showing that experience‐independent Ca2+ signalling during immature stages does not influence the acquisition of key features of mature hair cells.
Methods
Ethics statement
All animal work was performed at the University of Sheffield and University of Sussex (UK) and licensed by the Home Office under the Animals (Scientific Procedures) Act 1986 and was approved by the University of Sheffield Ethical Review Committee. For in vitro work, mice were culled by cervical dislocation, which is a Schedule 1 method. In vivo recordings (ABR and DPOAE measurements: see below) were conducted under anesthesia using ketamine (100 mg kg−1, Fort Dodge Animal Health, Fort Dodge, IA, USA) and xylazine (10 mg kg−1, Rompun 2%, Bayer, HealthCare LLC, NY, USA), which were administered by intraperitoneal injection as previously described (Ingham et al. 2011). At the end of the experiments, which lasted up to 40 min, mice were culled using Schedule 1 methods.
Tissue preparation
The study was performed on wild‐type and transgenic mice (CaV1.3−/− mice: Platzer et al. 2000) of either sex on the C57BL/6 background. Apical‐ and basal‐coil hair cells were mainly studied in acutely dissected organs of Corti from postnatal day 0 (P0) to P18, where the day of birth (P0) corresponds to embryonic day 19.5 (E19.5). For some experiments in which recordings were performed from embryonic OHCs, mice were paired overnight and checked for vaginal plugs the following morning. Assuming ovulation occurs midway through the dark cycle, the mid‐point of the light cycle of the day following mating is considered to be E0.5 (Marcotti et al. 2003a). The genotyping protocol for the CaV1.3−/− mice was performed as previously described (Platzer et al. 2000). Mice were killed by cervical dislocation and the organ of Corti dissected in extracellular solution composed of (in mm): 135 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 mgCl2, 0.7 NaH2PO4, 5.6 d‐glucose, 10 HEPES‐NaOH. Sodium pyruvate (2 mm), amino acids and vitamins were added from concentrates (Thermo Fisher Scientific, UK). The pH was adjusted to 7.5 (osmolality ∼308 mmol kg−1). For OHCs recorded in situ, the dissected organ of Corti was transferred to a microscope chamber, immobilized using a nylon mesh fixed to a stainless steel ring and viewed using an upright microscope (Olympus BX51, Japan; Leica, DMLFS, Germany; Nikon, Germany; Bergamo II System B232, Thorlabs Inc.). Hair cells were observed with Nomarski differential interference contrast (DIC) optics (×63 water immersion objective) or Dodt gradient contrast (DGC) optics (×60 water immersion objective) and either ×10 or ×15 eyepieces.
Single‐cell electrophysiology
For cell‐attached recordings (Fig. 1), the pipette contained (in mm): 140 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 mgCl2, 0.7 NaH2PO4, 5.6 d‐glucose, 10 HEPES‐NaOH (pH 7.5; 308 mmol kg−1). For whole‐cell recordings, which were used in all the other figures containing electrophysiological data, the pipette intracellular solution contained (in mm): 131 KCl, 3 mgCl2, 1 EGTA‐KOH, 5 Na2ATP, 5 HEPES‐KOH, 10 sodium phosphocreatine (pH was adjusted with 1 m KCl to 7.28; 294 mmol kg−1). For Ca2+ current recordings, the pipette intracellular solution contained (in mm): 125 CsCl, 3 mgCl2, 1 EGTA‐CsOH, 5 Na2ATP, 5 HEPES‐CsOH, 5 TEA, 5 4‐AP (pH 7.3, 294 mmol kg−1). Membrane currents and voltage responses were investigated either at room temperature (20–24°C) or at body temperature (∼35°C), using Optopatch (Cairn Research Ltd, UK) or Axopatch 200B (Molecular Devices, USA) amplifiers. Data acquisition was controlled by pClamp software using Digidata 1320A, 1440A or 1550 boards (Molecular Devices). Recordings were low‐pass filtered at 2.5 kHz (8‐pole Bessel), sampled at 5 khz and stored on computer for off‐line analysis (Origin: OriginLab, USA). Membrane potentials in whole‐cell recordings were corrected for the residual series resistance R s after compensation (usually 70–90%) and the liquid junction potential (LJP) of −4 mV measured between electrode and bath solutions. The extracellular application of a Ca2+‐free solution or solutions containing acetylcholine (Sigma‐Aldrich, UK) or strychnine (Tocris Bioscience, UK) was performed with a multi‐barrelled pipette positioned close to the patched cells.
Figure 1. Early postnatal OHCs show spontaneous Ca2+‐induced action potentials.

A and B, spontaneous inward currents recorded from P2 apical coil (Ac) and P0 basal coil (Bc) OHCs, respectively, using cell‐attached voltage clamp at body temperature (∼35°C). Recordings were performed using physiological 1.3 mm extracellular Ca2+. C, expanded view of a single spontaneous current from panel A. D, cell‐attached recordings of inward currents from a P3 apical OHC during the repeated application of a Ca2+‐free solution containing 0.5 mm EGTA, a condition that abolishes APs (Johnson et al. 2011b). E, example of cell‐attached recording from a basal‐coil OHC of a P1 CaV1.3 −/− mouse with no spontaneous currents. F–I, representative ΔF/F 0 traces from 7 apical (F and G) and basal (H and I) OHCs each of a P2 wild‐type (F and H) and a P1 CaV1.3−/− (G and I) mouse. Traces are computed as pixel averages of regions of interest (white squares) centred on OHCs (Ceriani et al. 2019). Scale bars: 20 µm.
The presence of electromotile activity in OHCs was estimated by applying a depolarizing voltage step from the holding potential of –84 mV to +36 mV and cell length was viewed with a Nikon FN1 microscope with ×75 magnification and images captured using a Flash 4.0 SCCD camera (Hamamatsu, Japan). Cell body movement was analysed using Fiji ImageJ software (Schindelin et al. 2012). Lines were drawn across the basal membrane of patched OHCs, perpendicular to the direction of cell motion, and a projected time‐based z‐stack of the pixels under the line was made. Cell movement was measured with Photoshop as a pixel shift and then converted to nanometres (290 pixels = 10 µm).
Non‐linear (voltage‐dependent) capacitance was measured from P11 hair cells using whole‐cell patch clamp recordings. In order to block most of the ion channels in hair cells, the pipette intracellular solution contained (in mm): 125 CsCl, 3 mgCl2, 1 EGTA‐CsOH, 5 Na2ATP, 5 HEPES‐CsOH, 5 tetraethylammonium (TEA), 5 4‐amynopyridine (4‐AP) (pH was adjusted with CsOH to 7.28; osmolality was 290 mmol kg−1). Real‐time changes in non‐linear membrane capacitance (C N‐L) were investigated using the capacitance track‐in‐mode of the Optopatch amplifier (Cairn Research Ltd, UK) during the application of a 4 kHz sine wave of 13 mV RMS. From the holding potential of −84 mV, hair cells were subjected to a voltage ramp from −154 mV to +96 mV over 2 s. The capacitance signal from the Optopatch amplifier was filtered at 250 Hz and sampled at 5 kHz.
Two‐photon confocal Ca2+ imaging
For calcium dye loading, acutely dissected preparations were incubated for 40 min at 37°C in DMEM/F12, supplemented with fluo‐4 AM (final concentration 10–20 µM; Thermo Fisher Scientific). The incubation medium also contained pluronic F‐127 (0.1%, w/v, Sigma Aldrich, UK), and sulfinpyrazone (250 µM) to prevent dye sequestration and secretion. Preparations were then transferred to the microscope stage and perfused with extracellular solution for 20 min before imaging to allow for de‐esterification. Ca2+ signals were recorded using a two‐photon laser‐scanning microscope (Bergamo II System B232, Thorlabs Inc., USA) based on a mode‐locked laser system operating at 800 nm, 80‐MHz pulse repetition rate, < 100‐fs pulse width (Mai Tai HP DeepSee, Spectra‐Physics, USA). Images were formed by a ×60 objective, 1.1 NA (LUMFLN60XW, Olympus, Japan) using a GaAsp photomultiplier tube (Hamamatsu) coupled with a 525/40 bandpass filter (FF02‐525/40‐25, Semrock). Images were analysed off‐line using custom‐built software routines written in Python (Python 2.7, Python Software Foundation) and Fiji ImageJ software (Schneider et al. 2012). Ca2+ signals were measured as relative changes of fluorescence emission intensity (∆F/F 0). ΔF = F – F 0, where F is fluorescence at time t and F 0 is the fluorescence at the onset of the recording
Immunofluorescence microscopy
Dissected inner ears from wild‐type and CaV1.3−/− mice (n = 4 for each set of experiment) were fixed with 4% paraformaldehyde in phosphate‐buffered saline (PBS, pH 7.4) for 5–20 min at room temperature. Cochleae were microdissected, rinsed three times for 10 min in PBS, and incubated for 1 h at room temperature in PBS supplemented with 5% normal goat or horse serum and 0.5% Triton X‐100. The samples were then incubated overnight at 37°C with the primary antibody in PBS supplemented with 1% of the specific serum. Primary antibodies were: mouse anti‐myosin 7a (1:1000, Developmental Studies Hybridoma Bank, No. 138‐1C), rabbit anti‐myosin 7a (1:200, Proteus Biosciences, No. 25‐6790), rabbit anti‐prestin (1:5000, kindly provided by Robert Fettiplace), mouse anti‐KCNQ4 (1:100, StressMarq, No. SMC‐309D), goat anti‐Oncomodulin (1:200, Swant, OMG4), rabbit anti‐SK2 (1:500, Sigma‐Aldrich, P0483), mouse anti‐BK (1:100, Antibodies, 75–408) and goat anti‐choline acetyltransferase (ChAT, 1:500, Millipore, AB144P). All primary antibodies were labelled with species appropriate Alexa Fluor secondary antibody for 1 h at 37°C. Samples were then mounted in VECTASHIELD. The z‐stack images were captured with a Nikon A1 confocal microscope equipped with Nikon CFI Plan Apo ×60 oil objective, which is part of the Light Microscope Facility at the University of Sheffield. Image stacks were processed with Fiji ImageJ Analysis software. Cell volume was estimated from the z‐stack images of the immunolabelled hair cells with the cell marker Myo7a using Imaris software (Oxford Instruments).
Auditory brainstem responses
Auditory brainstem responses (ABRs) were recorded from male and female wild‐type and CaV1.3−/− mice between P16 and P18. Recordings were performed in a soundproof chamber (MAC‐3 Acoustic Chamber, IAC Acoustic, UK) as previously described (Ingham et al. 2011). Briefly, stimuli were delivered to the ear by calibrated loudspeakers (MF1‐S, Multi Field Speaker, Tucker‐Davis Technologies, USA) placed 10 cm from the animal's pinna. Sound pressure was calibrated with a low‐noise microphone probe system (ER10B+, Etymotic, USA). Experiments were performed using customized software (Ingham et al. 2011) driving an RZ6 auditory processor (Tucker‐Davis Technologies). Response thresholds were estimated from the resulting ABR waveform and defined as the lowest sound level where any recognizable feature of the waveform was visible. Responses were measured for click and stimulus pure tones of frequencies between 3 and 42 kHz. Stimulus sound pressure levels were typically 0–95 dB SPL, presented in steps of 5 dB. The brainstem response signal was averaged over 256 repetitions. Tone bursts were 5 ms in duration with a 1 ms on/off ramp time, which was presented at a rate of 42.6 s−1.
Distortion product otoacoustic emissions
OHC function was assessed in vivo by measuring distortion product otoacoustic emissions (DPOAEs). Recordings were performed in a soundproof chamber (MAC‐3 Acoustic Chamber, IAC Acoustic, UK). DPOAEs were recorded at 2f1‐f2 in response to primary tones of frequency f1 and f2, where f2/f1 = 1.2. The f2 level (L2) was set from 20 to 80 dB with 10 dB increments, and the f1 level (L1) was set equal to L2. Frequency pairs of tones between f2 = 6 kHz and f2 = 36 kHz were presented directly into the ear canal by means of a metal coupler connected to two calibrated loudspeakers (MF1‐S, Multi Field Speaker, Tucker‐Davis Technologies, USA). The emission signals were recorded by a low‐noise microphone (ER10B+: Etymotic Research Inc, USA) connected to the coupler. Experiments were performed using BioSigRZ software driving an RZ6 auditory processor (Tucker‐Davis Technologies). The DPOAE thresholds were defined by the minimal sound level, where the DPOAEs were two times above the standard deviation of the noise. The determined DPOAE thresholds were plotted against the geometric mean frequency of f1 and f2. Stimulus sound pressure levels were typically 20–80 dB SPL, presented in steps of 10 dB. The response signal was averaged over 500 repetitions.
Statistical analysis
Statistical comparisons of means were made by Student's two‐tailed t test or, for multiple comparisons, analysis of variance (one‐way or two‐way ANOVA followed by Bonferroni's or Tukey's post hoc test) was applied. P < 0.05 was selected as the criterion for statistical significance. Mean values are quoted as means ± SD (ABR and DPOAE experiments) or means ± SEM (all other figures).
Results
Immature OHCs fire spontaneous Ca2+‐dependent action potentials
Using cell‐attached patch clamp, which preserves the in vivo intracellular milieu and Ca2+ levels, we recorded spontaneous action potentials (APs) in the form of biphasic capacitative currents (Fig. 1 A–C; see also Johnson et al. 2011b) in early postnatal apical (Fig. 1 A) and basal (Fig. 1 B) OHCs. We compared the AP activity of P2 apical and P0 basal OHCs because the development of the former lags behind that of basal cells by about 2 days (Helyer et al. 2005: see also Fig. 4). The mean spike frequency of apical OHCs (0.75 ± 0.13 Hz, n = 7) was significantly lower than that in basal cells (1.95 ± 0.34 Hz, n = 4, P = 0.0033, t test). However, the coefficient of variation (CV) was not significantly different (apical: 1.62 ± 0.18, n = 7; basal: 1.77 ± 0.28, n = 4, P = 0.6506; t test), and, being greater than one, is indicative of a bursting pattern of activity. Action potentials were mainly recorded from early postnatal OHCs and by P6 they were no longer present, which agrees with previous measurements of Ca2+ signals using 2‐photon imaging (Ceriani et al. 2019). Spontaneous AP activity was abolished by the perfusion of a Ca2+‐free solution (Fig. 1 D) and was absent in CaV1.3−/− mice (Fig. 1 E). Spontaneous, rapid Ca2+ signals were also recorded from control OHCs maintained in acutely dissected cochleae loaded with the Ca2+ indicator Fluo‐4 (Fig. 1 F and H). These Ca2+ signals represent the optical read‐out of OHC firing activity in wild‐type mice (see also Ceriani et al. 2019) and were absent in both apical and basal cells of CaV1.3−/− mice (Fig. 1 G and I).
Figure 4. Acquisition of I K,n in developing OHCs.
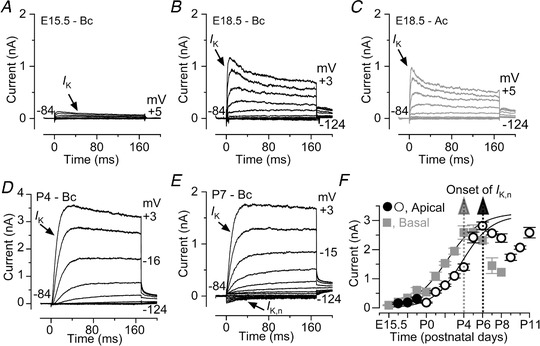
A–E, typical current responses from E15.5, E18.5, P4 and P7 OHCs recorded from wild‐type mice. Note that most of the examples are from basal‐coil OHCs apart from the recordings in panel C (see panel F below for details). Also note that the y‐axis in panel D covers a larger current range than that in all the other panels (A–C and E). Outward current was elicited by using depolarizing and hyperpolarizing voltage steps (10 mV increments) from –84 mV to the various test potentials shown by some of the traces. F, size of the total outward K+ current measured at 0 mV as a function of embryonic and early postnatal ages for OHCs positioned in the apical and basal cochlear region (including those shown in panels A–E). Note that the onset of I K,n occurs about 2 days earlier in basal OHCs. Numbers of basal OHCs measured at the various ages (E15.5–P8) are from left to right: 3, 4, 5, 3, 3, 4, 27, 15, 13, 3, 7, 4, 1; apical OHCs (E16.5–E18.5) 5, 4, 4 and (P0–P11) are from Marcotti & Kros (1999). Fits to the data are according to a sigmoidal logistic growth curve: where A is the size of the current, k is a slope factor and t half is the age where A is halfway between the maximal (A max) and minimal (A min) currents. t half was 2.2 days in basal and 3.8 days in apical OHCs.
OHCs from CaV1.3−/− mice retain healthy characteristics up to the onset of hearing
Previous light microscopy studies have shown that apical‐coil OHCs from CaV1.3−/− mice are present at P7 but largely absent at P14–P15, with their degeneration progressing from the apical to the basal region of the cochlea (Platzer et al. 2000; Glueckert et al. 2003). Therefore, we initially investigated the progression of OHC degeneration in the apical region used for the majority of our experiments, which corresponded to a frequency range in the adult mouse of ∼8–12 kHz (Müller et al. 2005; see also Ceriani et al. 2019). Cochleae from wild‐type and CaV1.3−/− mice between P10 and P14 were acutely dissected and DIC images were taken from the apical coil. The appearance (e.g. hair bundle and basolateral region) and number of apical‐coil OHCs was similar between the two genotypes up to P12 (Fig. 2 A, B and D). However, from just after the onset of hearing, P13–P14 apical‐coil OHCs from CaV1.3−/− mice exhibited an abrupt reduction in number compared to wild‐type mice (Fig. 2 C and D: P < 0.0001, Bonferroni's post hoc test, two‐way ANOVA). By contrast, the number of OHCs positioned in the basal coil of the cochlea, which corresponds to an approximate frequency range of ∼ 25–45 kHz (Müller et al. 2005; Ceriani et al. 2019), were not significantly different between wild‐type and CaV1.3−/− mice at P11 (P = 0.874) and after the onset of hearing (P14, P ≥ 0.999, Fig. 2 D).
Figure 2. The number and size of pre‐hearing OHCs is normal in CaV1.3−/− mice.

A and B, DIC images of a region of the mouse cochlear apical coil (∼8–12 kHz) showing the presence of IHCs and OHCs at P10 and P12 for wild‐type (left top panels) and CaV1.3−/− (right top panels) mice. Note that the hair bundles of IHCs are also visible. The presence of normal hair bundles in OHCs is shown in the bottom panels. C, DIC images from a wild‐type (left) and CaV1.3−/− (right) mice, as described in panels A and B, but from a cochlea just after hearing onset (P13). Note that OHCs began to disappear in CaV1.3−/− mice. D, number of OHCs present in a 200 µm region from the apical and basal coil of wild‐type and CaV1.3−/− mice as a function of postnatal development. Numbers of cochleae tested at the various ages from left to right are: wild‐type apical 6, 7, 6, 6; CaV1.3−/− apical 4, 6, 7, 5. * P < 0.0001; wild‐type basal 3, 3; CaV1.3−/− basal 4, 4. E, membrane capacitance (C m) measured from apical OHCs of wild‐type and CaV1.3−/− mice during development. Numbers of OHCs measured: wild‐type 3, 6, 5, 0, 2, 9, 23, 4; CaV1.3−/− 4, 4, 5, 7, 1, 2, 10, 14. * P<0.0001. F, resting membrane potential (V m) of wild‐type and CaV1.3−/− OHCs during development. * P = 0.0013. Numbers of OHCs measured: wild‐type 3, 4, 2, 4, 8, 6, 4; CaV1.3−/− 3, 2, 2, 4, 3, 7. P11 data points are from Ceriani et al. 2019.
We then investigated two crucial biophysical properties of OHCs, their membrane capacitance (C m), which provides an indication of the cell surface, and resting membrane potential (V m), by performing whole‐cell patch clamp recordings. These experiments were only performed in apical OHCs because of the difficulty of obtaining reliable recordings from basal OHCs after P8. The OHC membrane capacitance, which provides a quantifiable measure of the cell's surface area (C m: Fig. 2 E) was not significantly different between the two genotypes (P1–P13: P = 0.754, two‐way ANOVA), although a significant decrease in C m was present in OHCs from CaV1.3−/− mice at P13 (P < 0.0001, Bonferroni's post hoc test). We then performed current clamp experiments in order to investigate the resting membrane potential (V m) of OHCs as a function of age, which provides a reliable measure of the baseline physiological status of cells. We found that the resting V m was comparable between wild‐type and CaV1.3−/− OHCs up to P12, becoming significantly more depolarized only at P13 in the latter (Fig. 2 F, P = 0.0013, Bonferroni's post hoc test, two‐way ANOVA). Another key functional marker of OHCs is the presence of oncomodulin, which is a member of the parvalbumin family of Ca2+ binding proteins expressed in OHCs from early postnatal ages (Simmons et al. 2010). We found that oncomodulin was present in both apical and basal OHCs from P11 wild‐type and CaV1.3−/− mice (Fig. 3).
Figure 3. The Ca2+ binding protein oncomodulin is present in OHCs from wild‐type and CaV1.3−/− mice.
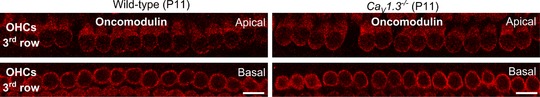
Confocal images taken from apical and basal coil OHCs of wild‐type (left panel) and CaV1.3−/− (right panel) mice at P11, which is after their onset of functional maturation at P8, but just before the onset of hearing (P12). Similar staining was seen in additional 4 mice for each genotype. Scale bar: 10 µm.
The above morphological and biophysical data, including the electromotile activity (see below), demonstrate that OHCs from CaV1.3−/− mice maintain a healthy functional profile for at least 5 days after their initial onset of maturation at the beginning of the second postnatal week. However, just after the onset of hearing at ∼P12, apical but not basal OHCs abruptly begin to degenerate.
Apical OHCs from CaV1.3−/− mice fail to acquire the full adult biophysical profile
Since the presence of early spontaneous AP activity has been shown to regulate the transcription of genes involved in cell development (e.g. Greer & Greenberg, 2008), we investigated whether OHCs from CaV1.3−/− mice, where Ca2+ spikes are absent, were able to acquire their normal adult‐like characteristics. OHC fate is determined during early embryonic development and from about embryonic day 14 (E14) they are recognizable within the sensory epithelium (Pujol et al. 1998). Using whole‐cell patch clamp recordings, we compared the developmental acquisition of the K+ currents in mouse OHCs (Fig. 4 A–F). Small outward K+ currents (I K) were recorded as early as E15.5 in basal and E16.5 in apical OHCs. Their amplitude gradually increased with age and reached a maximum at around P4 in basal and P6 in apical OHCs (Fig. 4 F), at about the time when OHCs lose their ability to fire spontaneous Ca2+ spikes. During the following few days the size of the immature I K gradually decreased (Fig. 4 F). At the same time OHCs began to express a negatively activating K+ current I K,n (Fig. 4 E), the main K+ current in mature mouse OHCs (Marcotti & Kros, 1999). This caused the overall outward K+ current to increase again (Fig. 4 F), reaching a mature‐like amplitude in apical OHCs at around P12 (Marcotti & Kros, 1999). A similar developmental change in the K+ currents is also likely to occur in basal OHCs but, as mentioned above, patch‐clamp recordings in basal cells became unreliable after P8.
During the first postnatal week, apical‐coil OHCs from both wild‐type and CaV1.3−/− mice were indistinguishable in terms of their basolateral membrane properties under whole‐cell patch clamp (Fig. 5 A, D and E). During the second postnatal week, wild‐type OHCs began to express I K,n, which becomes the major adult‐type K+ current by P12 as mentioned above (Fig. 5 C; see also Marcotti & Kros, 1999). On the other hand, OHCs (≥P7) from CaV1.3−/− mice retained the immature delayed rectifier as the main K+ current (Fig. 5 C), which was evident by the slower activating and inactivating time course (Marcotti & Kros, 1999). I K,n in mature OHCs from CaV1.3−/− mice was either absent or very small compared to that in wild‐type cells (Fig. 5 D, P < 0.0001, two‐way ANOVA). The failure in the normal up‐regulation of I K,n caused the total outward current to remain at a nearly constant size between P9 and P13 (Fig. 5 E, P = 0.628, one‐way ANOVA) but significantly lower than that in wild‐type OHCs (Fig. 5 E, P = 0.0003, two‐way ANOVA). Because OHCs from CaV1.3−/− mice retain an immature‐like current profile, the measured I K,n in these cells is likely to be contaminated by the inward rectifier K+ current I K1. This is because I K1 is normally expressed by immature OHCs and is strongly activated at the membrane potential used to estimate the size of I K,n (Marcotti et al. 1999). The persistence of I K1 in CaV1.3−/− OHCs would also explain why during pre‐hearing stages they exhibit a resting V m comparable to that of wild‐type cells (Fig. 2 F), since I K1 is active at the resting V m and fulfils a similar role to I K,n in setting the resting V m (Marcotti et al. 1999). Therefore we investigated the presence of KCNQ4 channels, which carry I K,n in OHCs (Kubisch et al. 1999; Marcotti & Kros, 1999), using immunohistochemistry experiments. In P11 wild‐type mice, KCNQ4 staining was present throughout the entire basolateral membrane of both apical and basal OHCs, although the latter were more intensely stained (Fig. 5 F, e.g. white arrowheads). In age‐matched CaV1.3−/− mice, KCNQ4 expression was much reduced in apical OHCs (Fig. 5 G, upper panel), supporting the electrophysiological data (Fig. 5 D), while it was easily detectable in basal OHCs (Fig. 5 G, lower panel).
Figure 5. The expression of the mature K+ current profile is impaired in CaV1.3 −/− OHCs.

A–C, current responses in wild‐type (left) and CaV1.3−/− (right) apical‐coil OHCs during immature stages (A, P1), at the onset of function when I K,n is first detected (B, P8: Marcotti & Kros, 1999) and at a more mature age when I K,n has almost reached its mature size in mice (C, P12: Marcotti & Kros, 1999). Note that even at P12 I K,n is very small and possibly contaminated by the inward rectifier I K1. D, size of the isolated I K,n as a function of postnatal age in wild‐type and CaV1.3−/− apical OHCs (measured as the deactivating tail currents at −124 mV from the holding potential of −84 mV). Numbers of cells from left to right: wild‐type: 3, 7, 5, 7, 0, 3, 15, 4; CaV1.3−/− 4, 4, 4, 7, 3, 2, 12, 11. Two‐way ANOVA with Bonferroni's post hoc test: * P = 0.0192 (P8); ** P = 0.0022 (P9), *** P < 0.0001 (at P11, P12 and P13). E, total steady‐state outward I K as a function of age in wild‐type and CaV1.3−/− OHCs (measured at 0 mV from the holding potential of −84 mV). *** P = 0.0002 (at P12 and P13). Number of OHCs investigated as in panel D. F and G, maximum intensity projections of confocal z‐stack images that were taken from apical (upper panels) and basal (lower panels) coil OHCs (P11) in wild‐type (F) and CaV1.3−/− (G) mice. Immunostaining for KCNQ4 is shown in white (arrows) and Myo7a is used as a cell marker (blue). Similar staining was seen in an additional 4 mice for each genotype. Scale bar: 10 µm.
Prestin expression and electromotility are unaffected by the absence of spontaneous AP activity
The maturation of OHCs is also associated with the expression of the motor protein prestin (Zheng et al, 2000; Liberman et al, 2002), which drives the somatic motility of mouse OHCs from about P7 onwards (Marcotti & Kros, 1999; Abe et al. 2007). In line with the normal morphological appearance throughout pre‐hearing stages (≤P12: Fig. 2), OHCs from CaV1.3−/− mice showed normal electromotile activity at P12 (Fig. 6). Under whole‐cell patch clamp conditions, stepping the membrane potential from −84 mV to +36 mV caused OHCs from both wild‐type and CaV1.3−/− mice to shorten (Fig. 6 A and B), as previously described (Marcotti & Kros, 1999; Abe et al. 2007). OHC movement was not significantly different between the two genotypes (P = 0.7881: Fig. 6 C and D). This was further investigated using non‐linear (voltage‐dependent) capacitance changes (C N‐L), which is the electrical signature of electromotility in OHCs. We found that the maximum size of C N‐L in P11 apical coil OHCs (Fig. 6 E–G) was comparable to that reported from P10–P12 cells (Abe et al. 2007), and was not significantly different between the two genotypes (P = 0.1537: Fig. 6 F), even when it was normalized to the OHC membrane capacitance (P = 0.2368: Fig. 6 G). The above electrophysiological results (Fig. 6 A–D: electromotility; Fig. 6 E–G: non‐linear capacitance) are in agreement with the normal expression of prestin in age‐matched OHCs (P11) from both wild‐type and CaV1.3−/− mice (Fig. 6 H). The expression of prestin was also comparable between the two genotypes even after the onset of hearing (P14: Fig. 6 I and J), in the few remaining apical OHCs.
Figure 6. Prestin expression and electromotility are normal in OHCs from CaV1.3 −/− mice.
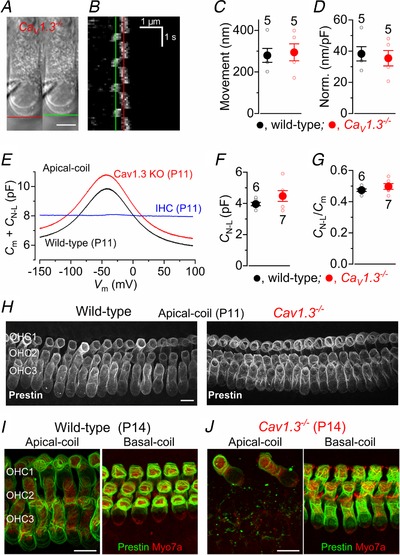
A, images showing the patch pipette attached to an apical‐coil OHC from a CaV1.3−/− mouse at P12. The red lines indicate the position of the OHC basal membrane before (left: at –84 mV) and during a depolarizing voltage step from –84 mV to +36 mV (right). Note that membrane depolarization causes OHCs to shorten. Scale bar, 5 µm. B, kymograph showing the movement of the subnuclear region of the OHC shown in panel A as a function of time in response to the repetitive voltage steps described above. The red line indicates the resting position of the OHC basal membrane and the green line indicates the shortening. C and D, average movement in response to the depolarizing voltage step was not significantly different between wild‐type and CaV1.3−/− apical‐coil OHCs (C, P = 0.79), even after normalization to the average OHC membrane capacitance (D, P = 0.68). E, examples of voltage‐dependent non‐linear capacitance (C N‐L) recorded in apical‐coil hair cells by applying a voltage ramp from –154 mV to +96 mV over 2 s. Note that the cell membrane capacitance (C m) was added to the measured C N‐L. C N‐L was present in the OHCs from both genotypes, but absent in the IHC. F and G, average C N‐L was not significantly different between wild‐type and CaV1.3−/− apical‐coil OHCs (F), even after normalization to the individual OHC membrane capacitance C m (G). C N‐L was calculated as the difference between the peak of the recording near −40 mV and the lowest value at positive membrane potentials. Recordings in A–G are at room temperature. Number of cells investigated is shown near the columns. H, maximum intensity projections of confocal z‐stacks taken from the apical cochlear region of wild‐type (left) and CaV1.3−/− (right) mice at P11 using antibodies against prestin (white). I and J, images obtained as in H, but from the apical (left panels) and basal (right panels) cochlear of wild‐type (I) and CaV1.3−/− (J) post‐hearing mice (P14). The hair‐cell marker Myo7a (red) was used to better identify the synaptic, basal portion of OHCs that does not contain prestin (green); this was more evident in basal OHCs. Note that the different angle of the basal OHCs gives the incorrect impression that wild‐type OHCs are smaller than those in the CaV1.3−/− mice. Prestin labelling was also present in the few remaining OHCs of CaV1.3−/− mice (J, left panel). Scale bars in H–J: 10 µm.
Efferent synapses do not fully develop in apical OHCs from CaV1.3−/− mice
OHC maturation is also associated with their ability to respond to the inhibitory efferent neurotransmitter acetylcholine (ACh) from around the end of the first postnatal week (Katz et al. 2004; Marcotti et al. 2004a), which coincides with the appearance of I K,n (Marcotti & Kros, 1999). ACh exerts an overall inhibitory effect because Ca2+ that enters through α9α10nAChRs (Katz & Elgoyhen, 2014) then opens small‐conductance Ca2+ activated K+ channels (SK2 channels: Katz et al. 2004; Marcotti et al. 2004a), which leads to OHC hyperpolarization.
Wild‐type apical OHCs expressed nAChRs since they responded to the extracellular application of ACh with a large inward current at the holding potential of −90 mV, which was blocked by the specific α9α10‐nAChRs antagonist strychnine (–256 ± 53 pA, n = 4, at –90 mV: Fig. 7 A, top). The presence of SK2 channels was indicated by ACh‐induced outward current (200 ± 6 pA, n = 3: Fig. 7 A, bottom), when cells were held at −40 mV, that was removed by the perfusion of a Ca2+‐free solution, as previously described (Glowatzki & Fuchs, 2000; Oliver et al. 2000; Marcotti et al. 2004a). In apical OHCs from CaV1.3−/− mice, ACh elicited very small responses both at –90 mV (–6.6 ± 4.2 pA, n = 5: Fig. 7 B, top) and –40 mV (7.2 ± 4.9 pA, n = 5: Fig. 7 B, bottom), indicating that the expression of both nAChRs and SK2 channels is either absent or reduced. Immunolabelling experiments in apical coil OHCs confirmed that SK2 channels were present in wild‐type but almost completely absent in CaV1.3−/− mice (Fig. 7 C). Moreover, efferent cholinergic terminals on P11 OHCs, which were visualized by ChAT immunoreactivity, were very few and far between in CaV1.3−/− compared to wild‐type mice (Fig. 7 C), indicating that cholinergic fibres themselves are also strongly affected by the absence of CaV1.3 channels in apical OHCs. In the basal coil both the pre‐ and postsynaptic elements were present in most OHCs from CaV1.3−/− mice, although SK2 puncta appeared less prominent than in wild‐type mice (Fig. 7 D).
Figure 7. Cholinergic efferent synapses are differentially affected in apical and basal OHCs of CaV1.3−/− mice.
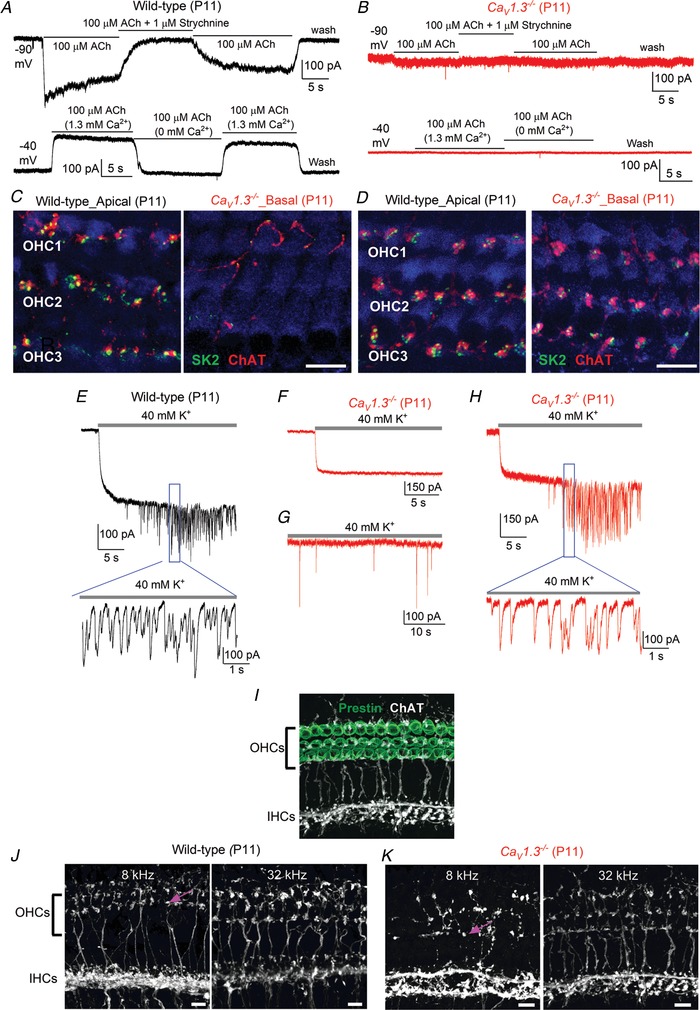
A, inward (top) and outward (bottom) currents in wild‐type apical‐coil OHCs elicited during the extracellular application of 100 µM extracellular ACh at –90 mV and –40 mV, respectively. Note that at –90 mV the current was reversibly blocked by 1 µM strychnine, indicating the direct involvement of α9α10‐nAChRs; at –40 mV, the outward current was prevented by the absence of Ca2+ in the extracellular solution, indicating the presence of SK2 channels. B, same experiments as in panel A but performed in apical‐coil OHCs from CaV1.3−/− mice. Note that ACh produced very little or no responses at both potentials. C and D, maximum intensity projections of confocal z‐stack images that were taken from mature P11 apical (C) and basal coil (D) of wild‐type and CaV1.3−/− mice. Immunostaining for SK2 channels (green) and ChAT, which is used to visualize the efferent olivocochlear innervation of OHCs (red); Myo7a (blue) was used as the hair‐cell marker. Scale bars: 10 µm. E–H, whole‐cell voltage‐clamp recordings obtained from mature OHCs in wild‐type (E) and CaV1.3−/− (F–H) mice during the superfusion of 40 mm extracellular K+. Lower panels in E and H show an expanded time scale of the area shown in the panels above. I–K, maximum intensity projections of confocal z‐stack images taken at two different frequencies along the cochlea (apical: 8 kHz; basal: 32 kHz) from P11 wild‐type (J) and CaV1.3−/− mice (K). Immunostaining for the OHC marker prestin (I: green) and ChAT (I: white), which is used to visualize the efferent terminals and fibres below the IHCs, tunnel crossing fibres (arrows), and terminals below the OHCs. In the cochlear apical coil of CaV1.3−/− mice (K) there were fewer ChAT‐labelled tunnel crossing fibres and OHC terminals than wild‐type mice (J). The ChAT‐labelled OHC terminals at 8 kHz were also disordered and larger than in the wild type. In the basal coil, the efferent innervation was visually comparable between the two genotypes. Scale bars: 10 µm.
In order to establish whether the few remaining efferent terminals on OHCs from CaV1.3−/− mice were functional, we perfused the cochlea with a high‐K+ extracellular solution, which depolarizes the efferent synaptic terminals, thereby triggering the release of ACh onto OHCs. Under whole‐cell patch clamp, the superfusion of 40 mm KCl caused a sustained, inward current in OHCs from wild‐type mice (Fig. 7 E), owing to a positive shift in the K+ reversal potential in these cells. This current was followed by high‐frequency synaptic currents (Oliver et al. 2000; Lioudyno et al. 2004), which were caused by the activation of nAChRs following the release of ACh from the efferent terminals onto OHCs. These spontaneous currents were recorded in all 7 control OHCs tested; in cells with stable recordings lasting > 1 min, the average amplitude of the inward currents was 227 ± 3 pA (n = 4, P11). In CaV1.3−/− mice, 8 out of 12 OHCs tested remained either silent (5 cells: Fig. 7 F) or showed a very minimal activity (3 cells: Fig. 7 G) during the application of 40 mm K+. The 4 remaining OHCs responded to high K+ with more spiking activity (Fig. 7 H), although the amplitude of the inward currents was significantly smaller than that measured in wild‐type OHCs (167 ± 6 pA, n = 4, P10–P11, P < 0.0001, t test) than that measured in wild‐type OHCs. We further assessed the cholinergic innervation with ChAT‐immunoreactivity (Fig. 7 I–K). Compared to wild‐type mice (Fig. 7 J), the apical cochlear coil from Cav1.3−/− mice showed highly disorganized ChAT‐labelled efferent fibres, but progressively less so towards the base (32 kHz: Fig. 7 K). CaV1.3−/ − mice also had very dense ChAT labelling below the IHCs, with tunnel crossing fibres showing an irregular pattern of large terminals on OHCs (8 kHz: Fig. 7 K).
The above experiments testing the efferent synapses and fibres indicate that the majority of apical coil OHCs from CaV1.3−/− mice do not acquire the characteristic cholinergic efferent innervation. The innervation of basal OHCs appeared less affected by the absence of CaV1.3 Ca2+ channels.
Basal OHCs from CaV1.3−/− mice exhibit a milder phenotype
The above data suggest that the absence of the CaV1.3 Ca2+ channels during early postnatal stages prevent the full maturation of apical OHCs. Although basal OHCs appeared to develop normally, these conclusions are only made from immunolabelling experiments, which do not provide a comprehensive physiological read‐out of OHC activity. Moreover, it has been shown that even basal OHCs will eventually degenerate in CaV1.3−/− mice before ∼P30 (Platzer et al. 2000). Therefore, we performed in vivo experiments to primarily test the function of OHCs. Initially, we tested the hearing ability of the CaV1.3−/− mice by recording auditory brainstem responses (ABRs), which provide a measure of the activity of the auditory neurons downstream of IHCs. We found that, compared to wild‐type mice, which had thresholds as low as 40 dB, CaV1.3−/− mice showed threshold above 95 dB for both click and pure‐tone evoked ABRs (Fig. 8 A and B), which is consistent with an absence of CaV1.3 Ca2+ channel‐induced neurotransmitter release from the IHCs (Brandt et al. 2003) and also with previous observations (Platzer et al. 2000; Eckrich et al. 2019). We then performed distortion product otoacoustic emissions (DPOAEs), which are read‐outs of amplification and cochlear non‐linearities, which are induced by the displacement of OHC stereociliary bundles during sound‐induced motion of the cochlear partition. We found that the DPOAE thresholds, for frequencies between 6 kHz and 30 kHz in P16–P22 CaV1.3−/− mice were significantly elevated compared to P16–P18 wild‐type mice (P < 0.0001, two‐way ANOVA). We found that DPOAE thresholds above noise floor were still present in the high‐frequency regions, although significantly elevated compared to those in wild‐type mice (12 kHz and 18 kHz: P < 0.0001; 24 kHz: P = 0.0055; Tukey's post hoc test).This result indicates that the biophysical properties of basal coil‐OHCs are also affected by the absence of the CaV1.3 Ca2+ channels, although to a lesser extent than those of apical cells.
Figure 8. DPOAE, but not ABR, thresholds are still present in the high‐frequency region of CaV1.3‐/− mice.
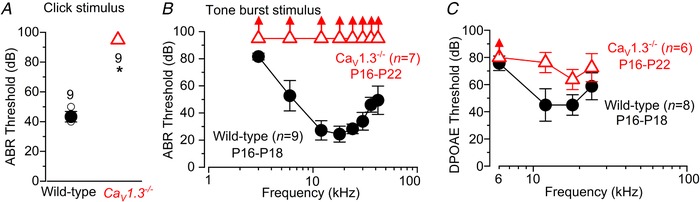
A and B, auditory brainstem responses (ABRs) for click (A) and tone burst stimuli (B) in P16–P18 wild‐type (circles) and P16–P22 CaV1.3−/− mice (triangles) as a function of frequency position along the cochlea (3, 6, 12, 18, 24, 30, 36, 42 kHz). CaV1.3−/− mice were profoundly deaf at all cochlear frequencies tested. C, DPOAE thresholds (6, 12, 18, 24 kHz) from P16–P18 wild‐type and P16–P22 CaV1.3−/− mice. Data are means ± SD.
IHCs from CaV1.3−/− mice have similar maturation defects to OHCs
It is well established that CaV1.3 Ca2+ channels contribute to ∼90% of the total Ca2+ current in apical coil IHCs and OHCs (Platzer et al. 2000; Brandt et al. 2003; Beurg et al. 2008). Since electrophysiological recording from mature basal‐coil IHCs can be reliably performed (e.g. Marcotti et al. 2004b), we investigated whether a differential regulation in development was also present between apical and basal IHCs of CaV1.3−/− mice (Fig. 9 A–K). Mature IHCs express the characteristic rapid‐activating Ca2+‐activated K+ current I K,f (Kros et al. 1998; Marcotti et al. 2004b) and, similar to OHCs, I K,n (Oliver et al. 2003; Marcotti et al. 2003a) from about the onset of hearing at P12. We found that, in agreement with previous findings (Brandt et al. 2003; Eckrich et al. 2019), P18 apical IHCs from CaV1.3−/− mice failed to acquire both I K,f and I K,n (Fig. 9 A–C and G). In P18 basal IHCs of CaV1.3−/− mice, I K,f was present but significantly reduced in size compared to that recorded in wild‐type cells (Fig. 9 D–G; P < 0.0002). These electrophysiological data are supported by immunolabelling experiments (Fig. 9 H and I) showing that the expression of the BK channels (carrying I K,f) at the neck region of IHCs (Pyott et al. 2004), was either absent (apical IHCs) or strongly reduced (basal IHCs) in CaV1.3−/− mice (Fig. 9 I) compared to wild‐type cells (Fig. 9 H). Similar to I K,f, I K,n was absent in apical IHCs of CaV1.3−/− mice, but present in basal cells albeit again significantly reduced compared to that recorded in IHCs of wild‐type mice (Fig. 9 D–F and J; P = 0.0037). We also found that the cell membrane capacitance C m is significantly reduced in both apical and basal IHCs of CaV1.3−/− mice compared to that measured in cells from wild‐type mice (Fig. 9 K: P > 0.001 for both apical and basal IHCs; Tukey's post hoc test); the reduced C m was also evident from the immunolabelling images (Fig. 9 H and I). When the size of I K,f and I K,n was normalized to the C m of IHCs, only the former was still significantly larger in basal IHCs of wild‐type compared to that of CaV1.3−/− mice (I K,f: P = 0.0002; I K,n: P = 0.6639).
Figure 9. Potassium current expressed in post‐hearing IHCs of CaV1.3−/− mutant mice.
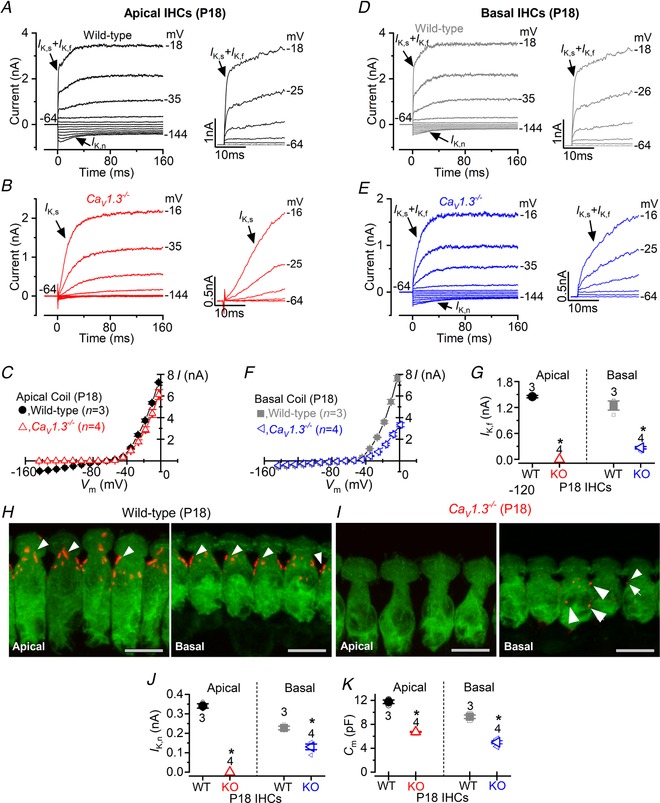
A and B, potassium currents recorded from P18 IHCs of the apical‐coil of the cochlea of wild‐type (A) and CaV1.3−/− mutant mice (B) using 10 mV depolarizing voltage steps from –64 mV to the various test potentials shown by some of the traces. The adult‐type currents (I K,f and I K,n) were only present in IHCs from wild‐type mice (A). IHCs from CaV1.3−/− mice retained the delayed rectifier current (B, I K) characteristic of immature cells (Marcotti et al. 2003a). The absence of the rapidly activating I K,f in CaV1.3−/− IHCs is also evident when comparing the activation time course of the total outward currents on an expanded time scale (see insets). C, average current‐voltage (I‐V) curves obtained from apical P18 IHCs of wild‐type and CaV1.3−/− mice. D and E, potassium currents recorded from P18 IHCs of the basal‐coil of the cochlea of wild‐type (D) and CaV1.3−/− mice (E) as described in panels A and B. Different from apical IHCs of CaV1.3−/− mice (B), basal cells retain I K,n and some I K,f (E). F, average current‐voltage curves obtained from P18 basal IHCs of wild‐type and CaV1.3−/− mice. G, average size of I K,f recorded from apical and basal IHCs of both genotypes. The isolated I K,f was measured as previously described (Marcotti et al. 2003a: current measured at 1.5 ms from the current onset and at the membrane potential of −25 mV). H and I, maximum intensity projections of confocal z‐stack images of IHCs taken from mature P18 wild‐type (H) and CaV1.3−/− mice (I) of both apical (left panels) and basal (right panel) coil of the cochlea. Immunostaining for BK channels (arrowheads) and Myo7a (IHC marker), which is used to visualize the IHCs. Note that BK puncta in IHCs from CaV1.3−/− mice were either absent in apical (I, left panel) or very few in basal cells (I, arrows in right panel). Scale bars: 10 µm. J, average size of I K,n recorded from apical and basal IHCs of both genotypes. The isolated I K,n was measured as previously described (Marcotti et al. 2003a: difference between the peak and steady‐state deactivating tail current at the membrane potential of −124 mV). K, average IHC membrane capacitance (C m) in both genotypes and as a function of cochlear position. In panels G, J and K, single cell value recordings (open symbols) are also plotted behind the average closed symbols. *Statistical significance (see Results).
A smaller C m was also seen in apical OHCs of CaV1.3−/− mice at P13 (Fig. 2 E), but information about basal OHCs is missing due to our inability to perform reliable electrophysiological recordings from these cells (see above). In order to have an indication of the size of OHCs in the basal coil, we estimated the hair cell volume by measuring pixel area from z‐stack immunofluorescence images (see Methods). Initially we confirmed the reliability of the above imaging method by comparing the measured cell volume with known surface‐area (C m) measurements recorded using patch clamp electrophysiology. Although the two techniques measure different cellular characteristics, they both provide an indication of cell size. We found that the volume of basal IHC was reduced by ∼33% between P11 CaV1.3−/− (301 ± 24 µm3, n = 25 cells) and age‐matched wild‐type mice (450 ± 22 µm3, n = 32, P < 0.0001), which is consistent with our C m measurements (∼46%: Fig. 9 K). We found that the volume of basal OHCs at P11 in CaV1.3−/− mice was also significantly reduced by ∼30% (109 ± 6 µm3, n = 83 cells) compared to that of age‐matched wild‐type mice (156 ± 4 µm3, n = 76, P < 0.0001), confirming that the reduced size of mature hair cells of CaV1.3−/− mice is a general characteristic associated with the absence of CaV1.3 Ca2+ channels.
These results indicate that the absence of CaV1.3 Ca2+ channels during pre‐hearing stages of development differentially affected the developmental acquisition of the mature biophysical profile of apical and basal IHCs, with the latter being less influenced by early Ca2+ signals, as seen for OHCs (see above). One common change observed in hair cells of CaV1.3−/− mice irrespective of their cochlear position was the reduced surface area.
The differential maturation of apical and basal hair cells in CaV1.3−/− mice is not due to tonotopic differences in the residual Ca2+ current
The above findings indicate that the maturation of both OHCs and IHCs from the basal cochlea of CaV1.3−/− mice was affected by the absence of CaV1.3 Ca2+ channels, but to a lesser extent compared to apical cells. Since ∼10% of the total Ca2+ current in apical hair cells has been shown to be carried by Ca2+ channels other than CaV1.3 (Platzer et al. 2000; Michna et al. 2003), we sought to investigate whether basal cells express a larger residual Ca2+ current that could explain the milder phenotype of these cells in CaV1.3−/− mice. Although the identity of the residual Ca2+ channels in auditory hair cells is still unknown (for a recent review see Pangrsic et al. 2018), it could involve CaV1.4 (Brandt et al. 2003) and/or CaV3.1 T‐type subunits (Inagaki et al. 2008; Levic & Dulon, 2012).
We performed whole‐cells recordings to measure the Ca2+ currents in P6 IHCs from CaV1.3−/− mice (Fig. 10 A–D), a time when spontaneous Ca2+‐dependent action potentials are present in IHCs of wild‐type mice (e.g. Marcotti et al. 2003b; Johnson et al. 2011b). Recordings were performed at body temperature and using physiological 1.3 mm extracellular Ca2+ (Johnson et al. 2005). We found that the residual Ca2+ current in IHCs of CaV1.3−/− mice showed no signs of inactivation during the 100 ms voltage steps, indicating that it is unlikely to be carried by T‐type Ca2+ channels (Zamponi et al. 2015). The size of the residual Ca2+ current was indistinguishable between the low‐ (apical) and high‐frequency (basal) cells (P = 0.994, Fig. 10 C), even when it was normalized to the respective C m (P = 0.239, Fig. 10 D). Although we did not investigate the size of the Ca2+ current in OHCs of CaV1.3−/− mice with electrophysiology, which in the presence of physiological 1.3 mm extracellular Ca2+ is likely to be in the order of a few picoamps (Michna et al. 2003), 2‐photon imaging experiments (Fig. 1 G and I) show that both apical and basal OHCs of CaV1.3−/− mice are completely deprived of any spontaneous Ca2+ signals. This indicates that, as for IHCs of CaV1.3−/− mice (Fig. 10), the residual Ca2+ current in OHCs is likely to be similar in the two cochlear locations, and as such is unlikely to contribute to the differential regulation of hair cell development.
Figure 10. Residual Ca2+ current in IHCs of CaV1.3−/− mice.
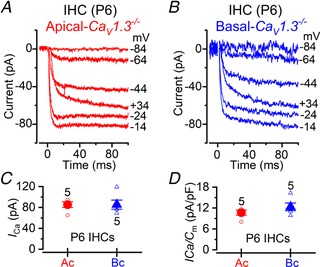
A and B, calcium currents recorded from apical (A) and basal (B) IHCs from P6 CaV1.3−/− mice. Currents were elicited by depolarizing voltage steps of 10 mV increments (100 ms in duration) starting from the holding potential of –84 mV. For clarity only some of the traces are shown. Actual test potentials, corrected for voltage drop across uncompensated R s, are shown next to the traces. Residual capacitative transients have been blanked. C, comparison of the peak I Ca in apical (circles) and basal (triangles) IHCs. D, same data as in panel C, but normalized to the IHC membrane capacitance (C m). Recordings were obtained near body temperature (34–37°C) and using 1.3 mm extracellular Ca2+.
Discussion
We have found that the intrinsic spiking activity driven by CaV1.3 Ca2+ channels in early postnatal hair cells differentially regulates their maturation along the cochlea. OHCs and IHCs acquire their characteristic mature profile from ∼P7 and ∼P12, respectively, with the upregulation of several basolateral membrane ion channels and, for OHCs, the motor protein prestin. Like IHCs (see below), immature OHCs transiently generate spontaneous Ca2+‐dependent APs under near‐physiological recording conditions. We found that these APs regulate the normal temporal acquisition of mature‐like basolateral membrane currents in low‐frequency apical hair cells. This includes I K,n, which is carried by KCNQ4 channels and the current associated with the inhibitory cholinergic efferent system (I ACh, carried by α9α10‐nAChRs, and I SK2) in OHCs, and I K,n and I K,f (BK channels) in IHCs (see also Brandt et al. 2003). OHC electromotility was not affected by the ablation of APs in vivo using CaV1.3−/− mice. Different from apical cells, the maturation of high‐frequency basal hair cells from CaV1.3−/− mice was found to be less dependent on Ca2+ APs, since mature‐like proteins were present in their basolateral membrane, but to a lesser degree compared to wild‐type mice. The hair cell size in CaV1.3−/− mice was significantly smaller than that of wild type, irrespectively of cochlear position. We propose that the development of low‐ and high‐frequency hair cells of the mammalian cochlea is differentially regulated during pre‐hearing stages, with apical hair cells being more strongly dependent on experience‐independent Ca2+ AP activity.
Action potentials in cochlear hair cells
In the mammalian cochlea, Ca2+‐dependent APs have been shown to occur spontaneously in immature IHCs (Kros et al. 1998; Johnson et al. 2011b, 2017; Sendin et al. 2014; Corns et al. 2018; Eckrich et al. 2018) when experiments are performed under near‐physiological recording conditions. Here we have shown that reliable and long‐duration recordings of AP activity in early postnatal OHCs require experimental conditions that preserve the in vivo intracellular milieu and Ca2+ levels (cell‐attached patch clamp). Under these conditions, APs in OHCs exhibit a higher frequency in the basal (∼2 Hz) than in the apical (∼0.8 Hz) turn of the cochlea, similar to previous observations from immature IHCs (Johnson et al. 2011b, 2017). These APs are driven by CaV1.3 Ca2+ channels, which represent > 90% of the Ca2+ channels expressed in hair cells (Platzer et al. 2000; Michna et al. 2003), but there are notable differences between the activity of IHCs and OHCs. While OHCs showed a bursting pattern irrespective of their cochlear location, IHCs exhibit an apex‐to‐base difference, with apical cells showing a bursting firing pattern and basal IHCs firing a more regular activity (Johnson et al. 2011b). These different AP characteristics between the two sensory cells could be due to the fact that only immature IHCs, but not OHCs, contribute directly to the firing activity of type I spiral ganglion neurons (Jones et al. 2007; Tritsch et al. 2010) and to the tonotopic refinement of sensory maps along the auditory pathway in the developing brainstem (Lippe, 1995; Clause et al. 2014). Furthermore, while AP activity in OHCs only occurs during the first few days after birth, that of IHCs persists throughout pre‐hearing stages of postnatal development (Johnson et al. 2012), reflecting the different roles of the two cell types in auditory pathway development.
Role of action potentials in the maturation of hair cells
Functionally mature OHCs possess both sensory and motor functions, which allows them to sharpen the frequency selectivity of IHCs along the cochlear partition (Ashmore, 2008). This requires specific biophysical and morphological properties that are rapidly acquired at the onset of maturation towards the end of the first postnatal week (Corns et al. 2014), 5 days before the IHCs mature at the onset of hearing at ∼P12 (Ehret, 1975). Crucial to their function is the acquisition of the negatively activating delayed rectifier K+ current I K,n, which flows through KCNQ4 channels (Kubisch et al. 1999), and in mice first appears at about P8 in OHCs (Marcotti & Kros, 1999) and at P12 in IHCs (Oliver et al. 2003; Marcotti et al. 2003a). OHC maturation is also associated with the onset of somatic motility at P7–P8 (Marcotti & Kros, 1999; Abe et al. 2007), which is driven by the motor protein prestin (Zheng et al. 2000; Liberman et al. 2002), and the formation of connections with cholinergic efferent and type II afferent fibres (Echteler, 1992; Simmons, 1994; Simmons et al. 1996). The medial olivocochlear (MOC) cholinergic efferent system represents the primary innervation of mature OHCs, and its correct function requires the expression of small conductance Ca2+‐activated K+ channels (SK2) and α9α10‐nAChRs on the postsynaptic membrane (Oliver et al. 2000; Lioudyno et al. 2004). Unlike the type I fibres contacting IHCs, type II afferent fibres seem to be activated only by sounds loud enough to cause acoustic trauma (Flores et al. 2015; Liu et al. 2015).
The development of sensory systems is normally influenced by transient periods of experience‐independent Ca2+ action potential activity occurring just before the onset of function (Katz & Shatz, 1996; Huberman et al. 2008; Blankenship & Feller 2010). This Ca2+‐dependent activity has also been shown to regulate changes in channel expression (Moody & Bosma, 2005). Here we show that immature OHCs transiently fire spontaneous Ca2+ APs during the first few days after birth, which can also be visualized as rapid Ca2+ signals under 2‐photon imaging (Fig. 1; see also Ceriani et al. 2019). In CaV1.3−/− mice, in which Ca2+ APs are abolished, we found impaired expression of the mature basolateral membrane ion channels that are crucial for hair cell function. In IHCs, the KCNQ4 and BK channels were absent in apical cells but expressed at a low level in basal IHCs. In apical OHCs, the KCNQ4 channels and those required postsynaptically for the efferent function (SK2 and α9α10‐nAChRs) were also largely reduced or absent. Although we could not provide a similar electrophysiological quantification for basal OHCs, DPOAE recordings (Fig. 8) indicated that the maturation of these cells is likely to be affected in CaV1.3−/− mice, although like basal IHCs, to a lesser extent than apical cells. It is well established that both the size of IHCs and OHCs in mice increases during development (Marcotti & Kros, 1999; Marcotti et al. 2003a), a process that is largely prevented in CaV1.3−/− mice irrespective of hair cell position along the cochlea. Since the size and kinetics of the residual Ca2+ current in IHCs of CaV1.3−/− mice are comparable between apical and basal cells, it is possible that the milder phenotype in high‐frequency IHCs is due to their development being under a ‘stronger’ activity‐independent genetic influence.
The substantially reduced number of efferent contacts on pre‐hearing OHCs of CaV1.3−/− mice, together with the reduced expression of the postsynaptic SK2 channels and α9α10‐nAChRs, could potentially be due to the absence of CaV1.3 channels in the auditory brainstem (Hirtz et al. 2011). However, this seems unlikely to be the case since the MOC neurons have been shown to make normal synaptic contacts with developing IHCs in P15 Cav1.3−/− mice (Glueckert et al. 2003). This indicates that the inability of OHC efferent synapses to mature is likely to be a direct consequence of an absence of AP activity during early stages of postnatal development. A reduction in the number of OHC synaptic ribbons has been linked to a decreased synchronization of AP activity between adjacent OHCs, which occurs when the electrical connectivity between non‐sensory cells in the cochlea is disrupted (Ceriani et al. 2019). The adult‐like characteristic of OHCs that was not influenced by spontaneous Ca2+ APs was their electromotility, suggesting that the expression of the motor protein prestin is most probably controlled by an intrinsic genetic programme. This is also supported by recent evidence that the transcription factor Ikzf2 is required for the normal expression of prestin (Chessum et al. 2018).
Although spontaneous Ca2+ AP activity has been shown to be crucial for hair‐cell maturation, genetic programmes seem to play a crucial role early on during their functional development. The combination of these two independent but highly regulated developmental mechanisms ensures that the highly specialized features of IHCs and OHCs are optimal to process sound information along the tonotopic axis of the mammalian cochlea.
OHCs of CaV1.3−/− mice degenerate with the onset of hearing
Our morphological (OHC size and bundle appearance) and physiological data (resting V m, electromotility and expression of the main Ca2+ binding protein oncomodulin) demonstrate that pre‐hearing OHCs from CaV1.3−/− mice, despite being ‘stuck’ at an intermediate developmental profile, are physiologically viable and healthy, and they only begin to rapidly degenerate upon the onset of hearing. A similar level of OHC degeneration, after the onset of hearing, has previously being reported for mutations in Tmc1 (Marcotti et al. 2006), one of the main candidates for the mechanoelectrical transducer channel (Kawashima et al. 2011; Corns et al. 2016; Ballesteros et al. 2018; Pan et al. 2018). Similar to our findings in CaV1.3−/− mice, an absent or mutated Tmc1 in OHCs does not affect their electromotility but does prevent them acquiring I K,n at P8, causing them to retain an immature basolateral current profile at hearing onset (Marcotti et al. 2006). I K,n plays a key role in mature OHCs since it provides the majority of the outward K+ current and is nearly fully activated (∼80%: Marcotti & Kros, 1999) at their resting V m (near −40 mV: Johnson et al. 2011a). As such, I K,n provides the major exit route for K+ ions that have entered OHCs via the opening of the mechanoelectrical transducer (MET) channels during sound stimulation. The reduced I K,n in OHCs of CaV1.3−/− mice would lead to intracellular K+ accumulation as soon as their mechanoelectrical transducer apparatus is stimulated by incoming sound, leading to their abrupt degeneration. In contrast to OHCs, IHCs of CaV1.3−/− mice do not degenerate until ∼P30 (Platzer et al. 2000), most likely because of their smaller resting MET current (Johnson et al. 2011a) and the fact that they still express a large outward K+ current.
We propose that the mammalian cochlea uses distinct mechanisms to orchestrate the maturation of the sensory epithelium during pre‐hearing stages of development. Intrinsic Ca2+‐dependent AP activity in OHCs is restricted to the first postnatal week and is required for the normal expression of the basolateral ion channels characteristic of mature apical, low‐frequency OHCs from ∼P7 onwards (I K,n, SK2 and α9α10‐nAChRs). Moreover, waves of Ca2+ activity in the non‐sensory greater epithelial ridge synchronize the AP firing of apical OHC, resulting in the refinement of their afferent innervation (Ceriani et al. 2019). We also found that low‐frequency, apical‐coil OHCs are more strongly dependent on experience‐independent Ca2+ AP activity than high‐frequency cells. Moreover, APs are not required for the functional acquisition of the highly specialized electromotile activity in OHCs, which is regulated by the transcription factor Ikzf2 (Chessum et al. 2018). The effect of Ca2+ activity on IHCs, which occurs throughout post‐natal prehearing stages (Johnson et al. 2011b, 2012), has functionally different consequences for the cochlea since it seems only to contribute to the maturation of the basolateral membrane properties of the cells themselves (Brandt et al. 2003; Johnson et al. 2013; Corns et al. 2018). However, similar to OHCs, the development of high‐frequency IHCs is also less affected by the absence of experience‐independent Ca2+ signals. The increase in hair cell size during development appears to be the only feature that is similarly regulated by early Ca2+ activity, irrespective of cell position along the cochlea. Finally, only the CaV1.3 Ca2+ channels, but not the channels responsible for the residual Ca2+ current in CaV1.3−/− mice (possibly CaV1.4: see Results section), exert a key role in the maturation of hair cells. Our results reveal that an extraordinary subtlety between genetic and physiological regulation is orchestrated to ensure the correct tonotopic differentiation of hair cells along the cochlea, allowing coherent functional development of the adult auditory system.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
The majority of the experiments were conducted at the University of Sheffield. Experiments for Fig. 4 were conducted at the University of Sussex and those for Fig. 7 I–K at Baylor University. J‐Y.J., F.C. A.H., S.L.J., P.Y., D.D.S., C.J.K. and W.M. were involved in the acquisition, analysis or interpretation of data for the work. J‐Y.J, F.C., C.J.K. and W.M. were involved in drafting the work or revising it critically for important intellectual content. W.M. conceived and designed the study. All authors approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Wellcome Trust to W.M. (102 892/Z/13/Z). C.J.K. was supported by the MRC (MR/K005561/1). S.L.J. is a Royal Society URF.
Acknowledgements
The authors thank Joerg Striessnig (University of Innsbruck) for providing the CaV1.3−/− mice, Robert Fettiplace (University of Wisconsin–Madison) for the prestin antibody, Michelle Bird (University of Sheffield) for her assistance with the transgenic mouse colonies, and Maria Pakendorf (University of Sheffield) for helping with the genotyping.
Biographies
Jing‐Yi Jeng received her MSc degree in biotechnology at National Tsing Hua University (Taiwan) and her PhD in biomedical science at The University of Sheffield (UK). Her research aims to understand the physiological mechanisms crucial for the development and ageing of the cochlear hair cells in the mammalian cochlea.

Federico Ceriani received his BSc and MSc in physics and PhD in neurobiology from the University of Padua (Italy). As a postdoctoral fellow, he was trained at the Venetian Institute of Molecular Medicine (VIMM) in Padua and at IBCN‐CNR in Rome (Italy). His research interests lie in the functional development of the hearing organ and signal processing by the auditory system. Currently, Drs Jeng and Ceriani are Postdoctoral Research Associates within the Hearing Research Group (https://www.sheffield.ac.uk/hearing) in the Department of Biomedical Science at The University of Sheffield (UK).

J.‐Y. Jeng and F. Ceriani contributed equally to the work.
Edited by: Ian Forsythe & Florian Lesage
Linked articles: This article is highlighted in a Perspectives article by Ashmore. To read this article, visit https://doi.org/10.1113/JP279215.
This is an Editor's Choice article from the 1 January 2020 issue.
References
- Abe T, Kakehata S, Kitani R, Maruya S, Navaratnam D, Santos‐Sacchi J & Shinkawa H (2007). Developmental expression of the outer hair cell motor prestin in the mouse. J Membr Biol 215, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore J (2008). Cochlear outer hair cell motility. Physiol Rev 88, 173–210. [DOI] [PubMed] [Google Scholar]
- Ashmore JF (1987). A fast motile response in guinea‐pig outer hair cells: the cellular basis of the cochlear amplifier. J Physiol 388, 323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros A, Fenollar‐Ferrer C & Swartz KJ (2018). Structural relationship between the putative hair cell mechanotransduction channel TMC1 and TMEM16 proteins. Elife 7, e38433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C & Dulon D (2008). Calcium‐ and otoferlin‐dependent exocytosis by immature outer hair cells. J Neurosci 28, 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG & Feller MB (2010). Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci 11, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Striessnig J & Moser T (2003). CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci 23, 10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell WE, Bader CR, Bertrand D & de Ribaupierre Y (1985). Evoked mechanical responses of isolated cochlear outer hair cells. Science 227, 194–196. [DOI] [PubMed] [Google Scholar]
- Ceriani F, Hendry A, Jeng J‐Y, Johnson S, Stephani F, Olt J, Holley MC, Mammano F, Engel J, Kros CJ, Simmons DD & Marcotti W (2019). Spontaneous and coordinated Ca2+ activity of cochlear sensory and non‐sensory cells drives the maturation of OHC afferent innervation. EMBO J 38, e99839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessum L, Matern MS, Kelly MC, Johnson SL, Ogawa Y, Milon B, McMurray M, Driver EC, Parker A, Song Y, Codner G, Esapa CT, Prescott J, Trent G, Wells S, Dragich AK, Frolenkov GI, Kelley MW, Marcotti W, Brown SDM, Elkon R, Bowl MR & Hertzano R (2018). Helios is a key transcriptional regulator of outer hair cell maturation. Nature 563, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rűbsamen R & Kandler K (2014). The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corns LF, Bardhan T, Houston O, Olt J, Holley MC, Masetto S, Johnson SL & Marcotti W (2014). Functional development of hair cells in the mammalian inner ear In Development of Auditory and Vestibular Systems, eds Romand R. & Varela‐Nieto I, pp. 155–188. Academic Press, New York. [Google Scholar]
- Corns LF, Johnson SL, Kros CJ & Marcotti W (2016). Tmc1 point mutation affects Ca2+ sensitivity and block by dihydrostreptomycin of the mechanoelectrical transducer current of mouse outer hair cells. J Neurosci 36, 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corns LF, Johnson SL, Roberts T, Ranatunga KM, Hendry A, Ceriani F, Safieddine S, Steel KP, Forge A, Petit C, Furness DN, Kros CJ & Marcotti W (2018). Mechanotransduction is required for establishing and maintaining mature inner hair cells and regulating efferent innervation. Nature Comm 9, 4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P (1992). The active cochlea. J Neurosci 12, 4575–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echteler SM (1992). Developmental segregation in the afferent projections to mammalian auditory hair cells. Proc Natl Acad Sci U S A 89, 6324–6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckrich S, Hecker D, Sorg K, Blum K, Fischer K, Münkner S, Wenzel G, Schick B & Engel J (2019). Cochlea‐specific deletion of Cav1.3 calcium channels arrests inner hair cell differentiation and unravels pitfalls of conditional mouse models. Front Cell Neurosci 13, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckrich T, Blum K, Milenkovic I & Engel J (2018). Fast Ca2+ transients of inner hair cells arise coupled and uncoupled to Ca2+ waves of inner supporting cells in the developing mouse cochlea. Front Mol Neurosci 11, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G (1975). Masked auditory thresholds, critical ratios, and scales of the basilar membrane of the housemouse (Mus musculus). J Comp Physiol 103, 329–341. [Google Scholar]
- Flores EN, Duggan A, Madathany T, Hogan AK, Márquez FG, Kumar G, Seal RP, Edwards RH, Liberman MC & García‐Añoveros J (2015). A non‐canonical pathway from cochlea to brain signals tissue‐damaging noise. Curr Biol 25, 606‐‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E & Fuchs PA (2000). Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288, 2366–2368. [DOI] [PubMed] [Google Scholar]
- Glueckert R, Wietzorrek G, Kammen‐Jolly K, Scholtz A, Stephan K, Striessnig J & Schrott‐Fischer A (2003). Role of class D L‐type Ca2+ channels for cochlear morphology. Hear Res 178, 95–105. [DOI] [PubMed] [Google Scholar]
- Goutman JD, Elgoyhen AB & Gómez‐Casati ME (2015). Cochlear hair cells: the sound‐sensing machines. FEBS Lett 589, 3354–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL & Greenberg ME (2008). From synapse to nucleus: calcium‐dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. [DOI] [PubMed] [Google Scholar]
- Guinan JJ Jr (1996). Physiology of olivocochlear efferents In The Cochlea, eds Dallos P, Popper AN. & Fay RR, pp. 435–502. Springer, New York. [Google Scholar]
- Guinan JJ, Warr WB & Norris BE (1983). Differential olivocochlear projections from lateral vs medial zones of the superior olivary complex. J Comp Neurol 221, 358–370. [DOI] [PubMed] [Google Scholar]
- He DZ, Evans BN & Dallos P (1994). First appearance and development of electromotility in neonatal gerbil outer hair cells. Hear Res 78, 77–90. [DOI] [PubMed] [Google Scholar]
- Helyer RJ, Kennedy HJ, Davies D, Holley MC & Kros CJ (2005). Development of outward potassium currents in inner and outer hair cells from the embryonic mouse cochlea. Audiol Neurootol 10, 22–34. [DOI] [PubMed] [Google Scholar]
- Hirtz JJ, Boesen M, Braun N, Deitmer JW, Kramer F, Lohr C, Müller B, Nothwang HG, Striessnig J, Löhrke S & Friauf E (2011). Cav1.3 calcium channels are required for normal development of the auditory brainstem. J Neurosci 31, 8280–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Feller MB & Chapman B (2008). Mechanisms underlying development of visual maps and receptive fields. Annu Rev Neurosci 31, 479–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki A, Ugawa S, Yamamura H, Murakami S & Shimada S (2008). The CaV3.1 T‐type Ca2+ channel contributes to voltage‐dependent calcium currents in rat outer hair cells. Brain Res 1201, 68–77. [DOI] [PubMed] [Google Scholar]
- Ingham NJ, Pearson S & Steel KP (2011). Using the Auditory Brainstem Response (ABR) to determine sensitivity of hearing in mutant mice. Curr Protoc Mouse Biol 1, 279–287. [DOI] [PubMed] [Google Scholar]
- Johnson SL, Beurg M, Marcotti W & Fettiplace R (2011a). Prestin‐driven cochlear amplification is not limited by the outer hair cell membrane time constant. Neuron 70, 1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Ceriani F, Houston O, Polishchuk R, Polishchuk E, Crispino G, Zorzi V, Mammano F & Marcotti W (2017). Connexin‐mediated signaling in nonsensory cells is crucial for the development of sensory inner hair cells in the mouse cochlea. J Neurosci 37, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ & Marcotti W (2011b). Position dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci 14, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Kennedy HJ, Holley MC, Fettiplace R & Marcotti W (2012). The resting transducer current drives spontaneous activity in prehearing mammalian cochlear inner hair cells. J Neurosci 32, 10479–10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W & Kros CJ (2005). Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol 563, 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Wedemeyer C, Vetter DE, Adachi R, Holley MC, Elgoyhen AB & Marcotti W (2013). Cholinergic efferent synaptic transmission regulates the maturation of auditory hair cell ribbon synapses. Open Biol 3, 130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Leake PA, Snyder RL, Stakhovskaya O & Bonham B (2007). Spontaneous discharge patterns in cochlear spiral ganglion cells before the onset of hearing in cats. J Neurophysiol 98, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E & Elgoyhen AB (2014). Short‐term plasticity and modulation of synaptic transmission at mammalian inhibitory cholinergic olivocochlear synapses. Front Syst Neurosci 8, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gomez‐Casati ME, Knipper M, Vetter DE, Fuchs PA & Glowatzki E (2004). Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci 24, 7814–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC & Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Ge´le´oc GS, Kurima K, Labay V, Lelli A, Asai Y, Makishima T, Wu DK, Della Santina CC, Holt JR & Griffith AJ (2011) Mechanotransduction in mouse inner ear hair cells requires transmembrane channel like genes. J Clin Invest 121, 4796–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP & Rüsch A (1998). Expression of a potassium current in inner hair cells during development of hearing in mice. Nature 394, 281–284. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lütjohann B, El‐Amraoui A, Marlin S, Petit C & Jentsch TJ (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446. [DOI] [PubMed] [Google Scholar]
- Levic S & Dulon D (2012). The temporal characteristics of Ca2+ entry through L‐type and T‐type Ca2+ channels shape exocytosis efficiency in chick auditory hair cells during development. J Neurophysiol 108, 3116–3123. [DOI] [PubMed] [Google Scholar]
- Liberman MC (1980). Efferent synapses in the inner hair cell area of the cat cochlea: an electron microscopic study of serial sections. Hear Res 3, 189–204. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S & Zuo J (2002). Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–304. [DOI] [PubMed] [Google Scholar]
- Lioudyno M, Hiel H, Kong JH, Katz E, Waldman E, Parameshwaran‐Iyer S, Glowatzki E & Fuchs PA (2004). A “synaptoplasmic cistern” mediates rapid inhibition of cochlear hair cells. J Neurosci 24, 11160–11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippe WR (1995). Relationship between frequency of spontaneous bursting and tonotopic position in the developing avian auditory system. Brain Res 703, 205–213. [DOI] [PubMed] [Google Scholar]
- Liu C, Glowatzki E & Fuchs PA (2015). Unmyelinated type II afferent neurons report cochlear damage. Proc Natl Acad Sci U S A 112, 14723–14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Adams JC & Liberman MC (2003). Olivocochlear innervation in the mouse: immunocytochemical maps, crossed versus uncrossed contributions and transmitter colocalization. J Comp Neurol 455, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Erven A, Johnson SL, Steel KP & Kros CJ (2006). Tmc1 is necessary for normal functional maturation and survival of inner and outer hair cells in the mouse cochlea. J Physiol 574, 677–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Géléoc GS, Lennan GW & Kros CJ (1999) Transient expression of an inwardly rectifying potassium conductance in developing inner and outer hair cells along the mouse cochlea. Pflugers Arch 439, 113–122. [DOI] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Holley MC & Kros CJ (2003a). Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol 548, 383–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL & Kros CJ (2004a). A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol 560, 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL & Kros CJ (2004b). Effects of intracellular stores and extracellular Ca2+ on Ca2+‐activated K+ currents in mature mouse inner hair cells. J Physiol 557, 613–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Rusch A & Kros CJ (2003b). Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol 552, 743–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W & Kros CJ (1999). Developmental expression of the potassium current I K,n contributes to maturation of mouse outer hair cells. J Physiol 520, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michna M, Knirsch M, Hoda JC, Muenkner S, Langer P, Platzer J, Striessnig J & Engel J (2003). Cav1.3 (α1D) Ca2+ currents in neonatal outer hair cells of mice. J Physiol 553, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody WJ & Bosma MM (2005). Ion channel development, spontaneous activity, and activity‐dependent development in nerve and muscle cells. Physiol Rev 85, 883–941. [DOI] [PubMed] [Google Scholar]
- Müller M, von Hünerbein K, Hoidis S & Smolders JW (2005). A physiological place‐frequency map of the cochlea in the CBA/J mouse. Hear Res 202, 63–73. [DOI] [PubMed] [Google Scholar]
- Oliver D, Klöcker N, Schuck J, Baukrowitz T, Ruppersberg JP & Fakler B (2000). Gating of Ca2+‐activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26, 595–601. [DOI] [PubMed] [Google Scholar]
- Oliver D, Knipper M, Derst C & Fakler B (2003). Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ‐type potassium channels. J Neurosci 23, 2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Plinkert P, Zenner HP & Ruppersberg JP (1997). Sodium current expression during postnatal development of rat outer hair cells. Pflugers Archiv 434, 772–778. [DOI] [PubMed] [Google Scholar]
- Pan B, Akyuz N, Liu XP, Asai Y, Nist‐Lund C, Kurima K, Derfler BH, György B, Limapichat W, Walujkar S, Wimalasena LN, Sotomayor M, Corey DP & Holt JR (2018). TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron 99, 736–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Singer JH & Koschak A (2018) Voltage‐gated calcium channels: key players in sensory coding in the retina and the inner ear. Physiol Rev 98, 2063–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott‐Fischer A, Stephan K, Bova S, Chen H, Zheng H & Striessnig J (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Pujol R, Lavigne‐Rebillard M & Lenoir M (1998) Development of sensory and neural structures in the mammalian cochlea In Development of the Auditory System, eds Rubel EW, Popper AN. & Fay RR, pp. 146–192. Springer, New York. [Google Scholar]
- Pyott SJ, Glowatzki E, Trimmer JS & Aldrich RW (2004). Extrasynaptic localization of inactivating calcium‐activated potassium channels in mouse inner hair cells. J Neurosci 24, 9469–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P & Cardona A (2012). Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendin G, Bourien J, Rassendren F, Puel JL & Nouvian R (2014). Spatiotemporal pattern of action potential firing in developing inner hair cells of the mouse cochlea. Proc Natl Acad Sci U S A 111, 1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DD (1994). A transient afferent innervation of outer hair cells in the postnatal cochlea. Neuroreport 5, 1309–1312. [PubMed] [Google Scholar]
- Simmons DD, Mansdorf NB & Kim JH (1996). Olivocochlear innervation of inner and outer hair cells during postnatal maturation: evidence for a waiting period. J Comp Neurol 370, 551–562. [DOI] [PubMed] [Google Scholar]
- Simmons DD, Tong B, Schrader AD & Hornak AJ (2010). Oncomodulin identifies different hair cell types in the mammalian inner ear. J Comp Neurol 518, 3785–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E & Bergles DE (2007). The origin of spontaneous activity in the developing auditory system. Nature 450, 50–55. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Rodríguez‐Contreras A, Crins TT, Wang HC, Borst JG & Bergles DE (2010). Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat Neurosci 13, 1050–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz CJ, Lehar M, Hiel H, Glowatzki E & Fuchs PA (2012). Synaptic transfer from outer hair cells to type II afferent fibers in the rat cochlea. J Neurosci 32, 9528–9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD & Dallos P (2000). Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155. [DOI] [PubMed] [Google Scholar]