

Abstract
Pyropia haitanensis (Bangiales, Rhodophyta), a major economically important marine crop, is also considered as an ideal research model of Rhodophyta to address several major biological questions such as sexual reproduction and adaptation to intertidal abiotic stresses. However, comparative genomic analysis to decipher the underlying molecular mechanisms is hindered by the lack of high‐quality genome information. Therefore, we integrated sequencing data from Illumina short‐read sequencing, PacBio single‐molecule sequencing and BioNano optical genome mapping. The assembled genome was approximately 53.3 Mb with an average GC% of 67.9%. The contig N50 and scaffold N50 were 510.3 kb and 5.8 Mb, respectively. Additionally, 10 superscaffolds representing 80.9% of the total assembly (42.7 Mb) were anchored and orientated to the 5 linkage groups based on markers and genetic distance; this outcome is consistent with the karyotype of five chromosomes (n = 5) based on cytological observation in P. haitanensis. Approximately 9.6% and 14.6% of the genomic region were interspersed repeat and tandem repeat elements, respectively. Based on full‐length transcriptome data generated by PacBio, 10,903 protein‐coding genes were identified. The construction of a genome‐wide phylogenetic tree demonstrated that the divergence time of P. haitanensis and Porphyra umbilicalis was ~204.4 Ma. Interspecies comparison revealed that 493 gene families were expanded and that 449 were contracted in the P. haitanensis genome compared with those in the Po. umbilicalis genome. The genome identified is of great value for further research on the genome evolution of red algae and genetic adaptation to intertidal stresses.
Keywords: comparative genomic analysis, genome annotation, genome assembly, Pyropia haitanensis, repeat annotation, whole‐genome sequencing
1. INTRODUCTION
Red algae (Rhodophyta) are an ancient eukaryotic group that extended back to 1.6–1.0 billion years ago according to the observation of the cellular and subcellular structures of multicellular rhodophytes Rafatazmia and Ramathallus in fossils using synchrotron radiation X‐ray tomographic microscopy (Bengtson, Sallstedt, Belivanova, & Whitehouse, 2017). Red algae comprise a monophyletic lineage of ~7,200 photosynthetic species, which belong to the Archaeplastida (Plantae) derived from primary endosymbiosis (Yoon, Müller, Sheath, Ott, & Bhattacharya, 2006). The secondary and tertiary endosymbiosis of red algae have given rise to the most abundant, species‐rich and ecologically significant groups of algae and other eukaryotes present on Earth today, such as cryptophytes, haptophytes, apicomplexans, stramenopiles and dinoflagellates (Archibald, 2012; Hoek, Mann, Jahns, & Jahns, 1995; Reyes‐Prieto, Weber, & Bhattacharya, 2007). Genomic studies on red algae will provide valuable information on the evolution of oxygenic photosynthesis. Unfortunately, only a limited number of whole‐genome data sets for red algae have been reported, including those for the hot‐spring alga Cyanidioschyzon merolae, the mesophilic alga Porphyridium purpureum, the extremophilic alga Galdieria sulphuraria, as well as the multicellular red seaweeds Chondrus crispus, Gracilariopsis chorda and Porphyra umbilicalis (Bhattacharya et al., 2013; Brawley et al., 2017; Collén et al., 2013; Lee et al., 2018; Nozaki et al., 2007). The genomic information of Pyropia haitanensis would help to reveal the adaptation mechanisms of intertidal seaweeds and help to reconstruct the evolutionary history of red algae.
In Rhodophyta, several species of the genus Pyropia (previously named Porphyra, and commonly called “nori”) are well known for their economic value in the seaweed industry, such as P. haitanensis, P. yezoensis and P. tenera (Sutherland et al., 2011). According to the FAO's statistics, nori production in the year 2016 was ~1.8 million tons in fresh weight with a commercial value over 1.5 billion USD (http://www.fao.org/fishery/factsheets/en). Pyropia haitanensis is a native species distributed along the coastline of south China. This species is cultivated at a large scale with the highest annual production among all the nori species. The current total annual harvest of P. haitanensis is ~88,000 tons (dry weight), which accounts for approximately 75% and more than 50% of the total nori production in China and the world, respectively (Guo et al., 2018). With the aid of a high‐quality genome of P. haitanensis, modern molecular genetic techniques such as QTL mapping and GWAS will be used to identify the key loci of the important economic traits such as productivity, taste and colour, which undoubtedly will enhance the efficiency of molecular breeding of this economically important marine crop.
Pyropia haitanensis naturally inhabits a niche in the upper region of the intertidal zone (Sahoo, Tang, & Yarish, 2002). Routine tidal turning periodically exposes it to the air, and it inevitably experiences the drastic changes in environmental factors such as osmotic pressure, temperature, light and UV radiation (Blouin, Brodie, Grossman, Xu, & Brawley, 2011). P. haitanensis can survive even after losing 85%–95% of its cellular water (Wang, Mao, Kong, Cao, & Sun, 2015). The thriving nature of P. haitanensis suggested that long‐term evolutionary selection has made this species highly adaptable to the combined harsh stresses of the intertidal region. Thereby, this species is considered a model of intertidal red seaweed for physiology and genetic research on stress tolerance. Due to its distinctive evolutionary position in the red algal clade, P. haitanensis might harbour different genetic mechanisms of stress tolerance from those of high plants, which are probably derived from green algae. The genome information of P. haitanensis is a valuable source for the identification of unique genetic signatures involved in environmental adaptation.
Furthermore, genome sequences of P. haitanensis with relatively higher integrity and completeness are unavailable, which has been one of the major constraints to improve research on the physiology, cytology, genetics and genomics of Pyropia. Currently, the development of high‐throughput sequencing technologies for sequencing DNA, RNA and proteins has reduced sequencing time and cost, etc. Hitherto, there are already four generations. Every sequencing generation and its relevant sequencing platforms have advantages and disadvantages. Thus, it is necessary to assess their limitations and applications. Second‐generation sequencing is currently the most common because of its higher throughput, but the short‐read lengths and amplification biases have become disadvantages (Ari & Arikan, 2016). Single‐molecule real‐time (SMRT) is another sequencing technology that is currently in use, which can overcome the short‐read lengths and biases without any amplification step (Roberts, Carneiro, & Schatz, 2013). The appearance of an optical map can further place short reads on genomic fragments, even those totalling several millions of bases (Neely, Deen, & Hofkens, 2011). Hence, in this study, the combined techniques of Illumina short‐read sequencing, PacBio single‐molecule sequencing and BioNano optical mapping were used to assemble the genome of P. haitanensis. Subsequently, gene prediction, repeat annotation, functional annotation, gene family expansion and contraction, and phylogenetic relationship were determined according to standard procedures to elucidate the gene repertoire of P. haitanensis.
2. MATERIALS AND METHODS
2.1. Sample information
A laboratory‐cultured genetically pure line, Pyropia haitanensis PH40 (♀), was used in this study to eliminate the interference caused by genotypic differences. The original thallus was collected from a nori farm in Putian, Fujian Province, China. The material was first identified by amplification of its 18S rRNA gene as described in a previous study (Müller, Sheath, Vis, Crease, & Cole, 1998), as well as by its morphologies. Single somatic cells were enzymatically isolated from the thallus, and the allele homozygous sporophytes (conchocelis) were obtained after the haploid doubling spontaneously. The genetically homogenous gametophytes were then developed from the homozygous sporophytes and cultured for DNA and RNA sample collection. Another strain PH37 (♂) used in this study was also harvested from Putian, Fujian Province, China, and purified with the same method mentioned above. The gametophytes were cultured in a light incubator under the following conditions: 20 ± 1°C with 50–60 μmol photons·m−2·s−1 illumination during a 12 h:12 h light:dark cycle. The culture medium of Provasoli's enriched seawater (PES) (Starr, 1987) was refreshed every five days. To remove surface bacteria from P. haitanensis gametophytes, the thalli were harvested and mixed with quartz sands. Physical vibration was carried out in a homogenizer (Precellys 24), followed by several rounds of washing to remove the polysaccharides and bacteria from the surface of gametophytes. Subsequently, the samples were collected and immediately frozen in liquid nitrogen for total genomic DNA extraction using the CTAB method (Yang, Wang, Liu, & An, 1999).
2.2. Libraries construction
Five micrograms and 10 μg of genomic DNA were used to construct Illumina TruSeq paired‐end sequencing libraries (500‐bp insert sizes) and mate pair libraries (5 kb in size), respectively, according to the manufacturer's instructions. Meanwhile, a total of 10 μg of DNA was used to construct a 20‐kb library using the PacBio Pacific Biosciences SMRT Bell Template Kit 1.0. To further carry out optical map construction, 2 μg of purified high molecular weight (HMW) genomic DNA was isolated and labelled according to standard BioNano protocols with the single‐stranded nicking endonuclease BspQI. To assist in the genome annotation of P. haitanensis, total RNAs isolated from various stressful conditions (osmotic pressure, temperature, illumination, etc.) were equally mixed together to prepare the transcriptome sequencing libraries for SMRT platforms following the manufacturer's instructions. For SMRT sequencing, full‐length RNA libraries were constructed according to the manufacturer's instructions with minor modifications. To avoid overamplification of small fragments, we optimized the amplification cycle at 14 in a preliminary test. Then, three gel fractions, containing fragments >3, 2–3 and 1‐2 kb, were collected and purified using the QIAquick Gel Extraction Kit. The extracted products were amplified using the 5′ Primer IIA and purified using 0.5 × AMPure beads (#A63880; Beckman, http://www.beckmancoulter.com) for subsequent sequencing.
2.3. Genome sequencing and assembly
To estimate the genome size of P. haitanensis, the low‐quality reads and sequences aligning to the chloroplast (Accession no: KC464603) and mitochondrion (NC_017751) genomes of P. haitanensis were removed using the NGS QC Toolkit and Bowtie 2 (parameters: ‐very‐sensitive; version: 2.0.2) (Langmead, Trapnell, Pop, & Salzberg, 2009). Different K‐mer frequencies were calculated by Jellyfish and genome size (Luo et al., 2012). For genome assembly, subreads from PacBio were used to assemble the nuclear genome of P. haitanensis using the RS_HGAP_Assembly.3 protocol in smrt analysis v2.3.0 with default parameters (Chin et al., 2013). Then, mate pair data sets were aligned to the above‐assembled contigs using SSPACE (Boetzer, Henkel, Jansen, Butler, & Pirovano, 2010). Meanwhile, PacBio long reads were mapped to the scaffold sequences using BLASR, and the gaps that resulted from the scaffolds were filled using PBJelly2 with default parameters (English et al., 2012). Finally, Quiver was run again to polish the accurate consensus at the base level.
To improve the assembly, optical maps of the BioNano system were further used for scaffolding. A labelled DNA sample was loaded onto the Saphyr Chip nanochannel array, and the stretched DNA molecules were then imaged with the BioNano Saphyr system. Raw image data were converted into bnx files, and AutoDetect (BioNano Genomics) software generated basic labelling and DNA length information. Access (BioNano Genomics) software was used to filter and remove <150 Kb low‐quality reads, and then, IrySolve (BioNano Genomics) was used to carry out the assembly of BioNano's genome maps and the ‘Hybrid Scaffold' between genome maps from BioNano and sequence maps. Further gap filling using the reads that not used in the last step was achieved by RefAligner (BioNano Genomics). To remove the potential contamination of bacterial sequences in the current assembly, we applied a postprocessing step. We cut each scaffold into 100 bp overlapping 1‐Kb windows and blasted them against the NT database using BLASTn. The blast results were further analysed using MEGAN to search for bacterial hits. Scaffolds that met the following three criteria were considered to be bacterial contamination and removed from the final genome: (a) over 60% of windows in the scaffold had best hits as bacterial sequences with identity >70%; (b) the sequencing depth was <5; and (c) there was no cDNA support in these ‘bacterial windows.’ To assess the quality of the assembled genome, K‐mer frequency distribution, the full‐length transcriptome sequencing data map rate and Benchmarking Universal Single‐Copy Orthologs (BUSCO) analysis were used.
2.4. Genetic map construction and scaffold anchoring
To construct a genetic map of P. haitanensis, the gametophytic blades of PH40 (♀) and PH37 (♂) were selected as parents for crossing experiments. The blades from these two pure lines were cocultured in a flask until carposporangia appeared. Then, the fertilized female blade was selected and cultured until reproductive cells were released. Subsequently, the fertilized carpospores were cultured to generate heterozygous conchocelis. The heterozygote was then confirmed using two SSR markers in our laboratory. After confirmation, the heterozygous gametophytes (F1) were then developed from the homozygous conchocelis and used to establish double haploid populations (DH). Each individual F1 gametophyte was digested into single cells using snail enzymes. Then, a single cell from each gametophyte was picked out and cultured to conchocelis. The cultured conditions were the same as those described above. Finally, a population with 117 DH strains was established and used for genetic map construction. Genomic DNA from two parents and 117 offspring were extracted using the CTAB method. DNA quality was detected with 0.8% agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer. Then, 119 2b‐RAD libraries were constructed according to the protocols described by Wang et al. (Wang, Meyer, McKay, & Matz, 2012). These libraries were sequenced on an Illumina HiSeq system to generate single‐end reads with a length of 50bp. Subsequently, reads were trimmed to remove sequences with adapters, those without restriction sites and those containing ambiguous bases and of low‐quality value. Meanwhile, sequence reads from putative plastid and mitochondrial origins of P. haitanensis were also removed. The remaining reads were analysed using the RADtyping program v1.0 with default parameters (Fu et al., 2013) for genotyping. The markers that could be genotyped in at least 80% of offspring were used to calculate the genetic distance and draw linkage maps using JoinMap 4.0 at LOD 7.0 (Van Ooijen, 2006). The linkage group numbers were selected at a LOD threshold of more than 4.0. Meanwhile, genetic distances between markers and marker sequences were used to anchor scaffolds to the linkage groups using the R package.
2.5. Repeat elements
Repeat elements occupy a major proportion of the nuclear DNA in most eukaryotic genomes and have been demonstrated to have structural and functional roles (Biscotti, Olmo, & Heslop‐Harrison, 2015). repeatmodeler (version: 1.0.8) was used to analyse consensus sequences of interspersed repeats in genomes of P. haitanensis (Smit & Hubley, 2008). Consensus sequences that were shorter than 80 bp were discarded (Wicker et al., 2007). The remaining consensus sequences were used as the library in repeatmasker (version: open‐4‐0‐7) to predict interspersed repeat elements in the whole genome (Chen, 2004). Meanwhile, Tandem Repeats Finder (Benson, 1999) was used to identify tandem repeat sequences in P. haitanensis genome.
2.6. Gene prediction and functional annotation
After repeats' masking, we used a combination of de novo prediction, homology searches and transcript isoform based methods to predict gene structures of P. haitanensis. De novo prediction was performed using AUGUSTUS (Stanke et al., 2006). For homologous annotation, we queried the P. haitanensis genome scaffolds against a database containing protein sequences from five organisms (Chondrus crispus, Gracilariopsis chorda, Cyanidioschyzon merolae, Po. umbilicals and Porphyridium purpureum). At the same time, transcript isoforms of P. haitanensis were mapped to the genome using blast and then assembled by PASA (Haas et al., 2008). Finally, EVM was used to integrate these gene models from the above methods. To further detect the function of the protein‐coding genes in P. haitanensis, the predicted protein sequences were aligned against several public databases (NR, InterPro, GO, KOG, KEGG, CAZyme and Conserved Domains Database [CDD]).
2.7. Gene family expansion and contraction
To further examine the genome divergence and conservation among red algae, we carried out a phylogenetic analysis based on single‐copy orthologous groups using the P. haitanensis genome and other five red algal genomes to build orthologous genes using orthomcl (Li, Stoeckert, & Roos, 2003), with Cyanophora paradoxa as the outgroup species. Genome sequences were aligned using the program mafft version 5 (Katoh, Kuma, Toh, & Miyata, 2005) and were further trimmed using trimAl with the option “automated1” (Capella‐Gutiérrez, Silla‐Martínez, & Gabaldón, 2009). Maximum likelihood (ML) analyses were conducted using raxml‐8.2.4 (Stamatakis, 2014). The best model and parameter settings were chosen according to the Akaike information criterion using prottest 3.0 (Abascal, Zardoya, and Posada 2005). A Bayesian phylogenetic tree was constructed using mrbayes 3.2 under the same model (Huelsenbeck & Ronquist, 2001). Four incrementally heated Metropolis‐coupled Monte Carlo Markov chains were run for 10,000,000 generations for the concatenated data set, and runs were sampled every 1000th generation. Convergence and stationarity of the log‐likelihood and parameter values were assessed using tracer v.1.5 (Rambaut, Drummond, Xie, Baele, & Suchard, 2018). The initial 10% were discarded as burn‐in. A time‐calibrated phylogeny was inferred using a relaxed molecular clock method as implemented in beast v.1.8.3 (Drummond, Suchard, Xie, & Rambaut, 2012). We set the most recent common ancestor with a lognormal prior, an offset of 950 Ma, and a standard deviation of 25.0 based on the divergence of Florideophyceae and Bangiophyceae (Herron, Hackett, Aylward, & Michod, 2009; Yang et al., 2016).
3. RESULTS AND DISCUSSION
3.1. Material identification and Genome assembly
The material used in this study was identified as Pyropia haitanensis according to its morphology, life history, as well as its reproductive structure, etc (Figure 1). The blade was 15–16 cm in length and 2–3 cm in width, with a red to brown colour. Additionally, it had an umbilicate base, which can help the blade attach to substratum. The molecular marker and alignment results also supported identification of the specimen as P. haitanensis (Figure S1). Scanning electron microscopy showed that bacteria had been removed from the surface of the algae (Figure S2). And a total of ~22.1 Gb of raw sequence data were obtained using the Illumina platform for P. haitanensis. Based on calculation of the K‐mer frequency by Jellyfish, the estimated genome size of P. haitanensis was approximately 38.5 Mb (Table S1). For genome assembly, ~5.0 Gb of subreads from the PacBio RSII platform with a mean length of 5.7 kb were used to assemble the nuclear genome of P. haitanensis. A 59.7 Mb assembly was produced consisting of 1,839 contigs with an N50 of 510.3 kb. Then, the number of scaffolds built based on ~1.8 Gb of Illumina mate pair sequencing data was reduced to 1,168 and the length of N50 increased to 913.7 kb. Scaffolding using PacBio long reads allowed us to improve the assembly to 663 scaffolds (totalling 59.2 Mb) with a scaffold N50 of 912.3 kb. For optical map construction, a total of 93.8 Gb of molecular data were obtained (Table 1). Combined with optical mapping data, we finally yielded a P. haitanensis genome with a size of 53.3 Mb. Among the 195 scaffolds, 11 pseudomolecules had lengths larger than 0.4 Mb and covered 88.4% of the genome region. The contig N50 and scaffold N50 were 510.3 kb and 5.8 Mb, respectively, and the length of the longest scaffold was 7.6 Mb (Table 2). The average GC content of this genome was as high as 67.9%, which is the highest among all the published algal genomes. The phenomenon of high GC content was also found in the Bangiophyceae species Po. umbilicalis (65.8%) (Brawley et al., 2017) and green algae Chlamydomonas reinhardtii (64%) (Merchant et al. 2007). Compared with the assembly results of the published macroalgae, including Chondrus crispus (scaffold N50 = 240.0 kb), Po. umbilicalis (scaffold N50 = 202.0 kb) and Saccharina japonica (scaffold N50 = 252.0 kb), the assembly of P. haitanensis genome had the fewest scaffolds and the longest N50 and the highest contiguity and coverage (Ye et al., 2015).
Figure 1.
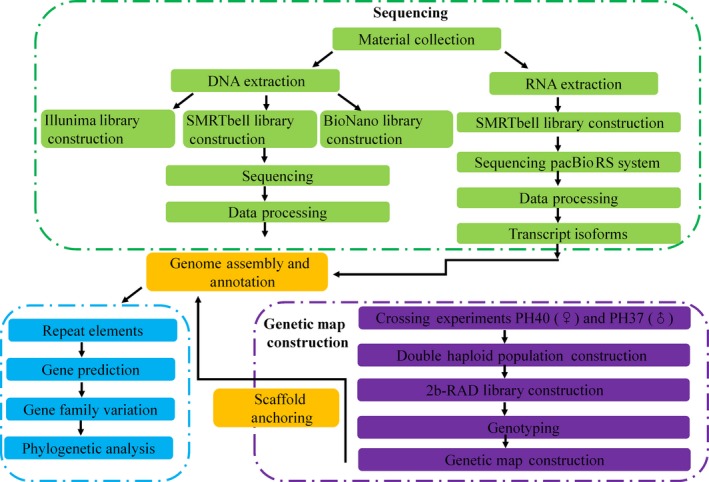
A workflow for the genome sequencing and genetic map construction
Table 1.
Genome and transcriptome sequencing information of Pyropia haitanensis
| Sequencing platforms | Library size | Data size (Gb) | Depth | |
|---|---|---|---|---|
| DNA library | Illumina | 500 bp | 22.1 | 220 |
| Illumina | 5 kb | 1.8 | 47 | |
| PacBio | 20 kb | 6.4 | 99 | |
| BioNano | ~ | 93 | 1,860 | |
| RNA library | PacBio | 1–2 kb | 1.5 | 12 |
| 2–3 kb | 1.3 | 12 | ||
| >3 kb | 1.5 | 12 |
Table 2.
Statistics of the final assembly of Pyropia haitanensis genome
| Contig | Scaffold | BioNano | |
|---|---|---|---|
| Total sequences | 1,497 | 230 | 195 |
| Total bases | 57,754,774 | 50,812,391 | 53,254,677 |
| Min sequence length | 504 | 740 | 60 |
| Max sequence length | 2,019,106 | 3,335,433 | 7,561,339 |
| Average sequence length | 38,580.3 | 220,923.4 | 273,100.9 |
| N50 length | 538,396 | 1,023,154 | 5,758,810 |
| N90 length | 14,603 | 143,036 | 158,429 |
| (G + C)s | 69.9% | 71.2% | 67.8% |
3.2. Anchor scaffolds by genetic maps
The genome sequencing of male and female parents and their offspring produced 32,327,297, 35,177,866 and 1,031,682,186 reads, respectively. These reads then were mapped to the genome for subsequent genotyping. The results showed that 1,367 SNPs were shared between the two parents. One hundred and twenty‐nine loci that met the linkage requirement were used to construct the genetic map. Finally, five linkage groups were constructed using these markers, with a number of markers ranging from 9 to 45. The length per group ranged from 88.6 cM to 284.0 cM, with an average of 171.4 cM. Based on the markers and genetic distance, 10 pseudomolecules representing 80.9% of the total assembly (42.7 Mb) were anchored and orientated to the 5 linkage groups (Figure 2). Among them, pseudomolecules 12, 26, 32 and 110 were anchored to one chromosome, and pseudomolecule 9 was mapped to one chromosome. Meanwhile, pseudomolecules 13 and 27 and pseudomolecules 80 and 201 were placed on two different chromosomes, respectively, based on the markers and their distance. The remaining pseudomolecule 140 was anchored to one chromosome. The number of linkage groups established in this study is consistent with the cytological observations (Tseng & Sun, 1989; Yan et al., 2008).
Figure 2.
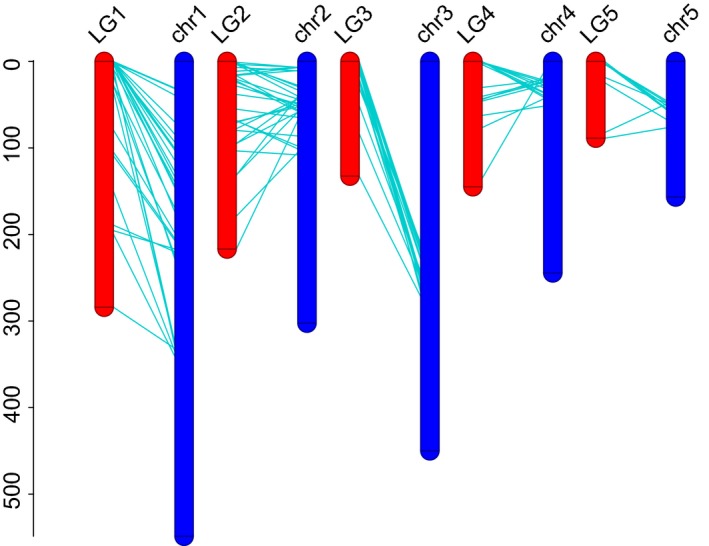
Anchor scaffolds from Pyropia haitanensis according to genetic maps. The red bar presents the linkage groups generated from genetic maps. The blue bar presents the chromosomes generated via genome assembly
3.3. Genome evaluation
To assess the quality of the assembled genome, three approaches were used. First, the final assembled genome size of this species (53.3 Mb) was similar to the size calculated based on the K‐mer frequency distribution (46.5 Mb). Second, we obtained a total of 17,383 unigenes from the PacBio system. Then, these transcriptome sequencing data were mapped to the current assembly by BLAT (Kent, 2002), and >87.2% of PacBio isoforms could be successfully aligned. Third, we performed Benchmarking Universal Single‐Copy Orthologs (BUSCO) analysis, and 85.5% of the eukaryotic single‐copy genes were detected in the P. haitanensis genome. This number is higher than the values in C. crispus (84.5%) and Po. umbilicalis (74.3%) (Figure S3). Interestingly, we also noticed that the 'complete' percentage of BUSCO in red algae was generally lower than those in other species. The reason for this possibly lies in independent evolution after primary endosymbiosis, leading to great genome diversity in red algae (e.g. reduction of the genome contents of the red algae (Qiu, Price, Yang, Yoon, & Bhattacharya, 2015)). The relative lack of red algal genome information in public databases might be another reason.
3.4. Repeat elements
For the repeat element analysis, the results showed that the repeat elements identified in P. haitanensis constituted 24.2% of the whole genome, including 14.6% as tandem repeat sequences and 9.6% as interspersed repeats. Among the tandem repeats, a total of 26,822 microsatellites were identified, accounting for 3.2% of the genome. In addition, 60,360 (8.1%) minisatellite and 3,586 (3.3%) satellite DNAs were identified. LTR elements represented the majority of the confirmed interspersed repeats, occupying 3.4% of the genome, while the DNA elements comprised 0.8% (Table 3, Figure 3). Among LTRs, 1,040 full‐length LTRs were predicted, 544 of which belonged to the Copia superfamily, 413 belonged to the Gypsy superfamily and 83 belonged to Caulimovirus superfamily. The remaining 76 LTRs were not full length and occupied 0.2% of the genome. When compared with closely related species, we noticed that the Po. umbilicalis genome had a substantial repeat element (43.9%) in its 87.7 Mb genome, including 17.7% DNA transposons (15.5 Mb) and 17.0% LTR elements (14.9 Mb) (Brawley et al., 2017). Comparison of the repeat landscape of the P. haitanensis genome and those in other species in red algae (Price et al., 2019) showed that the LTRs can be attributed to genome size variation.
Table 3.
Composition of repeat elements in genome of Pyropia haitanensis
| Class | Order | Superfamily | Number | Length (bp) | Percentage (%) |
|---|---|---|---|---|---|
| Interspersed repeats | LTR | Gypsy | 413 | 1,327,093 | 2.49 |
| Copia | 544 | 278,151 | 0.52 | ||
| Caulimovirus | 83 | 114,994 | 0.22 | ||
| Other LTR | 76 | 85,379 | 0.16 | ||
| DNA | CMC‐EnSpm | 83 | 80,307 | 0.15 | |
| PIF‐Harbinger | 331 | 253,169 | 0.48 | ||
| PiggyBac | 228 | 69,767 | 0.13 | ||
| Unknown | 10,009 | 2,874,529 | 5.40 | ||
| Tandem repeats | Microsatellite | 26,822 | 1,695,878 | 3.18 | |
| Minisatellite | 60,360 | 4,290,390 | 8.06 | ||
| Satellite | 3,586 | 1,776,700 | 3.34 |
Figure 3.
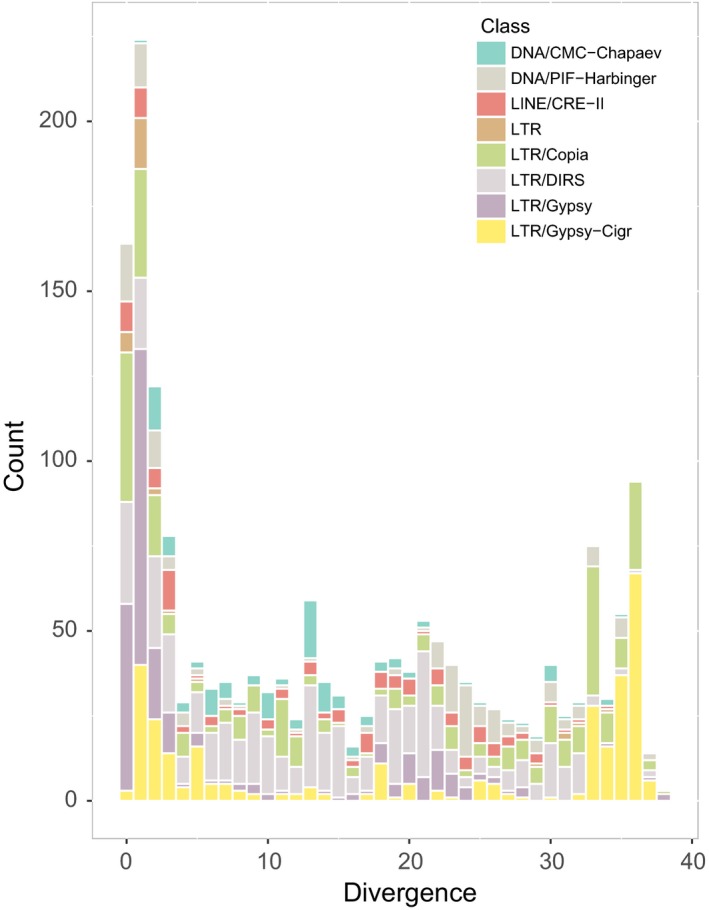
A repeat landscape of the Pyropia haitanensis genome showing the expansion and decline of transposable elements
3.5. Gene prediction
After repeats' masking, de novo prediction predicted 11,725 gene models for P. haitanensis. Based on the homologous protein database established from the five red algae mentioned above, 31,389 protein‐coding sequences were obtained. At the same time, we predicted 11,871 gene models using pasa software. Finally, EVM was used to integrate these gene models from the above methods to obtain a gene data set with 10,930 protein‐coding sequences (ORFs), which is comparable to the gene repertoire of other sequenced red algae genomes (Bhattacharya et al., 2013; Brawley et al., 2017; Collén et al., 2013; Lee et al., 2018; Nozaki et al., 2007). These protein‐coding genes in P. haitanensis were further employed to analyse their functions using several public databases. We identified 7,356 and 10,374 genes that showed homology to proteins in the NR and InterPro databases, respectively (Figure S4). A total of 3,147 genes were assigned to GO classifications. Based on KEGG analysis, we could annotate a total of 1,830 genes (Table S2) and a total of 317 KEGG metabolic pathways in the genome of P. haitanensis (Figure S5). Moreover, the CAZyme database annotation showed that a total of 303 genes in the P. haitanensis genome were associated with carbohydrate metabolism‐related enzymes (Table S3). In addition, 7,041 genes in P. haitanensis were assigned to CDD 1,295 superfamilies (Table S4).
3.6. Gene family expansion and contraction
To estimate the gene family expansion and contraction, the genome of P. haitanensis combined with five available red algae and an outgroup species was selected to define the orthologous genes. We identified 622 single‐copy orthologous genes within P. haitanensis and the other six species, which were used in phylogenetic analyses in the following study. Analysis suggested the divergence time of P. haitanensis and Po. umbilicals was 204.4 Ma (95% highest posterior density (HPD)=164.6–249.7 Ma), indicating that P. haitanensis was a more recently diverged lineage in the red algae (Figure 4).
Figure 4.
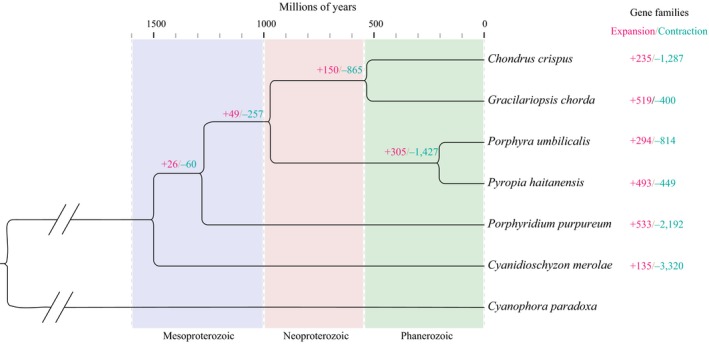
Phylogenetic analyses to reveal the evolutionary relationship and gene families and expansion in red algae. Six hundred and twenty‐two single‐copy orthologous genes within Pyropia haitanensis and six other species were used in phylogenetic analyses
A total of 493 orthologous groups (containing 2,514 genes) harboured more P. haitanensis paralogs than Po. umbilicals and were therefore defined as the expanded gene families. They mainly encoded ATP hydrolysis, nucleic acid metabolism, purine metabolism, cytoskeleton‐associated proteins, ion‐transporting proteins as well as E3 ubiquitin ligase, etc., according to their Pfam annotation (Tables S5 and S6). Meanwhile, 294 groups (containing 1,218 genes) with fewer P. haitanensis paralogs were defined as contracted gene families. These encoded phytochelatin synthase, sucrose transporter, cytochrome c oxidase copper chaperone, etc. Although the two closely related species are similar in morphology and physiology, the existence of large amounts of expanded and contracted gene families among them suggests that different environmental pressures have shaped their specific genetic contents to adapt to their individual habitats since they diverged from each other.
3.7. ROS‐ABA signalling pathway‐related genes in P. haitanensis
ROS is an important secondary messenger that is poised at the core of signalling pathway in plants maintaining the normal metabolic fluxes and different cellular functions and responding to environment stresses (Quigley et al., 2009). The production of ROS in cell originated from NADPH oxidases (NOX) located different organelles (cell wall, chloroplast and mitochondria) (Bedard & Krause, 2007). The NOX in cell wall is also considered as ROS‐generating respiratory burst oxidase (RBOH). In higher plants, RBOH is a family with more than ten members (Suzuki et al., 2011). We identified 10 members of RBOH in P. haitanensis, 8 in P. umbilicalis, 4 in C. crispus and 2 in Cyanidioschyzon merolae (Table 4). Compared to single‐cell red algae, RBOH in P. haitanensis endured significant expanding during evolution. The numbers of AOX and PTX in P. haitanensis are 2, with no significant difference with other red algae species. Under the downstream signal pathway activated by ROS, MAPK cascade is highly conserved and can be activated by phosphorylation (Xing, Ginty, & Greenberg, 1996). It plays major role in signal transduction of diverse stress responses even in combination of many stresses. The activation of MAPK cascade firstly is inhibited by MAPK repressor while induced by ROS (Son et al., 2011). The dual‐specificity protein tyrosine phosphatase (DSPTP) is MAPK repressor in ROS pathway (Martell, Angelotti, & Ullrich, 1998). Only 1 was identified, P. haitanensis; however, 8 and 5 was identified in single‐cell red algae species, P. purpureum and C. merolae, respectively. When the MAPK cascade was activated,the phosphorylation event can further activated many downstream factors, including transcript factors (TFs) etc. At present, MYB44, HSFA and ERF factors were identified to be activated by MAPK and involved in many stress and development process. We identified 16 MYB family TFs in P. haitanensis, including 12 MYB‐like, respectively. Yet, only 1 HSFA was identified in P. haitanensis. There are no significant differences in the numbers of these two‐type TFs in all red algae species studied. It was noting that ERF factor did not exist in either specie, which is an important TFs in ethylene signalling pathway.
Table 4.
ROS‐ABA signalling pathway‐related genes in Pyropia haitanensis and other red algae
| Gene name | Gene function | P. haitanensis | Porphyra umbilicalis | Chondrus crispus | Porphyridium purpureum | Cyanidioschyzon merolae |
|---|---|---|---|---|---|---|
| ROS production | ||||||
| RBOH | NADPH oxidase |
ph10359.t1 ph07364.t1 ph06070.t1 ph07507.t1 ph08568.t1 ph05196.t1 ph03740.t1 ph11172.t1 ph06827.t1 ph03938.t1 |
ccri|XP_005718545.1 ccri|XP_005719187.1 ccri|XP_005718335.1 ccri|XP_005716000.1 |
ppur|evm.model.contig_2134.3 ppur|evm.model.contig_2149.17 ppur|evm.model.contig_2146.22 ppur|evm.model.contig_3670.1 ppur|evm.model.contig_502.2 |
||
| AOX | in mitochondria | ph03278.t1 | OSX69369.1 | ccri|XP_005719100.1 | ppur|evm.model.contig_2288.11 | Cm|XP_005536259.1 |
| PTX | in plastid | ph07793.t1 | OSX69826.1 | ccri|XP_005712075.1 | ppur|evm.model.contig_4450.5 | Cm|XP_005536398.1 |
| ABA regulatory net | ||||||
| PYR1/PYL/PCAR | N | N | N | N | N | N |
| PP2C | type−2C protein phosphatase | ph10951.t1 | OSX76330.1 | ccri|XP_005719405.1 | ppur|evm.model.contig_3479.1 | Cm|XP_005536535.1 |
| ph09239.t1 | OSX79480.1 | ccri|XP_005711405.1 | ppur|evm.model.contig_510.16 | Cm|XP_005538832.1 | ||
| ph02078.t1 | OSX71532.1 | ccri|XP_005719125.1 | ppur|evm.model.contig_3807.1 | Cm|XP_005535984.1 | ||
| ph11536.t1 | OSX77048.1 | ccri|XP_005712925.1 | ppur|evm.model.contig_4456.15 | Cm|XP_005535913.1 | ||
| ph07863.t1 | OSX81030.1 | ccri|XP_005711323.1 | ppur|evm.model.contig_2501.2 | |||
| ph10321.t1 | OSX77620.1 | ppur|evm.model.contig_3441.20 | ||||
| ph02405.t1 | OSX69983.1 | ppur|evm.model.contig_441.27 | ||||
| ph06642.t1 | OSX71152.1 | ppur|evm.model.contig_3468.6 | ||||
| ph08933.t1 | ppur|evm.model.contig_528.2 | |||||
| ppur|evm.model.contig_2082.9 | ||||||
| ppur|evm.model.contig_3620.3 | ||||||
| ppur|evm.model.contig_4590.3 | ||||||
| OST1 | Protein OPEN STOMATA kinase | ph00419.t1 | OSX79527.1 | ccri|XP_005711343.1 | ppur|evm.model.contig_2031.6 | |
| ph03789.t2 | OSX79650.1 | ccri|XP_005713325.1 | ||||
| ccri|XP_005716962.1 | ||||||
| ccri|XP_005718769.1 | ||||||
| SLAC1 | slow anion channel_associated | ph09254.t1 | OSX76312.1 | ccri|XP_005718439.1 | ppur|evm.model.contig_498.15 | |
| G protein‐coupled receptor (GPCR) | ph09960.t1 | OSX76732.1 | ccri|XP_005716830.1 | ppur|evm.model.contig_4450.2 | Cm|XP_005539542.1 | |
| ph00460.t1 | OSX76731.1 | ccri|XP_005711645.1 | ppur|evm.model.contig_522.10 | Cm|XP_005537601.1 | ||
| ph10367.t1 | OSX70306.1 | ccri|XP_005711658.1 | ppur|evm.model.contig_431.16 | Cm|XP_005535191.1 | ||
| OSX68793.1 | ||||||
ABA signalling pathway plays important in response to environmental stress, especially drought stress (Davies, Kudoyarova, & Hartung, 2005). The turning on of this pathway is dependent on the ABA receptor binding to ABA. Currently, the ABA receptor widely studied including PYR1/PYL/PCAR component. Its binding to ABA can inhibit PP2C, further inhibit OST1 kinase and activate MAPK. After that, the downstream response factors were activated. In addition, OST1 can activate the slow anion channel‐associated (SLAC). We did not identify the presence of PYR1/PYL/PCAR type receptor in either red algae, but identified G protein receptor (GPCR), which is another receptor binding to ABA. The number of GPCR in P. haitanensis is 3. There are 10 PP2C in P. haitanensis, yet only 5 in P. umbilicalis, which indicated this gene family endured expanding in P. haitanensis. OST1 (1) and SLAC (2) were also identified in different red algae with no significant difference in numbers. Numerous reports highlight the importance of the ROS‐ABA signalling pathway in responding to drought stress in higher plants (Cruz de Carvalho, 2008; Golldack, Li, Mohan, & Probst, 2014). These stress factors in the intertidal zones make Pyropia highly environmentally tolerant for different stress, including osmotic stress, temperature stress and light stress (Hwang, Chung, & Oh, 1997). Therefore, we speculated that the expanded genes in ROS‐ABA signalling pathway were closely related to the ability of environmental adaptation in P. haitanensis.
4. CONCLUSIONS
In this study, we reported a high‐quality nuclear genome of Pyropia haitanensis, a red algal species of great economic, ecological and research value. We adopted multiple sequencing techniques to achieve an assembly with high contiguity and coverage. The investigation of genome characteristics and functional features yields further insights regarding the phylogenetic diversity of P. haitanensis. This genome will not only be a fundamental resource for deciphering the molecular mechanisms underlying the developmental processes of P. haitanensis and environmental adaptation mechanisms of intertidal seaweeds, but also help to reconstruct the evolutionary history of red algae.
AUTHOR CONTRIBUTIONS
Y.X.M. and D.M.W. conceived the study. C.M., X.Z.Y. and P.P.S. performed the experiments. K.P.X., G.Q.B., Y.L., F.N.K., X.H.T., Y.G. and G.Y.D. analysed and interpreted the assembly and annotations. K.P.X. and G.Q.B. performed the comparative genome analysis. C.M. and K.P.X. wrote the manuscript with input from all authors.
Supporting information
Cao M, Xu K, Yu X, et al. A chromosome‐level genome assembly of Pyropia haitanensis (Bangiales, Rhodophyta). Mol Ecol Resour. 2020;20:216–227. 10.1111/1755-0998.13102
Min Cao, Kuipeng Xu, Xinzi Yu, Guiqi Bi contributed equally to this work.
Funding information
This work was financially supported by the National Key R&D Program of China (2018YFD0900106), the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2018SDKJ0302‐5), the Fundamental Research Funds for the Central Universities (201762016) and the Project of National Infrastructure of Fishery Germplasm Resources (2018DKA30470). These funding bodies had no role in the study design, analysis, decision to publish or preparation of the manuscript.
Contributor Information
Dongmei Wang, Email: wangdm@ouc.edu.cn.
Yunxiang Mao, Email: yxmao@ouc.edu.cn.
DATA AVAILABILITY STATEMENT
The DNA sequencing data have been deposited into the NCBI Sequence Read Archive under the BioProject: PRJNA503796.
REFERENCES
- Abascal, F. , Zardoya, R. , & Posada, D. (2005). ProtTest: Selection of best‐fit models of protein evolution. Bioinformatics, 21 (9): 2104–2105. [DOI] [PubMed] [Google Scholar]
- Archibald, J. M. (2012). The evolution of algae by secondary and tertiary endosymbiosis. Advances in botanical research 64, 87–118. [Google Scholar]
- Ari, Ş. , & Arikan, M. (2016). Next‐generation sequencing: Advantages, disadvantages, and future In Hakeem K. R., Tombuloğlu H., & Tombuloğlu G. (Eds). Plant omics: Trends and applications (pp. 109–135). Cham: Springer. [Google Scholar]
- Bedard, K. , & Krause, K. H. (2007). The NOX family of ROS‐generating NADPH oxidases: Physiology and pathophysiology. Physiological Reviews, 87(1), 245–313. 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- Bengtson, S. , Sallstedt, T. , Belivanova, V. , & Whitehouse, M. (2017). Three‐dimensional preservation of cellular and subcellular structures suggests 1.6 billion‐year‐old crown‐group red algae. PLoS Biology, 15(3), e2000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Research, 27(2), 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya, D. , Price, D. C. , Chan, C. X. , Qiu, H. , Rose, N. , Ball, S. , … Yoon, H. S. (2013). Genome of the red alga Porphyridium purpureum . Nature Communications, 4, 1941 10.1038/ncomms2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti, M. A. , Olmo, E. , & Heslop‐Harrison, J. P. (2015).Repetitive DNA in eukaryotic genomes. [DOI] [PubMed]
- Blouin, N. A. , Brodie, J. A. , Grossman, A. C. , Xu, P. , & Brawley, S. H. (2011). Porphyra: A marine crop shaped by stress. Trends in Plant Science, 16(1), 29–37. 10.1016/j.tplants.2010.10.004 [DOI] [PubMed] [Google Scholar]
- Boetzer, M. , Henkel, C. V. , Jansen, H. J. , Butler, D. , & Pirovano, W. (2010). Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics, 27(4), 578–579. 10.1093/bioinformatics/btq683 [DOI] [PubMed] [Google Scholar]
- Brawley, S. H. , Blouin, N. A. , Ficko‐Blean, E. , Wheeler, G. L. , Lohr, M. , Goodson, H. V. , & Marriage, T. N. (2017)., Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta). Proceedings of the National Academy of Sciences, 114(31), E6361–E6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J. M. , & Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large scale phylogenetic analyses. Bioinformatics, 25 (15): 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 5(1), 4–10. [DOI] [PubMed] [Google Scholar]
- Chin, C.‐S. , Alexander, D. H. , Marks, P. , Klammer, A. A. , Drake, J. , Heiner, C. , … Korlach, J. (2013). Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nature Methods, 10(6), 563 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Collen, J. , Porcel, B. , Carre, W. , Ball, S. G. , Chaparro, C. , Tonon, T. , … Boyen, C. (2013). Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proceedings of the National Academy of Sciences of the United States of America, 110(13), 5247–5252. 10.1073/pnas.1221259110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz de Carvalho, M. H. (2008). Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signaling & Behavior, 3(3), 156–165. 10.4161/psb.3.3.5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, W. J. , Kudoyarova, G. , & Hartung, W. (2005). Long‐distance ABA signaling and its relation to other signaling pathways in the detection of soil drying and the mediation of the plant's response to drought. Journal of Plant Growth Regulation, 24(4), 285 10.1007/s00344-005-0103-1 [DOI] [Google Scholar]
- Drummond, A. J. , Suchard, M. A. , Xie, D. , & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29(8), 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, A. C. , Richards, S. , Han, Y. I. , Wang, M. , Vee, V. , Qu, J. , … Gibbs, R. A. (2012). Mind the gap: Upgrading genomes with Pacific Biosciences RS long‐read sequencing technology. PLoS ONE, 7(11), e47768 10.1371/journal.pone.0047768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X. , Dou, J. , Mao, J. , Su, H. , Jiao, W. , Zhang, L. , … Bao, Z. (2013). RADtyping: An integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations. PLoS ONE, 8(11), e79960 10.1371/journal.pone.0079960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golldack, D. , Li, C. , Mohan, H. , & Probst, N. (2014). Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Frontiers in Plant Science, 5, 151 10.3389/fpls.2014.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Gu, X. , Jiang, Y. , Zhu, W. , Yao, L. , Liu, Z. , … Wang, L. (2018). Antagonistic effect of laver, Pyropia yezonensis and P. haitanensis, on subchronic lead poisoning in rats. Biological Trace Element Research, 181(2), 296–303. 10.1007/s12011-017-1050-y [DOI] [PubMed] [Google Scholar]
- Haas, B. J. , Salzberg, S. L. , Zhu, W. , Pertea, M. , Allen, J. E. , Orvis, J. , … Wortman, J. R. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology, 9(1), R7 10.1186/gb-2008-9-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron, M. D. , Hackett, J. D. , Aylward, F. O. , & Michod, R. E. (2009). Triassic origin and early radiation of multicellular volvocine algae. Proceedings of the National Academy of Sciences of the United States of America, 106(9), 3254–3258. 10.1073/pnas.0811205106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek, C. , Mann, D. , Jahns, H. M. , & Jahns, M. (1995). Algae: An introduction to phycology. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Huelsenbeck, J. P. , & Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17(8), 754–755. 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Hwang, M. S. , Chung, I. K. , & Oh, Y. S. (1997). Temperature responses of Porphyra tenera Kjellman and P. yezoensis Ueda (Bangiales, Rhodophyta) from Korea. Algae, 12(3), 207–207. [Google Scholar]
- Katoh, K. , Kuma, K. I. , Toh, H. , & Miyata, T. (2005). MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Research, 33(2), 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J. (2002). BLAT‐the BLAST‐like alignment tool. Genome Research, 12(4), 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. , & Salzberg, S. L. (2009). Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biology, 10(3), R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. M. , Yang, E. C. , Graf, L. , Yang, J. H. , Qiu, H. , Zelzion, U. , … Yoon, H. S. (2018). Analysis of the draft genome of the red seaweed Gracilariopsis chorda provides insights into genome size evolution in Rhodophyta. Molecular Biology and Evolution, 35(8), 1869–1886. 10.1093/molbev/msy081 [DOI] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C. J. , & Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Research, 13(9), 2178–2189. 10.1101/gr.1224503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , … Wang, J. (2012). SOAPdenovo2: An empirically improved memory‐efficient short‐read de novo assembler. Gigascience, 1(1), 18 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell, K. J. , Angelotti, T. , & Ullrich, A. (1998). Dual‐specificity protein tyrosine phosphatases. Molecules and Cells, 8(1), 2–11. [PubMed] [Google Scholar]
- Merchant, S. S. , Prochnik, S. E. , Vallon, O. , Harris, E. H. , Karpowicz, S. J. , Witman, G. B. , …, Marshall, W. F. (2007). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science, 318(5848), 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, K. M. , Sheath, R. G. , Vis, M. L. , Crease, T. J. , & Cole, K. M. (1998). Biogeography and systematics of Bangia (Bangiales, Rhodophyta) based on the Rubisco spacer, rbc L gene and 18S rRNA gene sequences and morphometric analyses. 1. North America. Phycologia, 37(3), 195–207. [Google Scholar]
- Neely, R. K. , Deen, J. , & Hofkens, J. (2011). Optical mapping of DNA: Single‐molecule‐based methods for mapping genomes. Biopolymers, 95(5), 298–311. [DOI] [PubMed] [Google Scholar]
- Nozaki, H. , Takano, H. , Misumi, O. , Terasawa, K. , Matsuzaki, M. , Maruyama, S. , … Kuroiwa, T. (2007). A 100%‐complete sequence reveals unusually simple genomic features in the hot‐spring red alga Cyanidioschyzon merolae . BMC Biology, 5(1), 28 10.1186/1741-7007-5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, D. C. , Goodenough, U. W. , Roth, R. , Lee, J.‐H. , Kariyawasam, T. , Mutwil, M. , … Bhattacharya, D. (2019). Analysis of an improved Cyanophora paradoxa genome assembly. DNA Research, 26(4), 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, H. , Price, D. C. , Yang, E. C. , Yoon, H. S. , & Bhattacharya, D. (2015). Evidence of ancient genome reduction in red algae (Rhodophyta). Journal of Phycology, 51(4), 624–636. 10.1111/jpy.12294 [DOI] [PubMed] [Google Scholar]
- Quigley, M. , Conley, K. , Gerkey, B. , Faust, J. , Foote, T. , Leibs, J. , Ng, A. Y. (2009).ROS: an open‐source Robot Operating System. In ICRA workshop on open source software (Vol. 3, No. 3.2, p. 5).
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Prieto, A. , Weber, A. P. , & Bhattacharya, D. (2007). The origin and establishment of the plastid in algae and plants. Annual Review of Genetics, 41, 147–168. [DOI] [PubMed] [Google Scholar]
- Roberts, R. J. , Carneiro, M. O. , & Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biology, 14(6), 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo, D. , Tang, X. , & Yarish, C. (2002). Porphyra–the economic seaweed as a new experimental system. Current Science, 83(11), 1313–1316. [Google Scholar]
- Smit, A. F. , & Hubley, R. (2008). RepeatModeler Open‐1.0. Retrieved from http://www.repeatmasker.org [Google Scholar]
- Son, Y. , Cheong, Y. K. , Kim, N. H. , Chung, H. T. , Kang, D. G. , & Pae, H. O. (2011). Mitogen‐activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways?. Journal of Signal Transduction, 2011, Article ID 792639, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. . (2014). RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9): 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. , & Morgenstern, B. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Research, 34(suppl_2), W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, R. C. (1987). UTEX‐The culture collection of algae at the University of Texas at Austin. Journal of Phycology, 23(3), 1–78. [Google Scholar]
- Sutherland, J. E. , Lindstrom, S. C. , Nelson, W. A. , Brodie, J. , Lynch, M. D. , Hwang, M. S. , & Farr, T. (2011). A new look at an ancient order: Generic revision of the Bangiales (Rhodophyta) 1. Journal of Phycology, 47(5), 1131–1151. [DOI] [PubMed] [Google Scholar]
- Suzuki, N. , Miller, G. , Morales, J. , Shulaev, V. , Torres, M. A. , & Mittler, R. (2011). Respiratory burst oxidases: The engines of ROS signaling. Current Opinion in Plant Biology, 14(6), 691–699. [DOI] [PubMed] [Google Scholar]
- Tseng, C. K. , & Sun, A. (1989). Studies on the alternation of the nuclear phases and chromosome numbers in the life history of some species of Porphyra from China. Botanica Marina, 32(1), 1–8. [Google Scholar]
- Van Ooijen, J. (2006). JoinMap®4 Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, The Netherlands.
- Wang, L. , Mao, Y. , Kong, F. , Cao, M. , & Sun, P. (2015). Genome‐wide expression profiles of Pyropia haitanensis in response to osmotic stress by using deep sequencing technology. BMC Genomics, 16(1), 1012 10.1186/s12864-015-2226-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Meyer, E. , McKay, J. K. , & Matz, M. V. (2012). 2b‐RAD: A simple and flexible method for genome‐wide genotyping. Nature Methods, 9(8), 808. [DOI] [PubMed] [Google Scholar]
- Wicker, T. , Sabot, F. , Hua‐Van, A. , Bennetzen, J. L. , Capy, P. , Chalhoub, B. , … Schulman, A. H. (2007). A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics, 8(12), 973 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- Xing, J. , Ginty, D. D. , & Greenberg, M. E. (1996). Coupling of the RAS‐MAPK pathway to gene activation by RSK2, a growth factor‐regulated CREB kinase. Science, 273(5277), 959–963. [DOI] [PubMed] [Google Scholar]
- Yan, X. H. , He, L. H. , Huang, J. , Song, W. L. , Ma, P. , & Aruga, Y. (2008). Cytological studies on Porphyra haitanensis Chang et Zheng (Bangiales, Rhodophyta). Journal of Fisheries of China, 1, 131–137. [Google Scholar]
- Yang, E. C. , Boo, S. M. , Bhattacharya, D. , Saunders, G. W. , Knoll, A. H. , Fredericq, S. , … Yoon, H. S. (2016). Divergence time estimates and the evolution of major lineages in the florideophyte red algae. Scientific Reports, 6, 21361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Wang, Q. , Liu, M. H. , & An, L. J. (1999). A simple method for extracting total DNA of seaweeds. Biotechnol, 9(4), 39–42. [Google Scholar]
- Ye, N. , Zhang, X. , Miao, M. , Fan, X. , Zheng, Y. I. , Xu, D. , … Zhao, F. (2015). Saccharina genomes provide novel insight into kelp biology. Nature Communications, 6, 6986 10.1038/ncomms7986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, H. S. , Müller, K. M. , Sheath, R. G. , Ott, F. D. , & Bhattacharya, D. (2006). Defining the major lineages of red algae (RHODOPHYTA) 1. Journal of Phycology, 42(2), 482–492. 10.1111/j.1529-8817.2006.00210.x [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA sequencing data have been deposited into the NCBI Sequence Read Archive under the BioProject: PRJNA503796.