

Abstract
The aim of this study was to characterize the effects of upadacitinib, a Janus kinase 1 inhibitor, on in vivo activity of different cytochrome P450 (CYP) enzymes using a cocktail approach. Healthy subjects (n = 20) received single oral doses of the modified Cooperstown 5+1 cocktail drugs (midazolam [CYP3A], caffeine [CYP1A2], warfarin + vitamin K [CYP2C9], omeprazole [CYP2C19], and dextromethorphan [CYP2D6]) without upadacitinib and on day 11 (midazolam) or 12 (all other probes) of a 15‐day regimen of upadacitinib 30 mg once daily (extended‐release formulation). Serial blood samples and 12‐hour urine samples were collected for assays of the probe substrates and select metabolites. The ratio (90%CI) of area under the plasma concentration‐time curve from time 0 to infinity (AUCinf) central values when the cocktail drugs were administered with upadacitinib relative to when administered alone were 0.74 (0.68‐0.80) for midazolam, 1.22 (1.15‐1.29) for caffeine, 1.11 (1.07‐1.15) for S‐warfarin, 1.07 (0.95‐1.22) for dextromethorphan, and 0.82 (0.72‐0.94) for omeprazole. The ratio (90%CI) was 1.09 (1.00‐1.19) for 5‐hydroxy‐omeprazole to omeprazole AUCinf ratio and 1.17 (0.97‐1.41) for dextromethorphan to dextrorphan 12‐hour molar urinary ratio. Upadacitinib 30 mg once daily (a dose that is twice the optimal dose in rheumatoid arthritis based on phase 3 results) has a limited effect on CYP3A activity (26% decrease in exposure of midazolam, a sensitive CYP3A substrate) and no relevant effects on CYP1A2, CYP2C9, CYP2C19, or CYP2D6 activity in vivo. No clinically relevant changes in plasma exposures are expected for drugs that are substrates for the evaluated CYP enzymes when coadministered with upadacitinib.
Keywords: upadacitinib, ABT‐494, cytochrome P450, drug‐drug interactions
Upadacitinib (ABT‐494) is a selective inhibitor of Janus kinase 1 (JAK1) that is being developed by AbbVie for treatment of rheumatoid arthritis (RA) and several other inflammatory diseases.1, 2, 3, 4, 5 The relative selectivity of upadacitinib for JAK1 compared with JAK 2 or 3 has the potential to improve its benefit to the risk profile compared with that of the less selective JAK inhibitors.6, 7 Upadacitinib demonstrated favorable efficacy and acceptable safety in phase 2 and 3 studies in subjects with RA4, 5, 8, 9 and in phase 2 studies in subjects with Crohn's disease and atopic dermatitis.10, 11 Upadacitinib was evaluated in phase 3 studies in subjects with RA at doses of 15 and 30 mg administered once daily using the extended‐release formulation.1, 2, 3, 4, 5, 12 Based on phase 3 results, upadacitinib 15‐mg once‐daily dose is the optimal dose in RA patients, as it provided maximum efficacy across different studies.1, 2, 3, 4, 5
Upadacitinib is metabolized by cytochrome P450 (CYP) 3A and, to a lesser extent, by CYP2D6, and more than 60% of an administered dose is recovered as unchanged upadacitinib in feces and urine, respectively.13, 14 In a clinical drug interaction study, administration of ketoconazole, a strong CYP3A inhibitor, increased upadacitinib area under the curve (AUC) by only 75%, whereas administration of multiple doses of rifampin, a broad CYP inducer and a strong inducer of CYP3A, decreased upadacitinib AUC by 60%, indicating that upadacitinib is not a sensitive substrate for CYP3A.14 Upadacitinib terminal elimination half‐life ranged from 6 to 16 hours over the range of doses evaluated in clinical studies.13, 15
Patients with RA or other chronic inflammatory diseases often have comorbidities and are likely to take several concomitant medications, many of which may be substrates for metabolism through CYP enzymes.16, 17, 18 Therefore, it is important to evaluate the effect of upadacitinib on CYP enzymes to determine whether there is a potential need for dose adjustment of concomitant medications. The effect of upadacitinib on the in vivo activities of specific probes for CYP enzymes has not been previously evaluated.
The use of CYP phenotyping cocktails is an efficient approach for evaluating the effect of a drug on the in vivo activity of multiple CYP enzymes simultaneously in a relatively small number of subjects in a single study. CYP phenotyping cocktails comprise multiple agents, each of which is a CYP form‐preferred sensitive substrate of one of the CYP enzymes. Several CYP phenotyping cocktails have been developed and validated to ensure lack of effect of each probe substrate on the pharmacokinetics of the other drugs in the cocktail.19 One of the commonly used cocktails is the Cooperstown 5+1 cocktail, which consists of intravenous midazolam (CYP3A substrate), caffeine (CYP1A2 substrate), warfarin (CYP2C9 substrate) + vitamin K, omeprazole (CYP2C19 substrate), and dextromethorphan (CYP2D6 substrate).20 A modified version of the Cooperstown 5+1 cocktail has previously used oral instead of intravenous midazolam, which enables assessment of the effect of the tested drug on intestinal as well as hepatic CYP3A activity.20, 21, 22, 23
The aim of this study was to characterize the effects of repeated doses of upadacitinib on the pharmacokinetics of specific probe substrates for CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A in healthy volunteers using a cocktail approach.
Methods
The study was conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocol and informed consent form were approved by the institutional review board (Vista Health System, Vista Medical Center East Institutional Review Board, Waukegan, Illinois), and participants provided written informed consent before any study‐related procedures were performed.
Study Design and Participants
This was a single‐center, open‐label, single‐arm, 2‐period study designed to evaluate the effect of coadministration of multiple doses of upadacitinib on probe substrates for different CYP enzymes using a modified Cooperstown 5+1 CYP phenotyping cocktail (Figure 1). Twenty subjects received a single oral dose of midazolam HCl 5 mg syrup on day 1, period 1, followed by a single oral dose of the other components of the CYP phenotyping cocktail (caffeine 200‐mg tablet, warfarin sodium 10‐mg tablet + vitamin K 10‐mg tablet, omeprazole 40‐mg delayed‐release capsule, and dextromethorphan HBr 30 mg liquid) on day 2, period 1. Following a 5‐day washout period, subjects received upadacitinib 30 mg once daily on days 1‐15 of period 2. Upadacitinib was coadministered with midazolam 5 mg on day 11 of period 2 and with the other components of the CYP phenotyping cocktail on day 12 of period 2. All CYP probe substrates were administered in the morning after a minimum 10‐hour fast and 4 hours before lunch.
Figure 1.
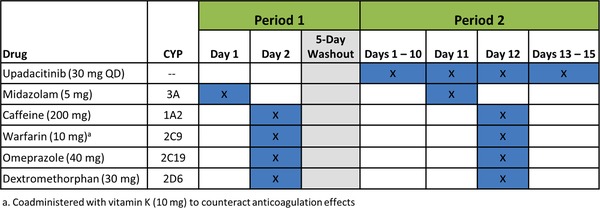
Study design.
Men and women aged between 18 and 55 years, inclusive, who had a body mass index between 18.0 and 29.9 kg/m2, inclusive, and were in good health based on results of a medical history, physical examination, vital signs assessment, laboratory profile, and a 12‐lead electrocardiogram (ECG) were eligible to enroll in the study. Female subjects had to be either permanently surgically sterile or postmenopausal. Subjects who had a history of diabetes or lymphoproliferative disease, evidence of immunosuppression, evidence of active or latent tuberculosis, or who used tobacco or nicotine‐containing products within 180 days of the first dose of study drug were not allowed to enroll. Subjects must not have had exposure to upadacitinib or other JAK inhibitors within the 3 months before the first dose of study drug and must not have used any medications known to inhibit or induce drug‐metabolizing enzymes within 30 days of the first dose of the study drug and through the end of the study.
Subjects were confined to the study site for approximately 23 days, beginning 1 day before day 1 of period 1 and ending after collection of the 96‐hour blood sample (relative to the dose of the CYP phenotyping cocktail on day 12) on day 16 of period 2. The meal content on the intensive pharmacokinetic sampling days (days 1 and 2 of period 1 and days 11 and 12 of period 2) was identical, and no breakfast was served on these days.
Pharmacokinetic Sampling and Bioanalytical Methods
The drug substrates and phenotyping metrics used to assess the in vivo activities of the different CYP enzymes are presented in Table 1. Serial blood samples for CYP probe substrate assays were collected by venipuncture into sodium heparin‐containing tubes just before dosing (0 hour) and for up to 96 hours after dosing. For all substrates and for 5‐hydroxy‐omeprazole, plasma concentrations were measured prior to dosing and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours after dosing. Additional blood samples were collected at 36 hours for caffeine, 48 and 72 hours for dextromethorphan and 48, 72, and 96 hours for warfarin to ensure sampling over a sufficient period for each study drug according to its half‐life. Urine samples for dextromethorphan and dextrorphan assays were collected into containers without preservatives at intervals of 0 to 6 hours and 6 to 12 hours on day 2 of period 1 and day 12 of period 2.
Table 1.
Phenotyping Probe Substrates and Metrics Used in the Study
| Enzyme | Probe Substrate | Phenotyping Metric |
|---|---|---|
| CYP1A2 | Caffeine 200 mg | Caffeine AUCinf |
| CYP2C9 | Warfarin 10 mg + vitamin K 10 mg | S‐Warfarin AUCinf |
| CYP2C19 | Omeprazole 30 mg | 5‐Hydroxy‐omeprazole to omeprazole AUCinf ratio |
| CYP2D6 | Dextromethorphan 30 mg |
Dextromethorphan AUCinf Dextromethorphan to dextrorphan molar urinary ratio |
| CYP3A | Midazolam 5 mg | Midazolam AUCinf |
Plasma samples for midazolam, omeprazole, 5‐OH‐omeprazole, and dextromethorphan were analyzed using validated assays developed by AbbVie Inc. (Lake County, Illinois). Plasma samples for S‐warfarin and caffeine as well as urine samples for dextromethorphan and dextrorphan used a validated assay developed by Pharmaceutical Product Development, LLC (Middleton, Wisconsin).
For the midazolam assay, a sample volume of 50 µL was combined with the internal standard (midazolam‐d4), and the analyte of interest extracted by protein precipitation using acetonitrile. A portion of the supernatant was evaporated to dryness under a stream of nitrogen. The resulting residue was reconstituted in acetonitrile:water (20:80, v/v), and the samples were submitted for analysis. Chromatographic separation was achieved using a X‐Bridge C18 column (3.5 µm, 2.1 × 50 mm; Waters Corporation, Milford, Massachusetts) and gradient conditions with mobile phase A consisting of 5/95/0.1 (v/v/v) Acetonitrile/water/formic acid and 100/0.1 (v/v) acetonitrile/formic acid as mobile phase B. An API 5500 mass spectrometer (AB Sciex, Framingham, Massachusetts) employing electrospray ionization in positive ion mode was used to monitor the analyte. Multiple‐reaction monitoring (MRM) transitions were m/z 326 → 291 for midazolam and 330 → 295 for the internal standard (midazolam‐d4). The lower limit of quantification (calibration range) was 0.0507 ng mL−1 (0.0507 to 20.0 ng mL−1), and interassay precision and accuracy/bias were ≤6.6% and between −0.8% and 4.5%, respectively.
For omeprazole and the 5‐OH‐omeprazole assay, a sample volume of 50 µL was combined with the internal standard (omeprazole‐d3 and 5‐OH‐omeprazole‐d3), and the analyte of interest extracted by protein precipitation using 0.1 M sodium carbonate and acetonitrile. A portion of the supernatant was evaporated to dryness under a stream of nitrogen. The resulting residue was reconstituted in methanol:water (10:90, v/v), and the samples were submitted for analysis. Chromatographic separation was achieved using a X‐Bridge C18 column (5 µm, 2.1 × 10 mm; Waters Corporation) and isocratic conditions with the mobile phase consisting of 0.1% (v/v) acetic acid and 10 mM ammonium acetate in 40/60 (v/v) methanol/water. An API 5500 mass spectrometer (AB Sciex) employing electrospray ionization in positive ion mode was used to monitor the analyte. MRM transitions were m/z 346 → 198 for omeprazole (349 → 198 for the internal standard omeprazole‐d3) and 362 → 214 for 5‐OH‐omeprazole (365 → 217 for the internal standard 5‐hydroxy‐omeprazole‐d3). The lower limit of quantification (calibration range) was 1.02 ng mL−1 (1.02 to 510 ng mL−1) for omeprazole and 0.993 ng mL−1 (0.993 to 496 ng mL−1) for 5‐hydroxy‐omeprazole. The interassay precision and accuracy/bias were ≤9.7% and between ‐0.9% and 4.4%, respectively, for omeprazole and ≤10.5% and between −0.5% and 6.7%, respectively, for 5‐hydroxy‐omeprazole.
For the S‐warfarin assay, a 200‐µL matrix aliquot was combined with 20 µL of 1000 ng/mL internal standard working solution. Analytes were isolated through liquid‐liquid extraction using organic solvent of methyl tert‐butyl ether and dichloromethane. The extracted organic solvent was evaporated under a nitrogen stream at approximately 45°C, and the remaining residue was reconstituted with 300 µL of reconstitution solution. The final extract was analyzed via high‐pressure liquid chromatography‐tandem mass spectrometry (HPLC‐MS/MS) detection using negative ion electrospray. The lower limit of quantification (calibration range) was 5.00 ng mL‐1 (5.00 to 1500 ng mL‐1), and interassay precision was ≤5.90%. Accuracy/bias was between −6.78% and 1.38%.
For caffeine assay, a 50‐µL matrix aliquot was combined with 25 µL of 2 µg/mL internal standard working solution. Analytes were isolated through liquid‐liquid extraction using organic solvent of chloroform and 2‐propanol. The extracted organic solvent was evaporated under a nitrogen stream at approximately 45°C, and the remaining residue was reconstituted with 400 µL of reconstitution solution. The final extract was analyzed via HPLC‐MS/MS detection using positive ion electrospray. The lower limit of quantification (calibration range) was 25.0 ng mL−1 (25.0 to 25 000 ng mL−1), and interassay precision and accuracy/bias were ≤8.33% and between −3.31% and 0.0173%, respectively.
For the dextromethorphan assay in plasma, a sample volume of 50 µL was combined with the internal standard (dextromethorphan‐d4), and the analyte of interest was extracted through liquid‐liquid extraction using organic solvent (50/50 [v/v] ethyl acetate/hexane). A portion of the supernatant was evaporated to dryness under a stream of nitrogen. The resulting residue was reconstituted in acetonitrile:water (20:80 v/v), and the samples were submitted for analysis. Chromatographic separation was achieved using an X‐Bridge C18 column (3.5 µm, 2.1 × 50 mm; Waters Corporation) and isocratic conditions with the mobile phase consisting of 20/80/0.1 (v/v/v) acetonitrile/water/formic acid. An API 5500 mass spectrometer (AB Sciex) employing electrospray ionization in positive ion mode was used to monitor the analyte. MRM transitions were m/z 272 → 215 for dextromethorphan and 275 → 215 for the internal standard (dextromethorphan‐d3). The lower limit of quantification (calibration range) was 0.0502 ng mL−1 (0.0502 to 10.0 ng mL‐1), and interassay precision and accuracy/bias were ≤5.9% and between −0.8% and 1.4%, respectively.
For the dextromethorphan and dextrorphan assay in urine, the analytes were extracted from 50 µL of urine incubated with beta‐glucuronidase solution and internal standards (dextromethorphan‐D3/dextrorphan‐D3/3‐methoxymorphinan‐D3). Extracts were analyzed by LC with MS/MS detection. The lower limit of quantification (calibration range), interassay precision and accuracy/bias were 0.00100 µg mL‐1 (0.00100‐1.00 µg mL‐1), 3.71%‐8.55%, and ‐4.65% to 1.27%, respectively, for dextromethorphan and 0.0200 µg mL‐1 (0.0200‐20.0 µg mL‐1), 2.51%‐5.91%, and −4.87% to −0.847%, respectively, for dextrorphan.
Genotyping and Phenotyping of CYP Enzymes
A single blood sample was collected from each subject for pharmacogenetic analyses. Polymorphisms for CYP2C9, CYP2C19, and CYP2D6 were screened as described in the DMET Plus Premier Pack Protocol (Affymetrix, Santa Clara, California). Blood samples were genotyped for the presence of the following alleles: CYP2C9, *2, *3, *5; CYP2D6, *2, *3, *4, *5, *6, *7, *8, *9, *10, *17, *29, *41, ×2; and CYP2C19, *2, *3, *4, *8, *10, *12. The metabolic phenotypes for each subject for CYP2C9, CYP2C19, and CYP2D6 were determined based on the genotyping results.24
Pharmacokinetic and Statistical Analyses
Pharmacokinetic parameters for the CYP probe substrates and metabolites were estimated using noncompartmental methods in Phoenix WinNonlin version 6.4 (Pharsight, A Certara Company, St. Louis, Missouri). Pharmacokinetic parameters included the maximum plasma concentration (Cmax), time to Cmax (Tmax), AUC up to the last measurable concentration (AUCt) and from time 0 to infinity (AUCinf), the terminal phase elimination half‐life (t1/2), and apparent oral clearance (CL/F). The 5‐hydroxy‐omeprazole metabolite‐to‐parent omeprazole AUCt and AUCinf ratios and the 12‐hour dextromethorphan‐to‐dextrorphan molar urinary ratio were calculated.
Statistical analyses were conducted using SAS version 9.3 (SAS Institute, Inc., Cary, North Carolina). To assess the effect of upadacitinib on the probe substrates, repeated‐measures analyses were performed for the natural logarithms of Cmax and AUC using data from periods 1 and 2. The bioavailability of the CYP probe substrates when administered with upadacitinib relative to that of the CYP probe substrates alone was assessed from the point estimates and the corresponding 90% confidence intervals for the difference of the least‐square means obtained from the repeated‐measures analyses of the natural logarithms of Cmax and AUC. Similar analyses were performed for the 5‐hydroxy‐omeprazole‐to‐omeprazole AUC ratio and the dextromethorphan‐to‐dextrorphan molar urinary ratio. A sensitivity analysis was conducted using only data from subjects who had extensive metabolizer phenotypes for CYP2C9 (for warfarin), CYP2C19 (for omeprazole and 5‐hydroxy‐omeprazole), and CYP2D6 (for dextromethorphan and dextrorphan). The size of the study was comparable to other reported cocktail drug interaction studies.21, 23
Safety Assessments
Safety was evaluated throughout the study based on adverse event monitoring, vital signs measurements, physical examinations, laboratory tests, and 12‐lead ECGs.
Results
Participants
Twenty healthy subjects (1 woman and 19 men) were enrolled in the study. The mean age was 33 years (range, 21‐54 years), and the mean body mass index was 25.4 kg/m2 (range, 19.6‐29.3 kg/m2). Nine subjects were white (45%), 10 subjects were black (50%), and 1 subject was an American Indian or Alaska native (5%).
Pharmacogenetics
Based on the pharmacogenetics analysis, 14 subjects were classified as extensive CYP2C9 metabolizers, 16 as extensive CYP2C19 metabolizers, and 13 as extensive CYP2D6 metabolizers. None of the subjects was a poor metabolizer for CYP2C9. One subject had a poor CYP2C19 metabolizer phenotype, and 4 had a poor CYP2D6 metabolizer phenotype. A sensitivity analysis excluding subjects who had a poor metabolizer phenotype was conducted, and results were consistent with the results from all subjects (Supplemental Table S1). Therefore, data from all subjects were included in the statistical analysis.
Pharmacokinetics
The individual plasma concentration‐versus‐time profiles for midazolam, caffeine, S‐warfarin, omeprazole, 5‐hydroxy‐omeprazole, and dextromethorphan when administered with and without upadacitinib 30 mg once daily are shown in Figure 2 and Supplemental Figure S1. With the exception of the slight decrease in midazolam plasma concentrations in the presence of upadacitinib, the profiles of the other probe substrates and their metabolites were similar when administered with and without upadacitinib.
Figure 2.
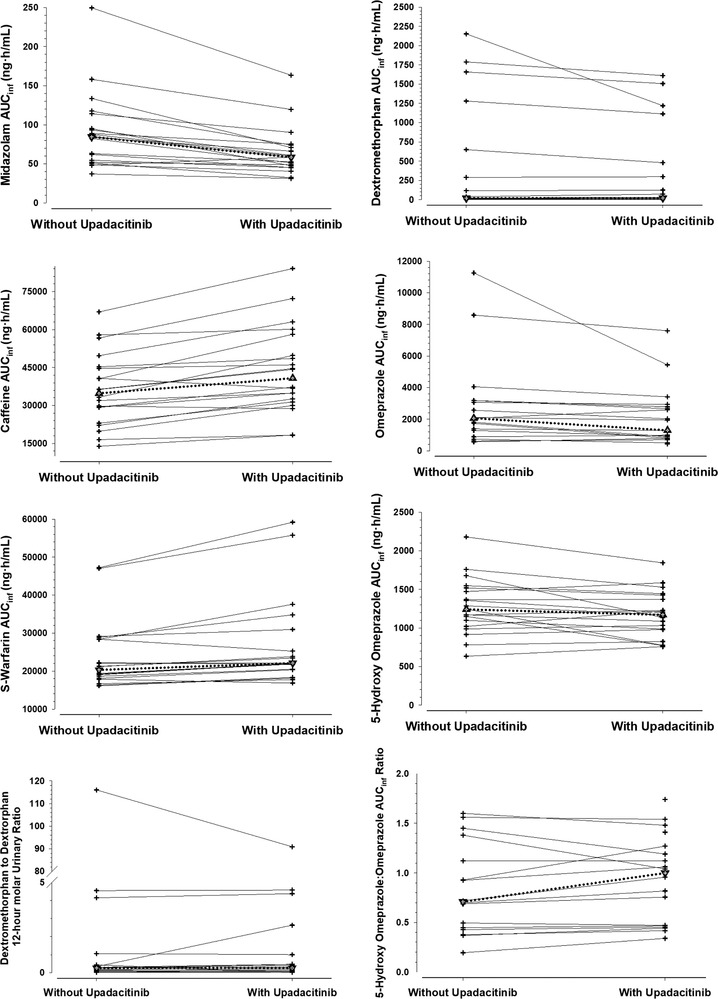
Individual plasma AUCinf and metabolic ratios for the CYP probe substrates when administered with or without upadacitinib 30 mg once daily.
The pharmacokinetic parameters and the ratios of the central values and 90% confidence intervals for the change in Cmax, AUC, and metabolic ratios for the CYP probe substrates and select metabolites in the presence of upadacitinib relative to without upadacitinib are presented in Table 2. Administration of multiple doses of upadacitinib 30 mg once daily slightly decreased midazolam (CYP3A) Cmax and AUCinf by 26% and had no relevant effect (point estimate for the ratio of central values between 0.8 and 1.25) on caffeine, S‐warfarin, omeprazole, 5‐hydroxy‐omeprazole, or dextromethorphan exposure or on the evaluated metabolic ratios (5‐hydroxy‐omeprazole‐to‐omeprazole AUC ratio and dextromethorphan‐to‐dextrorphan molar urinary ratio). An additional analysis of the effect of upadacitinib on 5‐OH‐omeprazole‐to‐omeprazole concentration ratio 2 hours after dosing (ratio for change [90%CI], 1.06 [0.93‐1.21]) was conducted, and the results are consistent with the AUC ratio (Table 2).
Table 2.
Effects of Upadacitinib 30 mg Once Daily on the Pharmacokinetic Parameters of CYP Probe Substrates
| Probe Substrate | Pharmacokinetic Parameter | Period 1 CYP Probes Alone (n = 20) | Period 2 CYP Probes + Upadacitinib 30 mg Once Daily (n = 20) | Central Value Ratio (90%CI)a |
|---|---|---|---|---|
| Midazolam (5 mg) | Cmax, ng/mL | 32.6 ± 14.1 | 23.9 ± 9.83 | 0.74 (0.68‐0.80) |
| AUCt, ng·h/mL | 87.5 ± 44.5 | 63.2 ± 29.6 | 0.74 (0.68‐0.80) | |
| AUCinf, ng·h/mL | 90.3 ± 49.0 | 64.6 ± 30.9 | 0.74 (0.68‐0.80) | |
| CL/F (L/h) | 68.1 ± 29.2 | 90.2 ± 32.7 | – | |
| Tmax, hb | 0.5 (0.5‐1.0) | 0.5 (0.5‐1.0) | – | |
| t1/2, hc | 4.65 ± 2.51 | 4.08 ± 2.33 | – | |
| Caffeine (200 mg) | Cmax, ng/mL | 4190 ± 826 | 4790 ± 1230 | 1.13 (1.05‐1.22) |
| AUCt, ng·h/mL | 35 500 ± 13 900 | 42 500 ± 16 100 | 1.21 (1.15‐1.28) | |
| AUCinf, ng·h/mL | 36 200 ± 14 300 | 43 800 ± 17 100 | 1.22 (1.15‐1.29) | |
| CL/F (L/h) | 6.53 ± 2.99 | 5.31 ± 2.26 | – | |
| Tmax, hb | 2.0 (1.0‐3.0) | 2.0 (1.0‐4.0) | – | |
| t1/2, hc | 4.52 ± 1.50 | 5.28 ± 1.50 | – | |
| Warfarin (10 mg) | S‐warfarin | |||
| Cmax, ng/mL | 630 ± 84.4 | 675 ± 121 | 1.07 (1.02‐1.11) | |
| AUCt, ng·h/mL | 18 600 ± 3860 | 20 400 ± 5010 | 1.09 (1.06‐1.12) | |
| AUCinf, ng·h/mL | 23 800 ± 9030 | 26 700 ± 11 800 | 1.11 (1.07‐1.15) | |
| CL/F (L/h) | 0.462 ± 0.122 | 0.422 ± 0.120 | – | |
| Tmax, hb | 2.0 (0.5‐4.0) | 2.0 (0.5‐3.0) | – | |
| t1/2, hc | 38.6 ± 8.80 | 39.7 ± 10.1 | – | |
| Omeprazole (30 mg) | Omeprazole | |||
| Cmax, ng/mL | 855 ± 544 | 731 ± 484 | 0.87 (0.72‐1.05) | |
| AUCt, ng·h/mL | 2520 ± 2750 | 2000 ± 1820 | 0.87 (0.77‐0.99) | |
| AUCinf, ng·h/mL | 2890 ± 2870 | 2080 ± 1860 | 0.82 (0.72‐0.94) | |
| CL/F (L/h) | 26.5 ± 20.5 | 34.9 ± 24.7 | – | |
| Tmax, hb | 3.0 (2.0‐4.0) | 3.0 (2.0‐6.0) | – | |
| t1/2, hc | 1.13 ± 0.400 | 1.14 ± 0.386 | – | |
| 5‐Hydroxy omeprazole | ||||
| Cmax, ng/mL | 435 ± 155 | 389 ± 116 | 0.92 (0.80‐1.067) | |
| AUCt, ng·h/mL | 1270 ± 359 | 1160 ± 292 | 0.92 (0.85‐0.99) | |
| AUCinf, ng·h/mL | 1290 ± 364 | 1180 ± 302 | 0.92 (0.86‐0.99) | |
| Tmax, hb | 3.0 (2.0‐6.0) | 3.0 (2.0‐6.0) | – | |
| t1/2, hc | 1.36 ± 0.293 | 1.37 ± 0.319 | – | |
| 5‐Hydroxy‐omeprazole to omeprazole AUCt Ratiob , e | 0.92 (0.19‐1.91) | 1.04 (0.32‐1.74) | 1.05 (0.97‐1.14) | |
| 5‐Hydroxy‐omeprazole to omeprazole AUCinf Ratiob , e | 0.71 (0.19‐1.60) | 1.00 (0.34‐1.74) | 1.09 (1.00‐1.19) | |
| Dextromethorphan (30 mg) | Cmax, ng/mL | 7.77 ± 10.1 | 7.64 ± 9.54 | 1.09 (0.98‐1.21) |
| AUCt, ng·h/mL | 250 ± 397 | 225 ± 347 | 1.10 (0.97‐1.25) | |
| AUCinf, ng·h/mL | 403 ± 706 | 328 ± 552 | 1.07 (0.95‐1.22) | |
| CL/F (L/h) | 4120 ± 5580 | 3390 ± 4560 | ||
| Tmax, hb | 3.5 (2.0‐8.0) | 3.5 (2.0‐6.0) | – | |
| t1/2, hc | 6.82 ± 4.68 | 7.26 ± 5.08 | – | |
| Dextromethorphan to dextrorphan molar urinary ratiob , f | 0.263 (0.0190‐116) | 0.263 (0.0289‐90.9) | 1.17 (0.97‐1.41) |
CYP, cytochrome P450; Cmax, maximum plasma concentration; AUC, area under the plasma concentration‐time curve from time 0 to the last measurable concentration (t) or infinity (inf); Tmax, time to Cmax; t1/2, terminal‐phase elimination half‐life
Data are presented as mean ± SD unless noted otherwise.
The point estimate is the antilogarithm of the difference (CYP probe substrate with upadacitinib minus CYP probe substrate alone) of the least‐squares means for logarithms.
Median (range).
Harmonic mean ± pseudo‐SD.
dThe percentage of AUC extrapolated relative to the overall AUCinf exceeded 20% for the majority of subjects; therefore, only AUCt is reported.
n = 19 for 5‐hydroxy‐omeprazole (plasma concentrations of 5‐hydroxy‐omeprazole could not be measured in 1 subject).
n = 17; 3 subjects did not have detectable dextrorphan in urine.
Safety
Administration of single doses of the CYP probe substrates alone or after multiple doses of upadacitinib 30 mg once daily was well tolerated by the healthy subjects in this study. Most adverse events were mild in severity and not considered related to upadacitinib or the probe substrates. One subject reported an adverse event of mild oropharyngeal pain, which was assessed as possibly related to upadacitinib. One subject reported mild nausea, and 1 subject reported mild diarrhea, which were assessed as possibly related to the probe substrates. No subject had a severe or serious adverse event, discontinued from the study because of an adverse event, or had a clinically relevant change in vital signs, laboratory values, or ECGs.
Discussion
This clinical study demonstrated lack of potential for upadacitinib to have a clinically relevant effect on the pharmacokinetics of concomitant medications that are substrates for different CYP enzymes (mainly CYP1A2, CYP2C9, CYP C19, CYP 2D6, and CYP3A). Repeated administration of 30‐mg once‐daily doses of upadacitinib (a dose higher than the optimal dose in RA) had a limited and non‐clinically relevant effect on the plasma exposure of midazolam, a sensitive CYP3A substrate, and no relevant effect on the exposure of sensitive in vivo substrates and markers for all the other evaluated CYP enzymes. These results were consistent with the predicted lack of clinically relevant effect of upadacitinib at the relevant doses and therapeutic exposures in RA on CYP enzymes based on in vitro data and physiologically based pharmacokinetic analyses (data on file at AbbVie).
At the time this study was conducted, phase 3 studies in RA were ongoing, and it was not confirmed yet which of the 2 doses being evaluated in phase 3 (15 mg once daily or 30 mg once daily using the extended‐release formulation) would be the optimal dose.25 Therefore, upadacitinib was administered in this study as repeated 30‐mg once‐daily doses, given that this was the highest dose being evaluated in phase 3 studies in patients with RA and thus represented the highest potential clinical dose in RA patients at that time.1, 2, 3, 4, 5 This is in agreement with regulatory guidance documents, which recommend using the highest clinical dose when evaluating the potential for a drug to be a perpetrator in a clinical drug interaction study.26, 27 Upadacitinib has no accumulation in plasma following multiple once‐daily dosing of the extended‐release formulation; therefore, steady‐state plasma concentrations are achieved after the first dose.28 In the current study, upadacitinib was administered for 10 days before being coadministered with the cocktail drugs to ensure that the maximal effect was characterized for potential CYP induction.29, 30 In addition, upadacitinib was administered alone for an additional 3 days after being coadministered with the CYP phenotyping cocktail to ensure that any potential effect of upadacitinib on CYP enzymes was sustained during washout of the CYP phenotyping cocktail. The current study used a modification of the Cooperstown 5+1 cocktail, which was originally validated with intravenous rather than oral midazolam.20 Therefore, to avoid any potential confounding effect of oral administration of midazolam on the remaining drugs in the CYP phenotyping cocktail, oral midazolam was administered separately from the other drugs of the cocktail. The washout interval of 5 days between the 2 study periods was deemed adequate based on results from prior studies using the same cocktail.20, 21 Among the evaluated probes substrates, S‐warfarin had the longest terminal t1/2 (approximately 40 hours; Table 2). S‐warfarin concentrations at the start of period 2 were very low (<5% of S‐warfarin Cmax), and therefore any S‐warfarin carryover did not meaningfully affect the study results or conclusions.
Typically, a drug is classified as a weak inhibitor if it increases the AUC of a sensitive index CYP substrate by ≥1.25‐ to ≤2‐fold.26 A drug is classified as a weak inducer if it decreases the AUC of a sensitive index CYP substrate by ≥20% to 50%.26 Therefore, an effect that is less than 25% increase or 20% decrease in the AUC of an index inhibitor is not considered a relevant inhibition or induction, respectively. Of all the sensitive probes and markers evaluated, only the effect of upadacitinib 30 mg once daily (a dose higher than the optimal dose in RA) on midazolam slightly exceeded these thresholds (26% decrease in midazolam exposure). This small effect is within or smaller than the expected variability in plasma exposure of the majority of drugs. Upadacitinib has no active major metabolites. Given that midazolam is a sensitive CYP3A substrate and that the effect of upadacitinib on midazolam exposure is relatively small, it is not expected that upadacitinib will have clinically relevant effects on plasma exposure of drugs metabolized by CYP3A or on the plasma exposure of nonactive metabolites formed by CYP3A. In a recent clinical study in healthy female subjects, coadministration of upadacitinib 30 mg once daily with the oral contraceptives ethinylestradiol and levonorgestrel, both of which are CYP3A substrates, had no effect on their exposure; the 90% confidence intervals for the ratios of changes in ethinylestradiol and levonorgestrel AUC and Cmax were within the equivalence boundaries of 0.8 to 1.25.31 This further supports the conclusion that the small effect of upadacitinib on midazolam exposure is not expected to be clinically relevant or to necessitate dose adjustment for concomitant medications that are substrates for CYP3A when coadministered with upadacitinib. Based on physiologically based pharmacokinetic modeling and as expected based on the results from this study, the optimal upadacitinib dose for the RA indication (15 mg once daily) is also not predicted to have any relevant effect on midazolam exposure (∼15% decrease in midazolam AUC, data on file at AbbVie).
Dextromethorphan plasma AUC ratio and dextromethorphan‐to‐dextrorphan molar urinary ratio are both often used as markers for CYP2D6 activity in drug interaction studies.20, 21, 23, 32, 33 The dextromethorphan‐to‐dextrorphan molar urinary ratio was the suggested CYP2D6 activity marker for the Cooperstown 5+1 cocktail.20 In the current study, the results were consistent between the 2 measures, but slightly more variable for dextromethorphan‐to‐dextrorphan molar urinary ratio than for dextromethorphan plasma AUC ratio (Table 2). The consistency of the results supports that either of the 2 markers can be used as a measure for CYP2D6 activity in cocktail studies, with dextromethorphan plasma AUC ratio having the advantage of eliminating the need for urine collection during the studies.
Induction of CYP3A as well as CYP2C enzymes is mediated through the pregnane X nuclear receptor.34 In most cases, the magnitude of the in vivo effect of inducers on sensitive CYP3A substrates is greater than that of inducers on CYP2C substrates.35 It is worth noting that omeprazole is known to be metabolized by CYP2C19 to 5‐OH‐omeprazole and by CYP3A to omeprazole sulfone.36 Therefore, changes in omeprazole exposures when evaluated with perpetrator drugs may be the result of effects on CYP3A, CYP2C19, or both. However, the ratio of 5‐OH‐omeprazole to omeprazole AUC is often used as a more selective marker of a drug effect on CYP2C19.21, 37 Following upadacitinib administration, the 5‐OH‐omeprazole to omeprazole ratio was unchanged, indicating a lack of any effect of upadacitinib on CYP2C19. The lack of effect of upadacitinib on CYP2C19 is in line with expectations based on the very small effect observed on CYP3A in vivo and the expectation of the effect on CYP2C enzymes to be weaker than that on CYP3A.
In the current study, all eligible subjects were enrolled regardless of their metabolic phenotype. One subject had a poor metabolizer phenotype for CYP2C19 and 4 subjects (of 20) had a poor‐metabolizer phenotype for CYP2D6. A sensitivity analysis excluding subjects with a poor‐metabolizer phenotype was conducted, and the results for the assessment of the effects of upadacitinib on CYP2C19 and CYP2D6 in the analysis excluding poor metabolizers were consistent with the results from all subjects (Supplemental Table S1).
This study was not conducted in rheumatoid arthritis patients, the target patient population. However, use of healthy volunteers is the standard for evaluating potential drug interactions,26 the mechanisms of which are not suspected to be disease specific. Use of healthy subjects eliminates some of the confounding factors that exist in patients because healthy subjects have generally lower variability in pharmacokinetics compared with patients, which can be attributed to younger age, lack of other concomitant medications, having normal renal and hepatic function, and less variability in body size per the standard study inclusion criteria. This study was not powered to demonstrate meeting the strict bioequivalence criteria in the presence versus absence of upadacitinib treatment, especially with the large number of different phenotyping metrics simultaneously evaluated in the cocktail approach.
Conclusions
At the 30‐mg once‐daily dose (a dose that is twice the optimal dose in RA), upadacitinib had a limited effect on midazolam exposure, which was not expected to be clinically relevant, and no relevant effect on the exposure of substrates for CYP1A2, CYP2C9, CYP2C19, or CYP2D6. The results of the study suggest that upadacitinib has a low potential to affect the pharmacokinetics of concomitantly administered medications that are metabolized by CYP enzymes.
Conflicts of Interest
AbbVie contributed to the study design, research, and interpretation of data, and the writing, review, and approval of the publication. All authors are employees of AbbVie and may hold AbbVie stock or stock options. The authors thank Allison Kitten, PhD, an employee of AbbVie, for medical writing support.
Supporting information
Figure S1. Individual plasma Cmax profiles for the CYP probe substrates when administered with or without upadacitinib 30 mg once daily.
Table S1. Effects of Upadacitinib 30 mg Once Daily on the Pharmacokinetic Parameters of CYP2C19 and CYP2D6 Probe Substrates Stratified by Poor‐Metabolizer Phenotype
Funding
This work was supported by AbbVie Inc.
Data‐Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (eg, protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan (SAP) and execution of a data‐sharing agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Dr. Ahmed. A. Othman is a Fellow of the American College of Clinical Pharmacology.
References
- 1. Van Vollenhoven R, Takeuchi T, Pangan AL, et al. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX‐naïve patients with active rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smolen J, Pangan AL, Emergy P, et al. A phase 3 randomised, placebo‐controlled, double‐blind study of upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate: SELECT‐MONOTHERAPY. Lancet. 2019;393(10188):2303‐2311. [DOI] [PubMed] [Google Scholar]
- 3. Fleischmann R, Pangan AL, Song IH, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase 3, double‐blind, randomized controlled trial. Arthritis Rhematol. 2019; http://10.1002/art.41032. [DOI] [PubMed] [Google Scholar]
- 4. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease‐modifying anti‐rheumatic drugs (SELECT‐BEYOND): a double‐blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513‐2524. [DOI] [PubMed] [Google Scholar]
- 5. Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10139):2503‐2512. [DOI] [PubMed] [Google Scholar]
- 6. Voss J, Graff C, Schwartz A, et al. Pharmacodynamics of a novel JAK1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects. Ann Rheum Dis. 2014;73(suppl 2):222. [Google Scholar]
- 7. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23(8):1067‐1077. [DOI] [PubMed] [Google Scholar]
- 8. Kremer JM, Emery P, Camp HS, et al. A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol. 2016;68(12):2867‐2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genovese MC, Smolen JS, Weinblatt ME, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68(12):2857‐2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sandborn W, Feagan B, Panes J, et al. Safety and efficacy of ABT‐494 (upadacitinib), an oral Jak1 inhibitor, as induction therapy in patients with Crohn's disease: results from celest. Gastroenterology. 2017;152(5):S1308‐1309. [Google Scholar]
- 11. Guttman‐Yassky E, Silverberg JI, Thaci D, et al. Primary results from a phase 2b, randomized, placebo‐controlled trial of upadacitinib for patients with atopic dermatitis [Abstract #6533]. American Academy of Dermatology Annual Meeting, San Diego, CA, USA. 16–20 February 2018.
- 12. AbbVie . A phase 3 study to compare ABT‐494 to abatacept in subjects with rheumatoid arthritis on stable dose of conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) who have an inadequate response or intolerance to biologic DMARDs (SELECT‐CHOICE) [ClinicalTrials.gov Identifier NCT03086343]. https://clinicaltrials.gov/ct2/show/NCT03086343. Accessed April 17, 2018.
- 13. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55(12):1547‐1558. [DOI] [PubMed] [Google Scholar]
- 14. Mohamed MF, Jungerwirth S, Asatryan A, Jiang P, Othman AA. Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high‐fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol. 2017;83(10):2242‐2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klünder B, Mittapalli RK, Mohamed M‐EF, Friedel A, Noertersheuser P, Othman AA. Population pharmacokinetics of upadacitinib using the immediate‐release and extended‐release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I–III clinical trials. Clin Pharmacokinet. 2019;58(8):1045‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Treharne GJ, Douglas KM, Iwaszko J, et al. Polypharmacy among people with rheumatoid arthritis: the role of age, disease duration and comorbidity. Musculoskeletal Care. 2007;5(4):175‐190. [DOI] [PubMed] [Google Scholar]
- 17. Bagatini F, Blatt CR, Maliska G, et al. Potential drug interactions in patients with rheumatoid arthritis. Rev Bras Reumatol. 2011;51(1):20‐39. [PubMed] [Google Scholar]
- 18. Cross RK, Wilson KT, Binion DG. Polypharmacy and Crohn's disease. Aliment Pharmacol Ther. 2005;21(10):1211‐1216. [DOI] [PubMed] [Google Scholar]
- 19. de Andres F, Llerena A. Simultaneous determination of cytochrome P450 oxidation capacity in humans: a review on the phenotyping cocktail approach. Curr Pharm Biotechnol. 2016;17(13):1159‐1180. [DOI] [PubMed] [Google Scholar]
- 20. Chainuvati S, Nafziger AN, Leeder JS, et al. Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the “Cooperstown 5+1 cocktail”. Clin Pharmacol Ther. 2003;74(5):437‐447. [DOI] [PubMed] [Google Scholar]
- 21. Goh BC, Reddy NJ, Dandamudi UB, et al. An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5+1 cocktail in patients with advanced solid tumors. Clin Pharmacol Ther. 2010;88(5):652‐659. [DOI] [PubMed] [Google Scholar]
- 22. Ma JD, Nafziger AN, Villano SA, Gaedigk A, Bertino JS Jr. Maribavir pharmacokinetics and the effects of multiple‐dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N‐acetyltransferase‐2, and xanthine oxidase activities in healthy adults. Antimicrob Agents Chemother. 2006;50(4):1130‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tran JQ, Othman AA, Wolstencroft P, Elkins J. Therapeutic protein‐drug interaction assessment for daclizumab high‐yield process in patients with multiple sclerosis using a cocktail approach. Br J Clin Pharmacol. 2016;82(1):160‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whirl‐Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mohamed M, Klunder B, Camp H, Othman A. Exposure‐response analyses of upadacitinib efficacy in phase 2 trials in rheumatoid arthritis and basis for phase 3 dose selection. Clin Pharmacol Ther. 2019 10.1002/cpt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. FDA . Clinical Drug Interaction Studies ‐ Study Design, Data Analysis, and Clinical Implications. Guidance for Industry. 2017. https://www.fda.gov/media/82734/download. Accessed July 16, 2019.
- 27. Kowdley KV, Lawitz E, Poordad F, et al. Phase 2b trial of interferon‐free therapy for hepatitis C virus genotype 1. N Engl J Med. 2014;370(3):222‐232. [DOI] [PubMed] [Google Scholar]
- 28. Mohamed MF, Zeng J, Marroum PJ, Song IH, Othman AA. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin Pharmacol Drug Dev. 2019;8(2):208‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamashita F, Sasa Y, Yoshida S, et al. Modeling of rifampicin‐induced CYP3A4 activation dynamics for the prediction of clinical drug‐drug interactions from in vitro data. PLoS One. 2013;8(9):e70330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42(9):819‐850. [DOI] [PubMed] [Google Scholar]
- 31. Mohamed MF, Trueman S, Feng T, Friedman A, Othman AA. The JAK1 inhibitor upadacitinib has no effect on the pharmacokinetics of levonorgestrel and ethinylestradiol: a study in healthy female subjects. J Clin Pharmacol. 2019;59(4):510‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Armani S, Ting L, Sauter N, et al. Drug interaction potential of osilodrostat (LCI699) based on its effect on the pharmacokinetics of probe drugs of cytochrome P450 enzymes in healthy adults. Clin Drug Investig. 2017;37(5):465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamazaki T, Desai A, Goldwater R, et al. Pharmacokinetic effects of isavuconazole coadministration with the cytochrome P450 enzyme substrates bupropion, repaglinide, caffeine, dextromethorphan, and methadone in healthy subjects. Clin Pharmacol Drug Dev. 2017;6(1): 54‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie W, Uppal H, Saini SP, et al. Orphan nuclear receptor‐mediated xenobiotic regulation in drug metabolism. Drug Discov Today. 2004;9(10): 442‐449. [DOI] [PubMed] [Google Scholar]
- 35. FDA . Reference for examples of clinical index inducers for P450‐mediated metabolisms. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/UCM518350.pdf; 2016. Accessed June 17, 2019.
- 36. Andersson T, Miners JO, Veronese ME, et al. Identification of human liver cytochrome P450 isoforms mediating omeprazole metabolism. Br J Clin Pharmacol. 1993;36(6):521‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baldwin RM, Ohlsson S, Pedersen RS, et al. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol. 2008;65(5):767‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Individual plasma Cmax profiles for the CYP probe substrates when administered with or without upadacitinib 30 mg once daily.
Table S1. Effects of Upadacitinib 30 mg Once Daily on the Pharmacokinetic Parameters of CYP2C19 and CYP2D6 Probe Substrates Stratified by Poor‐Metabolizer Phenotype