

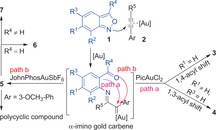
Abstract
We report a switchable synthesis of acylindoles and quinoline derivatives via gold‐catalyzed annulations of anthranils and ynamides. α‐Imino gold carbenes, generated in situ from anthranils and an N,O‐coordinated gold(III) catalyst, undergo electrophilic attack to the aryl π‐bond, followed by unexpected and highly selective 1,4‐ or 1,3‐acyl migrations to form 6‐acylindoles or 5‐acylindoles. With the (2‐biphenyl)di‐tert‐butylphosphine (JohnPhos) ligand, gold(I) carbenes experienced carbene/carbonyl additions to deliver quinoline oxides. Some of these epoxides are valuable substrates for the preparation of 3‐hydroxylquinolines, quinolin‐3(4H)‐ones, and polycyclic compounds via facile in situ rearrangements. The reaction can be efficiently conducted on a gram scale and the obtained products are valuable substrates for preparing other potentially useful compounds. A computational study explained the unexpected selectivities and the dependency of the reaction pathway on the oxidation state and ligands of gold. With gold(III) the barrier for the formation of the strained oxirane ring is too high; whereas with gold(I) this transition state becomes accessible. Furthermore, energetic barriers to migration of the substituents on the intermediate sigma‐complexes support the observed substitution pattern in the final product.
Keywords: acyl migrations, anthranils, epoxidations, gold carbenes, gold catalysis
The golden touch: Catalyst‐controlled, tunable [3+2] and [4+2] annulations were devised for the synthesis of acylindoles and quinoline derivatives. Six divergent compound classes—including 6‐acylindoles 3, 5‐acylindoles 4, quinoline oxides 5, 3‐hydroxylquinolines 6, quinolin‐3(4H)‐ones 7, and a polycyclic compound—were synthesized from anthranils 1 and ynamides 2 by selective acyl migration and epoxide formation via α‐imino gold carbene intermediates.
Introduction
Acyl groups are moieties of fundamental importance in organic chemistry. In contrast to classic Friedel–Crafts acylation reactions,1 the migration of an acyl group is an atom‐economic technique for preparing aryl aldehydes and ketones. In gold catalysis, propargylic esters undergo 1,2‐ or 1,3‐acyloxy migration by nucleophilic attack of the carbonyl oxygen atom at the gold‐activated C≡C triple bond, affording a gold carbene or an allene, which are both highly reactive for subsequent functionalization, and enable the generation of structural complexity.2 A gold‐triggered 1,3‐acyloxy shift and 1,5‐acyl shift cascade for preparing δ‐diketones was devised using more complex propargylic esters.3 Our group developed a gold‐catalyzed tandem 1,3‐acyloxy/1,5‐acyloxy migration for the synthesis of pyrrolidin‐2‐ones.4 1,2‐Acyl migrations for the synthesis of azepino[3,4‐b]indol‐1‐ones and dihydro‐γ‐carbolines were also explored by our group5a and the group of Liu et al.5b To date, although gold‐catalyzed 1,2‐ and 1,5‐acyl, as well 1,3‐ and 1,5‐acyloxy, migrations have been studied well, neither a 1,4‐ nor a 1,3‐acyl migration on a benzene ring has been described to the best of our knowledge.
The development of efficient synthetic methods through α‐imino gold carbene intermediates has become an important tool for the construction of heterocycles. Gold‐promoted nitrene transfer from nucleophilic nitrene equivalents, such as pyridine‐based aza‐ylides,6 azides,7 2H‐azirines,8 isoxazole derivatives,9 and sulfilimines10 to alkynes, generates α‐imino gold carbenes. These can efficiently undergo nucleophilic attack, C−H insertion, or cyclopropanation. However, no carbene/carbonyl additions of these gold carbenes to form epoxides have been reported in the literature. In 2015, the group of Ye et al.9a reported a formal [3+2] annulation of isoxazoles and ynamides for the synthesis of acylpyrroles (R=H, Scheme 1, entry 1). When R≠H, deacylated products are formed. Subsequently, diverse aza‐heterocycles have been prepared from isoxazole derivatives by labile N−O bond cleavage.9b–9t Anthranils, readily available and useful nitrene‐transfer reagents, have attracted considerable interest over the last three years. Either an attack at the nitrogen atom forms α‐imino gold carbenes and subsequent C−H insertions gives 7‐acylindoles (Scheme 1, entry 2),9b or this versatile reagent undergoes oxygen attack to give quinoline oxides through seven‐membered ring intermediates (Scheme 1, entry 3).9i Inspired by previous reports,9, 10, 11 we herein report the unprecedented site‐selective trapping of such α‐imino gold carbenes for a divergent synthesis of acylindoles or epoxides via selective acyl shifts and epoxidations (Scheme 1, entry 4).
Scheme 1.
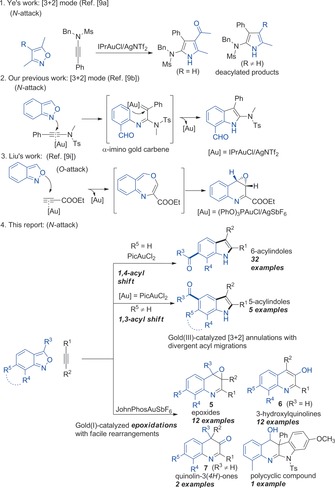
Gold‐catalyzed annulations of alkynes and isoxazole derivatives by N−O bond cleavage.
Results and Discussion
We initially envisioned the synthesis of 2‐aminoindole from 7‐methylanthranil 1 a and N‐phenylynamide 2 ac through a [3+2] C−H annulation (Scheme 2, path a). However, a quinoline oxide 5 b (confirmed by X‐ray crystallography)12 was formed instead via a carbene‐complex‐mediated epoxidation (Scheme 2, path b). An optimization of the reaction conditions was conducted with 7‐methylanthranil 1 a and N‐methylynamide 2 a in the presence of various catalysts LAuCl/AgX and solvents (Table 1). At room temperature and in toluene, IPrAuCl/AgNTf2 produced quinoline oxide 5 a in 20 % yield, along with the unexpected 6‐acylindole 3 a (13 % yield) via an unprecedented 1,4‐acyl migration (Table 1, entry 1). When PPh3 was replaced by (2‐biphenyl)di‐tert‐butylphosphine (JohnPhos), the conversion rates of 1 a were higher than 80 % and the ratios of 5 a/3 a increased dramatically (Table 1, entries 2–5). A change of the silver activators (AgX; X=OTs, OTf, BF4, SbF6) delivered 5 a/3 a with high selectivity (Table 1, entries 6–9), and JohnPhosAuCl/AgSbF6 performed best (Table 1, entry 9). Solvent screening revealed CH2Cl2 as the optimal choice (Table 1, entries 10–12), affording 5 a in 95 % NMR yield (88 % isolated yield on a 0.2 mmol scale). Interestingly, gold(III) catalysts precipitously reversed the reaction to give the totally unexpected 6‐acylindole 3 a as a major product (Table 1, entries 13–18). Best among the gold(III) catalysts and solvents was PicAuCl2,13 giving 3 a in 91 % NMR yield (87 % isolated yield on a 0.2 mmol scale) in CH2Cl2 (Table 1, entry 18). Alkyl‐substituted substrates only afforded products of a 1,2‐H shift.
Scheme 2.
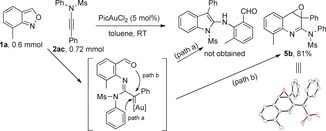
The initial design and the unexpected result.
Table 1.
Optimization of the reaction conditions[a,b].
|
Entry |
Catalyst |
Solvent |
Conversion 1 a [%] |
Yield 3 a [%] |
Yield 5 a [%] |
|---|---|---|---|---|---|
|
1 |
IPrAuCl/AgNTf2 |
toluene |
34 |
13 |
20 |
|
2 |
Ph3PAuCl/AgNTf2 |
toluene |
81 |
8 |
39 |
|
3 |
L1AuCl/AgNTf2 |
toluene |
87 |
11 |
50 |
|
4 |
L2AuCl/AgNTf2 |
toluene |
90 |
<5 |
29 |
|
5 |
L3AuCl/AgNTf2 |
toluene |
80 |
<5 |
51 |
|
6 |
L3AuCl/AgOTs |
toluene |
52 |
<5 |
26 |
|
7 |
L3AuCl/AgOTf |
toluene |
83 |
<5 |
64 |
|
8 |
L3AuCl/AgBF4 |
toluene |
84 |
<5 |
42 |
|
9 |
L3AuCl/AgSbF6 |
toluene |
87 |
<5 |
75 |
|
10 |
L3AuCl/AgSbF6 |
THF |
75 |
<5 |
56 |
|
11 |
L3AuCl/AgSbF6 |
MeCN |
97 |
9 |
77 |
|
12 |
L3AuCl/AgSbF6 |
CH2Cl2 |
>99 |
<5 |
95 (88)[c] |
|
13 |
NaAuCl4⋅2 H2O |
toluene |
92 |
69 |
<5 |
|
14 |
PicAuCl2 |
toluene |
>99 |
78 |
9 |
|
15 |
PicAuCl2 |
THF |
>99 |
42 |
16 |
|
16 |
PicAuCl2 |
MeCN |
>99 |
61 |
20 |
|
17 |
PicAuCl2 |
DCE |
>99 |
54 |
<5 |
|
18 |
PicAuCl2 |
CH2Cl2 |
>99 |
91 (87)[c] |
<5 |
|
19 |
AuBr3 |
CH2Cl2 |
86 |
60 |
16 |
|
| |||||
|
Acetonitrile (MeCN), 1,2‐dichloroethane (DCE), tetrahydrofuran (THF), tosylate (OTs), triflate (OTf), bis(trifluoromethylsulfonyl)imide (NTf2).
| |||||
[a] Reaction conditions: 1 a (0.10 mmol), 2 a (0.12 mmol), catalyst (0.005 mmol), solvent (1.0 mL, 0.1 m), RT. [b] The yield and conversion rate were determined by 1H NMR spectroscopy with 1,3,5‐trimethoxylbenzene as the internal standard. [c] Yields of isolated products (0.2 mmol).
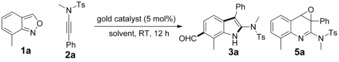
The scope for the synthesis of acylindoles was first investigated under the optimized reaction conditions (Table 1, entry 18). As shown in Scheme 3, anthranil 1 a reacted smoothly with a range of N‐sulfonyl ynamides 2 a–d at room temperature, affording the corresponding 6‐acylindoles 3 a–d in 87–94 % yields. While N‐arylynamides 2 j–n gave the desired products 3 j–n in moderate yields, N‐alkylynamides 2 e–i showed higher efficiency. Yields of 50–99 % were achieved with different ynamides 2 o–x bearing a wide range of functionalities (Me, tBu, OEt, F, Cl, Br, or CF3) at the ortho‐, meta‐, or para‐positions on the arene rings. Dimethoxy ynamide 2 y also underwent the transformation efficiently.12 Compared to phenyl‐substituted substrates, alkyl‐substituted substrates only afforded the products of a 1,2‐H shift; for example, the alkylynamide 2 ac gave the unsaturated amidine 8. The oxazolidinone‐derived ynamide 2 ab was well tolerated to produce 3 ab (66 %). 7‐Methoxyl‐ and 7‐chloroanthranil are appropriate substrates for this reaction. Disubstituted anthranils 1 d and 1 e were converted into the targets 3 ae and 3 af with 99 % yields. Unexpectedly, by blocking the 6‐position of anthranils, 1,3‐acyl migrations proceeded and yielded 5‐acylindoles 4 a–c 12 in 74–99 % yields. Acetyl also migrated successfully and 5‐acetylindole 4 d was obtained in 42 % yield. Notably the tricyclic product 4 e was delivered in 99 % yield.
Scheme 3.
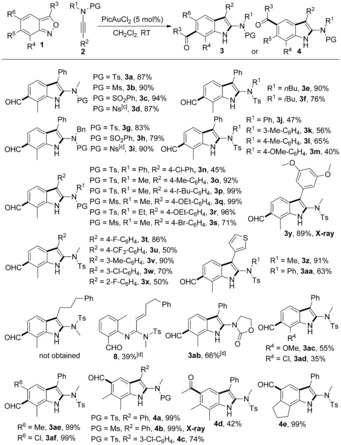
Scope for the synthesis of acylindoles.[a,b] [a] Reaction conditions: 1 (0.20 mmol, 1.0 equiv), 2 (0.24 mmol, 1.2 equiv), PicAuCl2 (3.9 mg, 5 mol %), CH2Cl2 (2 mL, 0.1 m). [b] Yield of isolated product. [c] 4‐nitrophenylsufonyl (Ns). [d] Reaction temperature=50 °C. Key: protecting group (PG), tosly (Ts), mesityl (Ms), nosyl (Ns).
We then explored the scope for the synthesis of quinoline derivatives by using JohnPhosAuCl/AgSbF6 as the best catalyst (Scheme 4). In the case of ynamides 2, electronically diverse aromatic and aliphatic substituents at the R1 position, and different protecting groups, were tolerated well to afford 5 a, 5 c–e, and 6 a–e in 56–97 % yields. Some of these quinoline oxides 5 underwent facile rearrangements14 to 3‐hydroxylquinolines 6 a–e.12 The polycyclic compound 10 was formed by an intramolecular nucleophilic ring opening reaction of epoxide 5 d (Scheme 5). Ynamides containing either an electron‐withdrawing group (F, CF3, or Cl) or an electron‐donating group (CH3 or OCH2CH3) on the arene ring (R2=Ar) were easily converted to epoxides 5 f–j and rearranged products 6 f–h. The epoxidation reaction was applied to phenyl ynamides (R2=phenyl) and heterocyclic ynamide (R2=thiophen‐3‐yl), and the terminal ynamide (R2=H) also reacted well (89 % of 3‐hydroxylquinoline 6 j). An alkyl ynamide delivered unsaturated amidine 8 in 47 % yield instead of the desired quinoline product. Various anthranils were then examined. Anthranil 1 j, without any substituents, gave epoxide 5 k in moderate yield. 7‐Chloro‐ and 7‐bromoanthranils led to the corresponding products 5 l (98 %) and 5 m (80 %). 3‐Methyl‐substituted anthranils 1 i and 1 g delivered quinolin‐3(4H)‐ones 7 a and 7 b through epoxidations and subsequent rearrangements (Scheme 5).
Scheme 4.
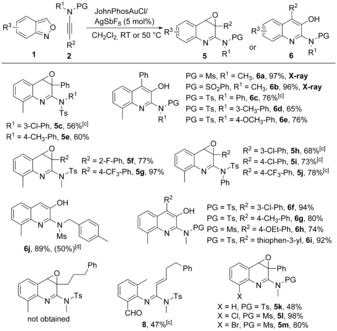
Scope for the synthesis of quinoline derivatives.[a,b] [a] General procedure: 1 (0.24 mmol, 1.2 equiv) and 2 (0.20 mmol, 1.0 equiv) were added to a well‐stirred mixture of JohnPhosAuCl (5.3 mg, 5 mol %) and AgSbF6 (3.4 mg, 5 mol %) in CH2Cl2 (2 mL, 0.1 m). [b] Yield of isolated product. [c] Catalyst (10 mol %), reaction temperature=50 °C. [d] The yield when using 5 mol % PicAuCl2 is shown in parentheses.
Scheme 5.
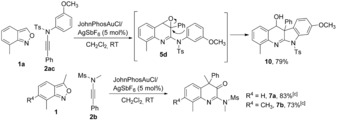
Synthesis of polycyclic compound 10 and quinolin‐3(4H)‐ones 7.[a,b] [a] Reaction conditions: 1 (0.24 mmol, 1.2 equiv), 2 (0.20 mmol, 1.0 equiv), JohnPhosAuCl (5.3 mg, 5 mol %), AgSbF6 (3.4 mg, 5 mol %), CH2Cl2 (2 mL, 0.1 m). [b] Yield of isolated product. [c] Catalyst (10 mol %), reaction temperature=50 °C.
Non‐polarized alkynes were also tested (Scheme 6). At higher temperatures, both PicAuCl2 and JohnPhosAuCl/AgSbF6 were able to catalyze the annulation of 1 a and phenylacetylene to 3‐hydroxylquinoline 6 k in 30 % yield in a slow reaction. 10 mol % Methanesulfonic acid (MsOH) was beneficial for substrate conversion and product formation. Diphenylacetylene also afforded 6 l in 60 % yield.
Scheme 6.
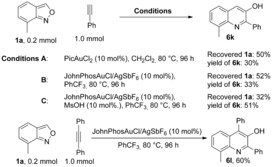
Tests with non‐polarized alkynes.[a] [a] Yield of isolated products.
The gram‐scale reaction between 1 a and 2 a (using only 2 mol % gold catalyst) gave 3 a in 83 % yield (Scheme 7). Even more useful compounds can be prepared from the obtained acylindoles and quinoline oxides. The NaBH4‐mediated reduction of 3 a afforded 6‐hydroxymethylindole 3 a′ in excellent yield, LiAlH4 reduced 3 a to 6,7‐dimethylindole 3 a′′′ in 74 % yield at higher temperature. The acyl group of 3 a could be converted to an alkynyl group through a Seyferth–Gilbert homologation. Treatment of quinoline oxide 5 b with 3 equivalents of LiAlH4 provided facile access to tetrahydroquinolin‐3‐ol 9 via reduction/deamination processes.
Scheme 7.
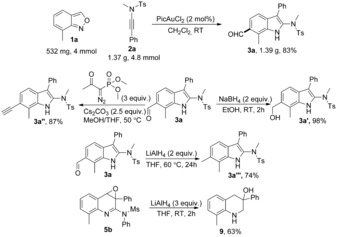
Gram‐scale synthesis and important transformations of 3 a and 5 b.
To gain more insight into the reaction mechanism,15 we conducted two crossover experiments (Scheme 8). With respect to the 1,4‐acyl migration, we conducted a crossover experiment using [D]‐1 a and 1 d reacting with 2 a. For the 1,3‐acyl migration process, the experiment employed a mixture containing an equimolar amount of [D]‐1 k and 1 f in the reaction with ynamide 2 a. A 1H NMR spectroscopy and mass spectrometry study of the obtained products [D]‐3 a, 3 ae, 4 a, and [D]‐4 e showed no scrambling of deuterium; both of these two acyl migration processes are intramolecular.
Scheme 8.
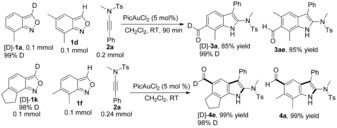
Crossover experiments.
Finally, we conducted a detailed computational study of the reaction mechanism at the M06‐D3/def2‐TZVP//M06/LANL2DZ,6‐31G(d) level in CH2Cl2. We commenced our DFT‐based mechanistic study by investigating which coordinating atom of the ligand on complex i is involved in the substitution reaction—a question that has not been studied in the many applications of i in catalysis. To this end, four trigonal bipyramidal transition structures TSi–TSiv were calculated for which the potential energies are reported underneath each structure in Figure 1.
Figure 1.
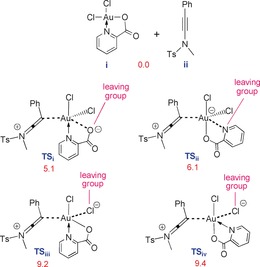
The potential energies (kcal mol−1) for transition structures TSi–TSiv.
Our calculations indicate that TSi is lower in energy than the other three transition states, suggesting that the substitution reaction should mostly occur through TSi to give intermediate iii (Figure 2) from which the remainder of the catalytic reaction proceeds. Once structure iii is formed, it is subjected to a nucleophilic attack by substrate iv to afford v through transition structure TSiii‐v, which lies only 6.8 kcal mol−1 above iii. The ensuing intermediate then undergoes N−O bond cleavage via transition structure TSv‐vi with an overall activation free energy of 14.4 kcal mol−1, leading to exergonic formation of intermediate vi.
Figure 2.
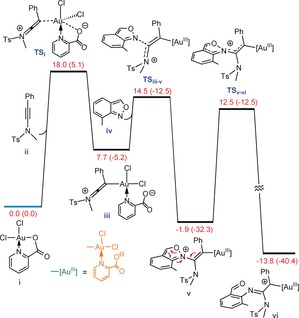
Free energy profile for the substitution reaction between i and ii, nucleophilic attack of iv at iii, and finally cleavage of the O−N bond in v forming vi. Gibbs free energies (potential energies) are in kcal mol−1.
Intermediate vi is a branching point for two pathways (Figure 3) as follows: 1) nucleophilic attack of the carbonyl group followed by a deauration reaction via TSvii (pathway A) and 2) nucleophilic attack of the aryl ring to afford intermediate ix (pathway B). Our calculations show that the carbonyl group is a better nucleophile than the aryl ring but formation of vii is a dead end, from which the deauration process is very energy consuming with ΔG ≠>30 kcal mol−1. This low reactivity toward C−C coupling (deauration with formation of a strained three‐membered oxirane ring) is ascribed to the high electrophilicity of the AuIII center. Thus, although formation of vii is feasible during the reaction, it undergoes a reverse reaction to give vi from which ix is formed via transition structure TSvi ‐ix.
Figure 3.
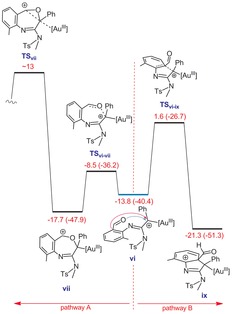
Free energy profile for competition between two pathways A and B. Gibbs free energies (potential energies) are in kcal mol−1.
Attempts to locate transition structure TSvii were unsuccessful. Thus, the energy of this transition structure was estimated by the scan of the potential energy surface, as illustrated in Figure 4.
Figure 4.
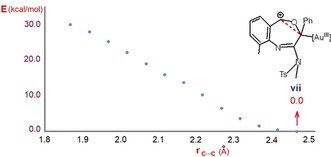
Scan of the potential energy surface for formation of the C−C bond starting from structure vii.
Intermediate ix is a branching point for two competitive pathways (Figure 5). The formyl group can migrate along either the anticlockwise (pathway C) or clockwise (pathway D) direction. Pathway C was calculated to be about 4.4 kcal mol−1 higher in energy than pathway D, implying that the formyl migration preferably takes place via pathway D. A potentially competing methyl shift would have a much higher barrier TSxi ‐xii′ to provide xii′. The preference for pathway D over C can be easily explained by the natural population analysis (NPA) of intermediate ix (Figure 5). The analysis shows that the partial charge of the carbon atom bonded to the sp2 nitrogen is much more electron deficient than other centers, resulting in the formyl group preferentially migrating along the clockwise direction.
Figure 5.
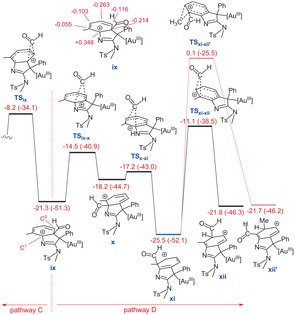
Free energy profile for competition between two pathways C and D. The NPA charges for intermediate ix are given in pink. Gibbs free energies (potential energies) are in kcal mol−1.
Once xii is formed, it can be consumed in two ways (Figure 6). It can be involved in another formyl migration (pathway E) or in a deprotonation process assisted by a base‐like substrate iv (pathway F). In agreement with the experiment, pathway F was determined to be more favorable. Indeed, this pathway is preceded by protodeauration (proton transfer from xiii to the sp2 nitrogen atom of xiv in a barrierless reaction) and finally produces the experimentally observed product. Based on these findings, we proposed a catalytic cycle for the acyl migration processes (Figure 7).
Figure 6.
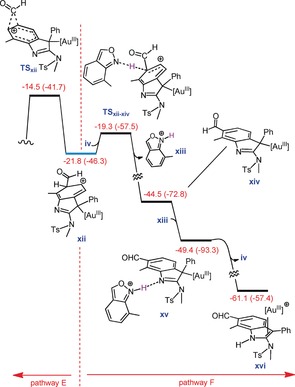
Free energy profile (kcal mol−1) for competition between two pathways E and F.
Figure 7.
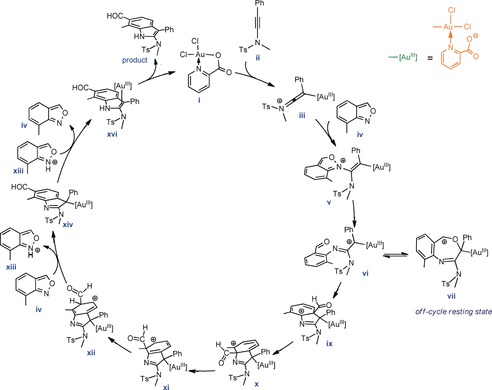
A proposed catalytic cycle derived from the DFT calculations.
At this juncture, we are interested in understanding why the gold(I) complex mainly gives the epoxide product, which contrasts with the behavior of the gold(III) complex. This difference stems from the fact that the gold(I) complex contains a less electrophilic center compared with the gold(III) complex. As a consequence, the deauration reaction that starts from intermediate xix, and proceeds via C−C coupling, takes place more easily. Our calculations support this claim by showing that the energies of TSxviii ‐xx and TSxvii‐xviii are comparable, resulting in both pathways A′ and B′ being competitive (Figure 8).
Figure 8.
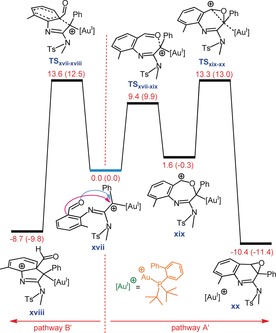
Free energy profile for competition between two pathways A′ and B′ where gold(I) is used as the catalyst. Gibbs free energies (potential energies) are in kcal mol−1.
Conclusion
In summary, catalyst‐controlled, tunable acyl migrations and epoxidations were discovered that occur via α‐imino gold carbene intermediates. This efficient, scalable, atom‐economic reaction shows high regioselectivity and broad scope, and it operates under mild conditions with easily available substrates. The method opens novel and concise routes to complicated acylindoles, quinoline oxide derivatives, polycyclic compounds, and related compounds—all of which are potentially useful in medicinal chemistry and drug discovery. A computational study fully supported the observed chemoselectivity for gold(III), as well as the selectivity switch when using gold(I) catalysts. The effects explaining these selectivities and selectivity switches will be inspiring for further research in the field.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
X.T. and L.S. are grateful to the CSC (China Scholarship Council) for a PhD fellowship. We gratefully acknowledge the generous allocation of computing time from the Australian National Computational Infrastructure and University of Tasmania, and the Australian Research Council (grant number DP180100904) for financial support.
X. Tian, L. Song, K. Farshadfar, M. Rudolph, F. Rominger, T. Oeser, A. Ariafard, A. S. K. Hashmi, Angew. Chem. Int. Ed. 2020, 59, 471.
Contributor Information
Prof. Dr. Alireza Ariafard, Email: alirezaa@utas.edu.au.
Prof. Dr. A. Stephen K. Hashmi, Email: hashmi@hashmi.de.
References
- 1.
- 1a. Olah G. A., Friedel–Crafts Chemistry, Wiley, New York, 1973; [Google Scholar]
- 1b. Sartori G., Maggi R., Chem. Rev. 2006, 106, 1077. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews, see:
- 2a. Day D. P., Chan P. W. H., Adv. Synth. Catal. 2016, 358, 1368; [Google Scholar]
- 2b. Kazem Shiroodi R., Gevorgyan V., Chem. Soc. Rev. 2013, 42, 4991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Wang S., Zhang G., Zhang L., Synlett 2010, 692; [Google Scholar]
- 2d. Marion N., Nolan S. P., Angew. Chem. Int. Ed. 2007, 46, 2750; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2806. For selected examples on metal-catalyzed acyloxy migrations, see: [Google Scholar]
- 2e. Sromek A. W., Kel'in A. V., Gevorgyan V. A., Angew. Chem. Int. Ed. 2004, 43, 2280; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2330; [Google Scholar]
- 2f. Mamane V., Gress T., Krause H., Fürstner A., J. Am. Chem. Soc. 2004, 126, 8654; [DOI] [PubMed] [Google Scholar]
- 2g. Shi X., Gorin D. J., Toste F. D., J. Am. Chem. Soc. 2005, 127, 5802; [DOI] [PubMed] [Google Scholar]
- 2h. Zhang L., J. Am. Chem. Soc. 2005, 127, 16804; [DOI] [PubMed] [Google Scholar]
- 2i. Johansson M. J., Gorin D. J., Staben S. T., Toste F. D., J. Am. Chem. Soc. 2005, 127, 18002; [DOI] [PubMed] [Google Scholar]
- 2j. Garayalde D., Gómez-Bengoa E., Huang X., Goeke A., Nevado C., J. Am. Chem. Soc. 2010, 132, 4720; [DOI] [PubMed] [Google Scholar]
- 2k. Rettenmeier E., Schuster A. M., Rudolph M., Rominger F., Gade C., Hashmi A. S. K., Angew. Chem. Int. Ed. 2013, 52, 5880; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5993; [Google Scholar]
- 2l. Yu Y., Yang W., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2013, 52, 7586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7735; [Google Scholar]
- 2m. Zhao C., Xie X., Duan S., Li H., Fang R., She X., Angew. Chem. Int. Ed. 2014, 53, 10789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10965; [Google Scholar]
- 2n. Qiu Y., Zhou J., Fu C., Ma S., Chem. Eur. J. 2014, 20, 14589; [DOI] [PubMed] [Google Scholar]
- 2o. Liu J., Chen M., Zhang L., Liu Y., Chem. Eur. J. 2015, 21, 1009. [DOI] [PubMed] [Google Scholar]
- 3. Lebœuf D., Simonneau A., Aubert C., Malacria M., Gandon V., Fensterbank L., Angew. Chem. Int. Ed. 2011, 50, 6868; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7000. [Google Scholar]
- 4. Hashmi A. S. K., Yang W., Yu Y., Hansmann M. M., Rudolph M., Rominger F., Angew. Chem. Int. Ed. 2013, 52, 1329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1368. [Google Scholar]
- 5.
- 5a. Hashmi A. S. K., Yang W., Rominger F., Adv. Synth. Catal. 2012, 354, 1273; [Google Scholar]
- 5b. Wang L., Xie X., Liu Y., Angew. Chem. Int. Ed. 2013, 52, 13302; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13544. [Google Scholar]
- 6.
- 6a. Davies P. W., Cremonesi A., Dumitrescu L., Angew. Chem. Int. Ed. 2011, 50, 8931; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9093; [Google Scholar]
- 6b. Li C., Zhang L., Org. Lett. 2011, 13, 1738; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Garzón M., Davies P. W., Org. Lett. 2014, 16, 4850; [DOI] [PubMed] [Google Scholar]
- 6d. Reddy R. J., Ball-Jones M. P., Davies P. W., Angew. Chem. Int. Ed. 2017, 56, 13310; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13495; [Google Scholar]
- 6e. Chatzopoulou E., Davies P. W., Chem. Commun. 2013, 49, 8617; [DOI] [PubMed] [Google Scholar]
- 6f. Hung H.-H., Liao Y.-C., Liu R.-S., Adv. Synth. Catal. 2013, 355, 1545. [Google Scholar]
- 7.
- 7a. Gorin D. J., Davis N. R., Toste F. D., J. Am. Chem. Soc. 2005, 127, 11260; [DOI] [PubMed] [Google Scholar]
- 7b. Lu B., Luo Y., Liu L., Ye L., Wang Y., Zhang L., Angew. Chem. Int. Ed. 2011, 50, 8358; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8508; [Google Scholar]
- 7c. Wetzel A., Gagosz F., Angew. Chem. Int. Ed. 2011, 50, 7354; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7492; [Google Scholar]
- 7d. Xiao Y., Zhang L., Org. Lett. 2012, 14, 4662; [DOI] [PubMed] [Google Scholar]
- 7e. Yan Z. Y., Xiao Y., Zhang L., Angew. Chem. Int. Ed. 2012, 51, 8624; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8752; [Google Scholar]
- 7f. Tokimizu Y., Oishi S., Fujii N., Ohno H., Org. Lett. 2014, 16, 3138; [DOI] [PubMed] [Google Scholar]
- 7g. Shu C., Wang Y.-H., Zhou B., Li X.-L., Ping Y.-F., Lu X., Ye L.-W., J. Am. Chem. Soc. 2015, 137, 9567; [DOI] [PubMed] [Google Scholar]
- 7h. Wu Y., Zhu L., Yu Y., Luo X., Huang X., J. Org. Chem. 2015, 80, 11407. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Zhu L., Yu Y., Mao Z., Huang X., Org. Lett. 2015, 17, 30; [DOI] [PubMed] [Google Scholar]
- 8b. Prechter A., Henrion G., dit Bel P. F., Gagosz F., Angew. Chem. Int. Ed. 2014, 53, 4959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5059; [Google Scholar]
- 8c. Pawar S. K., Sahani R. L., Liu R.-S., Chem. Eur. J. 2015, 21, 10843. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Zhou A.-H., He Q., Shu C., Yu Y.-F., Liu S., Zhao T., Zhang W., Lu X., Ye L.-W., Chem. Sci. 2015, 6, 1265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Jin H., Huang L., Xie J., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 804; [Google Scholar]
- 9c. Chen M., Sun N., Chen H., Liu Y., Chem. Commun. 2016, 52, 6324; [DOI] [PubMed] [Google Scholar]
- 9d. Jin H., Tian B., Song X., Xie J., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 12688; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12880; [Google Scholar]
- 9e. Sahani R. L., Liu R.-S., Angew. Chem. Int. Ed. 2017, 56, 1026; [Google Scholar]; Angew. Chem. 2017, 129, 1046; [Google Scholar]
- 9f. Zeng Z., Jin H., Xie J., Tian B., Rudolph M., Rominger F., Hashmi A. S. K., Org. Lett. 2017, 19, 1020; [DOI] [PubMed] [Google Scholar]
- 9g. Xu W., Wang G., Sun N., Liu Y., Org. Lett. 2017, 19, 3307; [DOI] [PubMed] [Google Scholar]
- 9h. Shen W.-B., Xiao X.-Y., Sun Q., Zhou B., Zhu X.-Q., Yan J.-Z., Lu X., Ye L.-W., Angew. Chem. Int. Ed. 2017, 56, 605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 620; [Google Scholar]
- 9i. Sahani R. L., Liu R.-S., Angew. Chem. Int. Ed. 2017, 56, 12736; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12910; [Google Scholar]
- 9j. Zeng Z., Jin H., Sekine K., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 6935; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7051; [Google Scholar]
- 9k. Jadhav P. D., Lu X., Liu R.-S., ACS Catal. 2018, 8, 9697; [Google Scholar]
- 9l. Zhao Y., Wang C., Hu Y., Wan B., Chem. Commun. 2018, 54, 3963; [DOI] [PubMed] [Google Scholar]
- 9m. Giri S. S., Liu R.-S., Chem. Sci. 2018, 9, 2991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9n. Tsai M.-H., Wang C.-Y., Raja A. S. K., Liu R.-S., Chem. Commun. 2018, 54, 10866; [DOI] [PubMed] [Google Scholar]
- 9o. Mokar B. D., Jadhav P. D., Pandit Y. B., Liu R.-S., Chem. Sci. 2018, 9, 4488; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9p. Zhu X.-Q., Yuan H., Sun Q., Zhou B., Han X.-Q., Zhang Z.-X., Lu X., Ye L.-W., Green Chem. 2018, 20, 4287; [Google Scholar]
- 9q. Xu W., Zhao J., Li X., Liu Y., J. Org. Chem. 2018, 83, 15470; [DOI] [PubMed] [Google Scholar]
- 9r. Pandit Y. B., Sahani R. L., Liu R.-S., Org. Lett. 2018, 20, 6655; [DOI] [PubMed] [Google Scholar]
- 9s. Zeng Z., Jin H., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 16549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16787; [Google Scholar]
- 9t. Song L., Tian X., Rudolph M., Rominger F., Hashmi A. S. K., Chem. Commun. 2019, 55, 9007. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Tian X., Song L., Rudolph M., Rominger F., Oeser T., Hashmi A. S. K., Angew. Chem. Int. Ed. 2019, 58, 3589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3627; [Google Scholar]
- 10b. Tian X., Song L., Rudolph M., Wang Q., Song X., Rominger F., Hashmi A. S. K., Org. Lett. 2019, 21, 1598; [DOI] [PubMed] [Google Scholar]
- 10c. Tian X., Song L., Han C., Zhang C., Wu Y., Rudolph M., Rominger F., Hashmi A. S. K., Org. Lett. 2019, 21, 2937; [DOI] [PubMed] [Google Scholar]
- 10d. Tian X., Song L., Rudolph M., Rominger F., Hashmi A. S. K., Org. Lett. 2019, 21, 4327. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Hashmi A. S. K., Frost T. M., Bats J. W., J. Am. Chem. Soc. 2000, 122, 11553; [Google Scholar]
- 11b. Li G., Zhang L., Angew. Chem. Int. Ed. 2007, 46, 5156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5248; [Google Scholar]
- 11c. Shapiro N. D., Toste F. D., J. Am. Chem. Soc. 2007, 129, 4160; [DOI] [PubMed] [Google Scholar]
- 11d. He W., Li C., Zhang L., J. Am. Chem. Soc. 2011, 133, 8482; [DOI] [PubMed] [Google Scholar]
- 11e. Wang Y., Ye L., Zhang L., Chem. Commun. 2011, 47, 7815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Kramer S., Skrydstrup T., Angew. Chem. Int. Ed. 2012, 51, 4681; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4759; [Google Scholar]
- 11g. Huang X., Peng B., Luparia M., Gomes L. F., Veiros L. F., Maulide N., Angew. Chem. Int. Ed. 2012, 51, 8886; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9016; [Google Scholar]
- 11h. Wang Y., Zarca M., Gong L.-Z., Zhang L., J. Am. Chem. Soc. 2016, 138, 7516; [DOI] [PubMed] [Google Scholar]
- 11i. Huple D. B., Ghorpade S., Liu R.-S., Adv. Synth. Catal. 2016, 358, 1348. [Google Scholar]
- 12.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201912334 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
- 13.For the benefits of N,O-ligands, see: Hashmi A. S. K., Weyrauch J. P., Rudolph M., Kurpejović E., Angew. Chem. Int. Ed. 2004, 43, 6545; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6707. [Google Scholar]
- 14.
- 14a. Maruoka K., Murase N., Bureau R., Ooi T., Yamamoto H., Tetrahedron 1994, 50, 3663; [Google Scholar]
- 14b. Kulasegaram S., Kulawiec R. J., Tetrahedron 1998, 54, 1361; [Google Scholar]
- 14c. Kita Y., Futamura J., Ohba Y., Sawama Y., Ganesh J. K., Fujioka H., J. Org. Chem. 2003, 68, 5917; [DOI] [PubMed] [Google Scholar]
- 14d. González M. J., González J., Vicente R., Eur. J. Org. Chem. 2012, 6140. [Google Scholar]
- 15.
- 15a. Hashmi A. S. K., Angew. Chem. Int. Ed. 2010, 49, 5232; [DOI] [PubMed] [Google Scholar]
- 15b. Hashmi A. S. K., Rudolph M., Siehl H.-U., Tanaka M., Bats J. W., Frey W., Chem. Eur. J. 2008, 14, 3703; [DOI] [PubMed] [Google Scholar]
- 15c. Hashmi A. S. K., Schwarz L., Choi J.-H., Frost T. M., Angew. Chem. Int. Ed. 2000, 39, 2285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary