

Summary
B‐cell and antibody responses to Plasmodium spp., the parasite that causes malaria, are critical for control of parasitemia and associated immunopathology. Antibodies also provide protection to reinfection. Long‐lasting B‐cell memory has been shown to occur in response to Plasmodium spp. in experimental model infections, and in human malaria. However, there are reports that antibody responses to several malaria antigens in young children living with malaria are not similarly long‐lived, suggesting a dysfunction in the maintenance of circulating antibodies. Some studies attribute this to the expansion of atypical memory B cells (AMB), which express multiple inhibitory receptors and activation markers, and are hyporesponsive to B‐cell receptor (BCR) restimulation in vitro. AMB are also expanded in other chronic infections such as tuberculosis, hepatitis B and C, and HIV, as well as in autoimmunity and old age, highlighting the importance of understanding their role in immunity. Whether AMB are dysfunctional remains controversial, as there are also studies in other infections showing that AMB can produce isotype‐switched antibodies and in mouse can contribute to protection against infection. In light of these controversies, we review the most recent literature on either side of the debate and challenge some of the currently held views regarding B‐cell responses to Plasmodium infections.
Keywords: classical and atypical memory B cells, long‐lived plasma cells, malaria, Plasmodium
1. INTRODUCTION
Malaria is a killer disease caused by infection with species of the protozoan parasite, Plasmodium. The most deadly of these parasites is Plasmodium falciparum, for which the estimates of morbidity and mortality in Africa were 219 million and 435 000, respectively, in 2017.1 Although several control methods have been employed with substantial success, malaria continues to place a heavy burden on the health systems and economies of the countries affected. Experts agree on the need for the development and subsequent deployment of an effective vaccine, which would be the most cost‐effective means of disease control and could even lead to elimination of malaria. However, despite decades of research in malaria vaccine development, a highly effective vaccine remains elusive. RTS,S/AS01, the most advanced malaria vaccine so far, has 30% efficacy of short‐lived protection.2
A better understanding of the host immune response to malaria, and particularly how to induce and maintain protective levels of circulating antibodies, would be highly valuable for producing effective vaccines which provide long‐lasting protection from malaria. Through extensive studies of humoral responses to immunization with model antigens and in acute viral infections, it is generally accepted that there are two types of long‐lived pathogen‐specific cells of the B‐cell lineage commonly persisting in the memory pool: long‐lived plasma cells, which secrete specific antibodies, in some cases for life; and memory B cells, which confer rapid and enhanced responses to secondary pathogen challenge. Follicular helper T‐cell (Tfh) and germinal center (GC) B‐cell responses are essential to generate isotype‐switched long‐lived plasma cells and memory B cells3 (Figure 1A). An understanding of whether and how the immune response is compromised, and of the true longevity of the memory compartments in Plasmodium infection is necessary for successful vaccine development.
Figure 1.
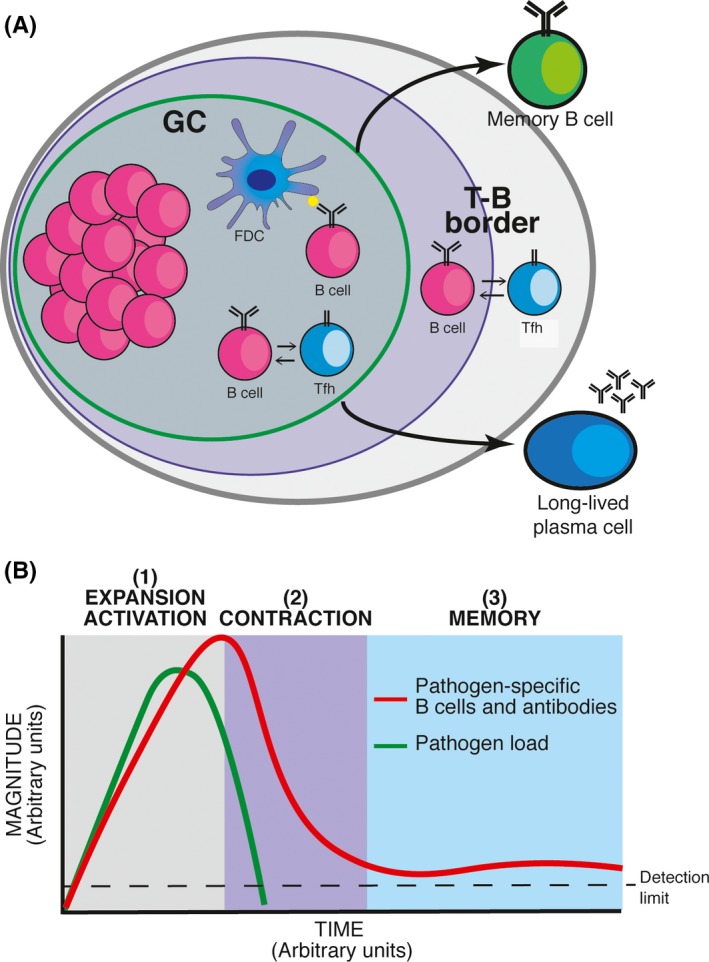
Pathogen‐specific B‐cell responses. (A) Schematic representation of the germinal center (GC) B‐cell response, which results in the generation of two arms of B‐cell memory, long‐lived plasma cells and memory B cells. (B) Schematic representation of the kinetics of B‐cell responses to pathogens, showing the expansion/activation phase (1), the contraction phase (2), and the memory phase (3). FDC: follicular dendritic cell; Tfh: follicular helper T cell
Protozoan parasites, such as Plasmodium, have complex life cycles and, in many cases, different cellular forms within the vertebrate host. The vast majority of protozoan parasites give rise to long‐lasting, and in some cases, lifelong, chronic infections, dramatically shaped by sophisticated immune evasion mechanisms which include a complex and diverse antigenic repertoire. Thus, protozoan parasites represent a substantial challenge to the immune system. Mechanisms that regulate B‐cell responses to Plasmodium species have gained increasing attention in recent years. It is now well‐established that B cells and antibodies are critical to control Plasmodium infection and to provide immunity to reinfection.4, 5, 6, 7, 8, 9, 10 Plasmodium‐specific antibodies, in particular of the IgG subclasses, act by inhibiting Plasmodium replication and cell invasion, opsonizing extracellular forms as well as infected red blood cells for their destruction by phagocytic cells, and promoting lysis by the complement.11, 12, 13
In contrast, Plasmodium infections trigger a series of temporary yet striking events that can potentially alter Plasmodium‐specific B‐cell responses. These include pronounced inflammation14 polyclonal B‐cell activation and hypergammaglobulinemia,15, 16, 17 alterations in splenic and bone marrow microarchitecture,18, 19, 20 and alteration in hematopoiesis.21, 22 Moreover, a series of field studies have suggested that B cell responses to Plasmodium spp. might be dysfunctional, with poor acquisition of long‐lasting B‐cell responses and accumulation of “exhausted” B cells.23, 24, 25 An intriguing subset of B cells expressing the transcription factor T‐bet, termed AMB, is expanded in subjects exposed to Plasmodium infection.23 B cells with similar phenotypical characteristics have been observed in response to other infections,23, 26, 27, 28 autoimmunity,29 and aging.30 Whether T‐bet+ AMB cells contribute to protection from infection or rather represent a dysfunctional B‐cell subset that leads to parasite persistence and pathology remains a focus of intense debate.
Here, we will review and challenge some currently held views regarding B‐cell responses to malaria, with a focus on the longevity of the circulating antibody response and potential roles of AMB in Plasmodium infection in humans and mice.
1.1. Are B‐cell responses to malaria short‐lived?
Immunological memory refers to long‐lived immunity sustained in the absence of pathogen re‐exposure. The B‐cell response to a pathogen presents three distinct phases (Figure 1B): (a) Expansion and activation: encounter with the pathogen results in the activation and extensive proliferation of B cells, leading to several fold increase in the frequency of pathogen‐specific B cells as well as the production of pathogen‐specific antibodies by plasmablasts and short‐lived plasma cells; (b) Contraction: as the pathogen load is controlled by the immune response or curtailed by drug treatment, both the frequency of pathogen‐specific B cells and the titer of pathogen‐specific antibodies drop significantly, (c) Memory: following pathogen clearance, a subset of pathogen‐specific long‐lived plasma cells and memory B cells survive the contraction phase; the former continue to produce pathogen‐specific antibodies, sustaining their circulating levels above background; the later recirculate through blood and secondary lymphoid organs readily armed for a second encounter with the same pathogen which initiated the response. Subsequent encounters with the same pathogen have a cumulative effect, resulting in increased precursor frequency of pathogen‐specific memory B cells with each round of exposure. Due to increased frequencies, reduced activation threshold, as well as isotype switching and affinity maturation resulting from GC reactions, the response of memory B cells is typically faster and of greater magnitude compared with that of naive B cells, and results in faster and greater production of antibodies of switched isotypes and increased affinity.31
While phases 1 and 2 can typically last for weeks to months, the memory phase can last for years to decades and even for life in the absence of pathogen re‐exposure.32, 33 Long‐term immunity is a feature of many systemic infections such as mumps, polio, yellow fever, smallpox, measles, and rubella.32, 34 Studies showed that detectable antibody titers to smallpox could be sustained for over 75 years after a single vaccination,35, 36 and smallpox‐specific memory B cells could be detected in the blood of vaccinees up to 60 years postvaccination.37 Amanna and colleagues performed a longitudinal analysis of antibody titers and memory B‐cell frequencies specific for viral antigens (vaccinia, measles, mumps, rubella, varicella–zoster virus, and Epstein–Barr virus) and nonreplicating antigens (tetanus and diphtheria).32 Both antibody responses and memory B‐cell responses were remarkably stable and long‐lived, with antibody responses half‐lives ranging from an estimated 50 years for varicella–zoster virus to more than 200 years for other viruses such as measles and mumps.32 Similar longevity of virus‐specific B‐cell memory was observed to natural exposure, as demonstrated by the identification of memory B cells specific for the 1918 pandemic strain of influenza virus circulating in the blood of survivors 90 years after primary exposure.33 In comparison, antibody responses to tetanus and diphtheria showed a shorter half‐life (11‐27 years) but still lasting at least a decade, and even over 50 years in some diphtheria cases.32, 35, 38
Plasmodium infection, with its complex life cycle, antigenic variation/polymorphism, and its ability to establish chronic infection could well influence development and longevity of B‐cell responses, and might not necessarily reflect the kinetics observed after acute viral or bacterial infections or vaccination. In fact, immunity to Plasmodium parasites is acquired much less efficiently than in most viral and bacterial infections, where a single infection is enough (sometimes) for life‐long protection.39
Plasmodium parasites present two very distinctive stages in the mammalian host (Figure 2A). An initial pre‐erythrocytic or liver stage, during which Plasmodium sporozoites invade hepatocytes. The liver stage can last between 2 and 7 days,40 depending on the parasite and host species, and does not lead to clinical disease. The liver stage is followed by a more prolonged erythrocytic stage in which Plasmodium merozoites invade red blood cells, giving rise to a continuous cycle of invasion, replication, and cell destruction followed by more invasion, leading to clinical manifestations which in some cases can be severe or even lethal. As mentioned, Plasmodium‐specific antibodies confer protection from Plasmodium blood‐stage infection in both human and mice.41 However, in humans, the presence of Plasmodium‐specific antibodies alone seems not sufficient to prevent reinfection.
Figure 2.
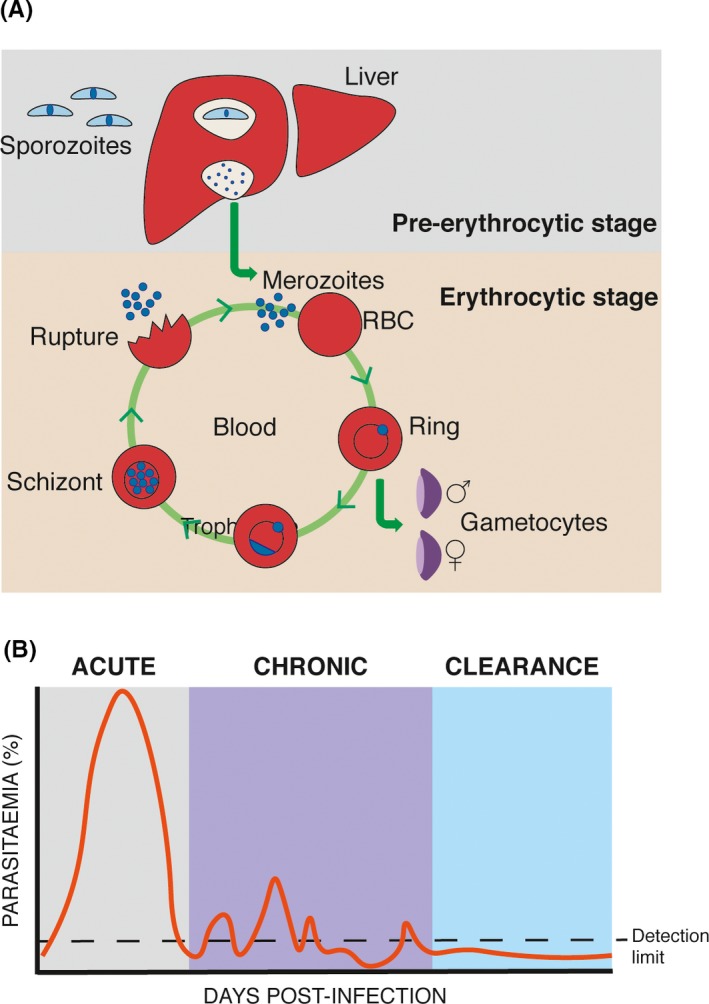
Plasmodium infection. (A) Schematic representation of pre‐erythrocytic (liver) and erythrocytic (blood) stages of Plasmodium spp. infection in the mammalian host. (B) Schematic representation of the course of erythrocytic P. chabaudi infection in C57BL/6 mice
In areas of stable transmission, Plasmodium‐associated life‐threatening disease is very much restricted to young children under 5 years of age and pregnant women.41, 42 Immunity to these severe clinical manifestations is acquired rapidly, generally after one or two infections, and appears to be maintained in the absence of boosting by reinfection.43 In contrast, immunity to infection and non‐severe disease takes several years to develop and requires continuous exposure to the parasite. These observations prompted the hypothesis that B‐cell responses to Plasmodium parasites might be dysfunctional or suboptimal, and that generation of long‐lasting humoral memory to the parasite might be defective. In support of this, a series of field studies showed that titers of Plasmodium‐specific antibodies drop rapidly after acute infection.44, 45, 46, 47 Moreover, persistent infection seems to be required to maintain high titers of Plasmodium‐specific antibodies, as reduction of transmission due to insecticide spraying or antimalarial treatment leads to a general reduction of Plasmodium‐specific antibodies, albeit not to reach background levels from naive populations. These field studies have an intrinsic limitation in that the source of antibodies cannot be unequivocally determined. Circulating antibodies can be produced either by short‐lived activated plasmablasts/plasma cells or from long‐lived plasma cells as part of a memory response. Thus, a decline in circulating antibodies does not necessarily represent a defect in long‐lived plasma cells. Measurements of the duration of antibody responses are further confounded by likely constant re‐exposure to the parasite in endemic areas. In order to control for these confounding factors, we resorted to study the different compartments of the B‐cell response to Plasmodium using mouse models of infection. We have exploited Plasmodium chabaudi infections in mice, which present a series of advantages. In particular, similar to human Plasmodium infections, P. chabaudi erythrocytic infections in mice give rise to an acute phase of infection followed by a distinctive chronic persistent infection with subpatent parasitemia which can last for up to 3 months, thus allowing to study the impact of low‐grade chronic infection on the B‐cell response (Figure 2B). Using this model, we showed that long‐lived plasma cells that secrete anti‐Plasmodium antibodies are generated and maintained in the later stages of malaria infections, and that these cells are maintained independently of the low‐grade chronic infection.48 In the same study, using in vitro cultures and ELISpot, we showed that a distinctive P. chabaudi‐specific memory B‐cell pool survives for a very prolonged period following complete parasite elimination.48 More recently, we confirmed this result using ex vivo flow cytometry in combination with natural mosquito transmission.49 Thus, a single Plasmodium infection in mice leads to the generation of long‐lived plasma cells and memory B cells specific to a dominant Plasmodium blood‐stage antigen, which are maintained above naive background level for very prolonged periods of time (presumably for life in mice) independently of the presence of a chronic infection.
These results in mice not necessarily contradict with the results observed in studies of human malaria. As described in Figure 1B, the B‐cell response goes through a dramatic expansion phase shortly after infection, followed by a contraction phase once clearance of the pathogen has begun. Thus, the drop of Plasmodium‐specific antibodies after the acute infection observed in some field studies might not necessarily be evidence of poor B‐cell longevity, but rather part of a contraction phase characteristic of short‐lived effector responses following reduction in parasite load or exposure to new antigenic variants (Figure 1B). In agreement with this, we have shown that Plasmodium‐specific antibody titers drop rapidly by several fold after P. chabaudi infection in mice, but are then sustained above background level for several months.48,50 Moreover, Plasmodium‐specific antibody levels after reinfection display kinetics consistent with secondary antibody responses.48, 50 Evidence of rapid boosting of antibody titers in response to Plasmodium re‐exposure has been largely documented in areas of seasonal malaria transmission, after outbreaks and in travelers.47, 51, 52, 53, 54, 55, 56 This is strongly indicative of secondary B cell responses driven by memory B cells. Moreover, although clinical immunity wanes in the absence of exposure, it has been documented that individuals, who are no longer exposed, experience significantly milder forms of the disease and lower levels of parasitemia when compared to fully naive individuals.57, 58, 59 Thus, immunological memory might indeed provide some level of protection. One possible interpretation of these data is that memory B cells contribute to protection from life‐threatening symptoms, while clinical immunity to infection might be largely conferred by short‐lived recently activated antibody‐producing cells sustained by persistent infection and constant re‐exposure. In contrast, the mechanisms that control severe disease might be entirely independent of B‐cell responses and antibodies. Immunity to severe symptoms might be related to a change in the type of immune response, such as a reduction in inflammatory responses or a switch away from a strong Th1 response, as we have shown in mouse models.60 In contrast, elimination of infection (mild or asymptomatic malaria) might require antibody responses to all variant forms, something we might expect to take time to develop.
Constant re‐exposure in areas of high transmission is a major confounder in estimating the longevity of B‐cell responses. Thus, evaluating Plasmodium‐specific B‐cell responses in historically infected individuals, but who are no longer exposed to malaria might be the most insightful way to measure longevity. In a study of adult Swedish residents who had traveled to malaria endemic areas, Plasmodium‐specific memory B cells were detected in 80% of these individuals. These responses were maintained for prolonged periods of time, lasting up to 16 years in the absence of re‐exposure to parasites in some cases. On the other hand, only 30% of travelers showed Plasmodium‐specific antibodies above naive levels; some displayed Plasmodium‐specific antibodies after at least a decade in the absence of re‐exposure.61 In other studies, migrants from endemic areas residing in Spain for long periods (>5 years) without continuous malaria exposure presented seropositivity of 32%‐98% for erythrocytic antigens,55 while 88% of migrants residing in France showed Plasmodium‐specific antibodies, in some cases for up to 4 years in the absence of re‐exposure.62 In studies of malaria outbreaks in Madagascar followed by drastic reductions in transmission, Migot et al showed that most exposed individuals maintained P. falciparum and P. vivax‐specific antibody and memory B cell responses above naive levels for as long as 3‐8 years.52, 53 In a study of subjects briefly exposed to a Plasmodium vivax malaria outbreak in Brazil, outside of the area in which malaria was endemic, Plasmodium‐specific antibody titers remained positive in 47% of cases after 7 years.63 In a study of adults with previous exposure living in an area of extremely low malaria transmission in Thailand, Wipasa and colleagues documented P. falciparum and P. vivax‐specific antibody titers and memory B cells which persisted for more than 7 years in the absence of re‐exposure.64 Several other studies have documented stable Plasmodium‐specific memory B cell56, 65, 66, 67, 68 and antibody levels69, 70, 71, 72 in non‐re‐exposed adults. Moreover, the magnitude and longevity of Plasmodium‐specific memory B‐cell responses was shown to be similar to the responses to other classic vaccine antigens such as diphtheria and tetanus toxoid,64, 65, 73, 74 suggesting that the immune system can indeed mount memory B‐cell responses to Plasmodium antigens to the same extent as to other antigens. Most of these studies have been carried out with adults. However, it seems that previously exposed children who had documented P. falciparum infections several years ago, but minimal exposure since, maintain Plasmodium‐specific memory B cells at similar levels as compared to those of persistently exposed children living in a separate but nearby endemic area.75
In contrast, several field studies, including ours, have shown that Plasmodium‐specific antibody responses can be substantially shorter‐lived than their cognate memory B‐cell responses, particularly in children.46, 47, 61, 75 Moreover, in some cases, Plasmodium‐specific antibody responses were shown to be considerably shorter than tetanus toxoid in the same individuals.46, 47, 56 This would either suggest that while memory B cells can be long‐lived, there might be specific problems in the maintenance of their cognate plasma cells, or that antibodies specific to Plasmodium antigens are mostly generated by short‐lived plasma cells not resident in survival niches such as the bone marrow. Similar results have been reported in HIV and HBV, where antigen‐specific memory B cells were found in circulation in the absence of their corresponding antibodies in contemporaneous plasma.76 The molecular and cellular basis for this observation is unclear. However, there are two schools of thought on the mechanisms for long‐term maintenance of plasma cells: (a) they could be intrinsically long‐lived or (b) are prone to decaying over time but replenished from the circulating memory B‐cell pool. In the latter case, it has been argued that this could be mediated by restimulation of memory B cells by either antigen retained in the system, or polyclonal stimulants including T cell cytokines and TLR ligands (bystander activation).46, 77 In addition, environmental factors such as nutritional status and co‐infections can impact longevity of the humoral response. In this sense, pre‐established long‐lived plasma cells seem to be in constant competition for their survival niches with newly recruited plasma cells77 and co‐infections with Plasmodium parasites and viruses seems to alter humoral responses to both viral78 and Plasmodium 79 antigens.
Age seems to be another important factor affecting longevity of the humoral response, as adults seem to make better long‐lived antibody responses to Plasmodium antigens than children.44, 45, 80 However, it is not clear yet if maturity of the immune system, rounds of re‐exposure, or a combination of both can explain this phenomenon. Finally, low‐transmission regions seem to favor the development of long‐term humoral immunity to malaria.64, 65, 72, 81 Thus, Plasmodium‐specific memory B cells seem to have a longevity similar to that of memory B cells specific to other antigens. On the other hand, Plasmodium‐specific antibody responses may be shorter lived. However, this is not a universal finding,62, 64, 65, 74, 82, 83 and is probably influenced by age of the host, nature of the antigen, intensity of transmission in specific regions, and natural host‐to‐host variations. Identifying the mechanisms that favor the generation of long‐lived plasma cells and consequently long‐lived antibody responses to Plasmodium parasites can critically contribute to the development of long‐lived protective malaria vaccines, and therefore should receive major attention.
1.2. Are atypical memory B cells “memory,” “effector,” anergic, protective or pathogenic?
An intriguing subset of B cells expressing the transcription factor T‐bet, termed AMB, has been shown to be expanded in blood of subjects exposed to Plasmodium infection.23 B cells with similar phenotypical characteristics have been observed in response to several other chronic infections including HCV, HIV, Mycobacterium tuberculosis, Toxoplasma gondii, and Leishmania infantum. 28, 84, 85, 86 Moreover, their involvement in immune responses seems to go beyond infections, as B‐cell subsets with very similar phenotypes have been shown to be expanded with age,30, 87 and suggested to be a driving force in autoimmune disorders.88 As these B cells have been identified independently by different research groups working in different fields, they have received a variety of alternative denominations, including “exhausted memory B cells,” “tissue‐like memory B cells,” “age‐associated B cells,” “double negative B cells,” and “T‐bet+” or “CD11c+T‐bet+ B cells.” In this review, we will use the denomination they were originally given in the field of malaria: AMB. Whether T‐bet+ AMB contribute to protection from malaria infection, or rather represent a dysfunctional B‐cell subset that leads to parasite persistence and pathology, remains a focus of intense debate.
AMB were first described in the context of malaria by Weiss and colleagues over 10 years ago.23 These cells showed a very similar phenotype to a FCRL4+ B‐cell subset that was described to be expanded in the blood of HIV‐infected individuals with high viral loads.26 More recently, malaria‐associated AMB were shown to preferentially express FCRL5 and FCRL3 but not FCRL4 as previously thought.24, 89 Similar to HIV, high‐circulating antigen load (parasitemia) seemed to favor the accumulation of this atypical B‐cell subset in malaria, thus suggesting chronic persistent infection may drive the expansion and accumulation of this B‐cell subset in peripheral blood.23 In the context of HIV, AMB were termed “exhausted” tissue‐like memory B cells, due to their similarity to a memory B‐cell subset found in human tonsillar tissues.90 In addition to FCRL5 and FCRL3, these cells express relatively high levels of other potentially inhibitory receptors including, CD22, CD85j, CD85k, LAIR‐1, CD72, and PD‐1, and show a trafficking receptor expression pattern consistent with a profile of migration to inflamed tissues, which includes CD11b, CD11c, and CXCR3. AMB are antigen‐experienced, class‐switched B cells, which lack the expression of GC markers. Further studies demonstrated the expression of the transcription factor T‐bet and the cytokine IFNγ by these cells, also characteristic of Th1 CD4+ T cells.84, 91, 92 Although they received the “memory” denomination, AMB do not express CD21 or the hallmark human memory B‐cell marker CD27, and have a substantially shorter life span than classical CD27+ memory B cells.26
Comparison of B‐cell profiles from children living in a rural community of P. falciparum transmission with those of age‐matched children living under similar conditions in a nearby community where P. falciparum transmission ceased over 5 years prior to the study shows that increases in AMB are driven by P. falciparum exposure, and not influenced by other factors commonly associated with malaria, such as coinfections and malnutrition.25 Moreover, temporary expansion of AMB in blood has been documented in response to human controlled malaria infections.93, 94 The appearance of AMB is strongly associated with high parasitemias or high exposure to the parasite, as individuals living in areas of high malaria transmission present higher frequencies of AMB than those in areas of moderate transmission.89 Similarly, repetitive Plasmodium episodes result in higher frequencies of AMB,92 and AMB frequencies are greater in children with persistent asymptomatic P. falciparum parasitemia compared with parasite‐free children.23 Conversely, AMB frequencies decline as parasitemias are reduced or eliminated and when there is no further exposure61, 74, 95, 96 supporting the view that the presence of a significant level of parasitemia over a period of time is necessary for the induction and maintenance of AMB cells.
The important question is—can these AMB cells be induced by Plasmodium antigens during the infection, and differentiate into antibody‐producing cells and respond to malaria antigens the same way as classical memory B cells? No studies on human AMB have so far directly demonstrated that they differentiate into plasma cells, which secrete antibodies. Single‐cell antibody cloning from circulating AMB obtained from asymptomatic semi‐immune adults showed that AMB B‐cell receptors (BCR) codified for P. falciparum ‐neutralizing antibodies, suggesting these cells could potentially contribute to the pool of P. falciparum‐ neutralizing antibodies detected in serum and play a protective role.97 High levels of secretory IgG transcripts from cloned AMB that match amino acid sequences of antibodies found in circulation support the idea that these cells secrete antibodies in vivo.97 However, in vitro restimulation of sorted AMB suggests otherwise; there is reduced Ca2+ mobilization, proliferation, cytokine production, and antibody secretion in response to BCR cross‐linking.24, 26 This has led to the hypothesis that AMB might represent an exhausted/dysfunctional/anergic B cell subset, which might not allow an effective antibody response to the parasite to develop and thus promoting chronic infection. However, in vitro assays to determine B‐cell capacity have relied on culture conditions developed for classical memory B cells. FCRL4+CD27‐CD21‐ AMB, contrary to classical memory B cells, proliferate poorly in response to either BCR ligation or Staphylococcus aureus stimulation, but can secrete high levels of immunoglobulins in response to IL‐2 and IL‐10 or to IL‐2, IL‐10, and CD40L.90 Thus, activation requirements for AMB might be different from those required by classical memory B cells. In addition, the high expression of activation, proliferation, and apoptosis markers on AMB24, 49, 97 might render these cells more sensitive to the manipulation required for cell sorting and in vitro restimulation.
The majority of the studies of AMB in human malaria have been performed on peripheral blood B cells, as the only accessible human lymphoid tissue. B cells and other immune cells normally initiate their response to antigen in lymphoid organs, whereas the blood contains mostly cells either circulating or trafficking to lymphoid organs, and it is not known how far peripheral blood represents B‐cell responses in lymphoid organs. In addition, the frequency of B cells in blood specific to any given antigen is very low, which makes difficult the investigation of their functional and developmental capacity. Therefore, we used our mouse model of mosquito‐transmitted P. chabaudi malaria to investigate Plasmodium‐specific memory B cells. Using a transgenic mouse strain with high frequencies of B cells specific to the 21‐kDa C‐terminal fragment of P. chabaudi merozoite surface protein 1 (MSP121),49 Plasmodium‐specific AMB could be tracked. We observed the expansion in the spleen of a distinctive P. chabaudi‐specific CD11b+CD11c+ B‐cell subset expressing similar surface markers as human AMB during the persistent stage of erythrocytic infection. In agreement with an AMB phenotype described in humans, these cells were class‐switched and expressed low levels of CD21 and high levels of mouse FCRL5, CD80, and CD273, the last two previously shown to be expressed on mouse memory B cells. Transcriptome analysis of these cells compared well with previously published data of human AMB transcripts. In agreement with data on human AMB, P. chabaudi‐specific CD11b+CD11c+ B cells expressed high levels of Tbx21 (T‐bet), Fcrl5, Ifng, Pdcd1 (PD1), Mki67, and class‐switched immunoglobulins, including Igha, Ighg1, Ighg2b, Ighg2c, and Ighg3. Moreover, similar to human AMB, P. chabaudi‐specific mouse AMB require parasite exposure to persist, as their numbers drop to background levels upon resolution of the persistent infection. P. chabaudi‐specific AMB also displayed high levels of the proapoptotic genes Bad, Bax, Fas, and Fasl, low level of the anti‐apoptotic gene Bcl2, and a transcriptome consistent with active replication and activation.49 In parallel, and in agreement with our previous data,48 a P. chabaudi‐specific classical memory B‐cell pool expressing different combinations of the mouse memory markers CD80, CD273, and CD73 together with high levels of Bcl2 developed and persisted long after resolution of the infection. Interestingly, mouse FCRL5 was highly expressed on all different subsets of P. chabaudi‐specific classical memory B cells, including IgM+.49 FCRL5 was also highly expressed on classical memory B cells generated through immunizations of mice.49, 98 Persistent infection therefore seems to drive expansion of P. chabaudi‐specific mouse AMB, but their existence does not prevent the generation of P. chabaudi‐specific classical memory B cells nor prevent resolution of the infection. These observations support the view that constant immune activation rather than impaired memory function leads to the accumulation of AMB in malaria. Nonetheless, although showing very high levels of IgG transcripts, the contribution of P. chabaudi‐specific AMB to the serum antibody pool and to protection from P. chabaudi infections remains to be demonstrated.
The occurrence of CD11b+CD11c+T‐bet+ AMB‐like B cells has been extensively described in mouse models of chronic viral and bacterial infections, and in many cases are shown to be a fully functional B cell subset able to contribute to protection from infection.99, 100, 101 The peak of CD11c+T‐bet+ B‐cell production in the spleen is detected early after Ehrlichia muris peak infection, and these cells persist thereafter in lower numbers but above background levels accompanying the persistent infection. These cells very much resemble the P. chabaudi‐specific AMB (ie, CD11b+CD11c+Tbet+CD273+CD73+CD80+Fas‐Lhi), with the exemption of being IgM+. Moreover, similar to P. chabaudi‐specific AMB, the CD11c+T‐bet+ B cells expanded in response to E. muris resembled plasmablasts, including expression of CD138.49, 101 In accordance, these cells give rise to antibody‐secreting cells that produced antibodies which recognized E. muris antigens.102, 103 Moreover, AMB‐like cells have also been shown to be the original source of protective virus‐specific antibodies in mice. A CD11b+CD11c+T‐bet+ AMB subset appears at the peak of murine gamma herpesvirus 68 (gHV68), lymphocytic choriomeningitis virus (LCMV), murine cytomegalovirus (MCMV), vaccinia, and Friend virus infections.100, 104 These cells are required for production of virus‐specific IgG2a and are critical to clear gHV68 infection.100, 104 A T‐bet+ AMB subset is also critical for the production of protective IgG2a and to control chronic LCMV cl13 infection.99 Thus, in the strong Th1‐biased context of intracellular bacterial and viral infections, AMB appear to be a source of protective antibodies.
A subset of CD11c+T‐bet+ AMB‐like cells, originally termed age‐associated B cells, have also been shown to expand in several mouse models of autoimmune disorders as well as in blood of subjects suffering autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, and Crohn's disease.88, 105, 106, 107, 108, 109 In mouse models of lupus‐like disease, AMB‐like cells expressed CD138, characteristic of antibody secreting plasma cells/plasmablasts, and directly contributed to the production of auto‐antibodies.110 Moreover, in the context of autoimmune disorders, AMB‐like cells showed potent antigen‐presenting capacity and production of proinflammatory cytokines.108, 110, 111 Similar to malaria, AMB‐like cells identified in autoimmunity showed signs of previous activation and proliferation in vivo, and defective Ca2+ signaling and poor proliferation in response to BCR stimulation in vitro.112, 113 Surprisingly, protective AMB‐like cells generated during E. muris infection also produced polyreactive autoantibodies.102, 103 This is intriguing and surprising, as these autoantibodies seem not to drive pathology in the E. muris infection model. A recent study showed that an AMB‐like CD11c+T‐bet+ B‐cell subset expanded in the spleen during acute blood stage Plasmodium yoelii 17XNL infection in mice and secreted autoantibodies specific to erythrocyte phosphatidylserine, which appear to drive anemia.114 This opens the intriguing possibility that AMB might contribute to malaria pathology through the production of autoantibodies.
Nonetheless, whether protective or pathogenic, the outstanding majority of data on AMB‐like cells obtained in viral and bacterial infections as well as autoimmune disorders is so far not compatible with the hypothesis that AMB are exhausted or anergic. It might be argued that these AMB‐like subsets generated in different immune scenarios, despite sharing an impressive number of key characteristics, are in fact intrinsically different from the AMB cells observed in malaria. Alternatively, although being the same subset, different immune scenarios might differentially affect their function, with malaria and HIV preferentially driving AMB to exhaustion unlike other Th‐1‐biased chronic infections such as LCMV. If the latter is the case, then those signals affecting AMB which are unique to malaria and not shared by other chronic infections or autoimmune disorders remain to be identified.
Together, most of these studies in human and experimental malaria as well as other infections suggest that AMB represent an effector B‐cell subset generated in, and sustained by, persistent antigen exposure. However, there are reports of relatively long AMB persistence in the apparent absence of malaria exposure,68 and long‐lived T‐bet+ memory B cells have been described in the E. muris infection model.115 Therefore, AMB in malaria and beyond represent an exciting field of study with many unanswered questions. What is the true nature of AMB? Do they contribute to protection, pathology, or both? Are they dysfunctional/exhausted/anergic? Do they contribute to the memory pool?
1.3. What are the signals required to generate long‐lasting B‐cell responses to plasmodium?
Although there is evidence of T‐independent B1 B cells, which bridge innate and adaptive responses, producing memory responses (B1 cells defined in mice by expression of IgMhiIgDloCD23−B220lo, and in humans by CD20+CD27+CD43+CD38lo/int), it is the adaptive response of B2 B cells (in mice B220+AA4.1−CD23+IgMintIgDhi, and humans CD20+CD27+CD43− B cells), and GC interactions which are critical for long‐lasting B‐cell responses in the majority of memory responses.31, 116, 117 Despite the importance of antibody responses in malaria, details on GC formation and activation/regulation of T cell help by follicular helper T‐cell in malaria are only starting to emerge. CD4+ T cells producing IL‐21, a characteristic cytokine of Tfh, and Tfh defined by expression of PD‐1 and CXCR5 are present in peripheral blood mononuclear cells from malaria‐exposed immune adults118, 119, 120 and in some cases, correlate with P falciparum‐specific IgG antibodies in children with acute P. falciparum malaria.121 Alterations in the GC B‐cell response to Plasmodium infection will very likely affect generation of long‐lasting B cell memory, but this is difficult to investigate fully using human peripheral blood cells, and we need to turn to experimental models to elucidate GC interactions in malaria. Plasmodium infections in mice trigger a robust GC response, and CD4+ T cells producing IL‐21, of which a substantial proportion have a Tfh cell phenotype, are required to generate GC B‐cell and IgG responses, and to resolve P. chabaudi and P. yoelii infections.10, 50, 122, 123, 124 Moreover, mice deficient in IL‐21 signaling are not immune to reinfection, and fail to generate P. chabaudi‐specific long‐lived plasma cells and memory B cells.10 P. chabaudi‐specific IgM responses are not altered in the absence of IL‐21 signaling, thus showing that IgM is not sufficient to clear the infection,10 and that IL‐21 is essential to generate long‐lasting class‐switched protective B cell responses. Although IL‐21 is not required to activate Tfh cells in P. chabaudi infections, Tfh cells are an important source of IL‐21 and are also essential to generate class‐switched antibody responses and clear the infection.10, 122
The pattern recognition receptor cyclic GMP‐AMP synthase (cGAS), ICOS, and IL‐10 signaling on B cells are all required for GC B‐cell and IgG responses,125 and this is the case also in experimental P. yoelii infections.123, 126, 127 Thus, these signals are probably also essential for generation of Plasmodium‐specific memory B cells. The signaling lymphocytic activation molecule (SLAM)‐associated protein (SAP), shown to be necessary for GC and B‐cell memory responses to immunizations and viral infections,128, 129 has only a partial impact on IgG and GC responses in P. chabaudi infections, but SAP does contribute to some control of chronic infection.122 A requirement for SAP interactions in immunity to reinfection in Plasmodium remains unexplored.
Although GC formation clearly takes place in experimental malaria, there is some indication that this may not be optimal as GC responses are enhanced by in vivo blockade of CTLA‐4, or PD‐L1 in combination with LAG‐3.130, 131 However, another interpretation is that GC formation during Plasmodium infection is normal, and that the blockade is simply overriding the normal mechanism of control of GC and B‐cell responses. Malaria is characterized by a strong Th1–like response, which affect B cell responses. The signature Th1 cytokine, IFN‐γ, is responsible for switching to IgG2a/c antibodies in mice, and for the human analogues IgG1 and IgG3,132, 133, 134 isotypes which activate complement and Fcγ receptors on macrophages bringing about pathogen killing and phagocytosis. These isotypes have been shown to clear viral infections,135, 136 and to correlate with protection against Plasmodium infections.137, 138
There are, however, seemingly opposing views on whether Th1 responses promote or impair B cells and GCs in experimental Plasmodium infections. On ‐one hand, Plasmodium‐specific Th1 CD4 T cells can support B‐cell responses.137, 138, 139 On the other hand, IFN‐γ and TNF‐α responses reduce activation of Tfh and GC B‐cell responses in Plasmodium berghei infections and P. yoelii infections in mice.124, 126, 140, 141 Similarly, a Th1‐polarized Tfh subset found expanded in blood from infected children shows reduced capacity to support B cell activation in vitro.119 These data suggest that while Th1 signaling is important for switching to potentially protective antibody isotypes, in excess it might also negatively limit normal development of the GC response. As yet, the direct impact of these signals on the longevity of memory B‐cell responses has not been demonstrated. However, and similar to viral and intracellular bacterial infections, acute P. chabaudi blood‐stage infection triggers a robust Th1 response,10 and yet we detect substantial GC formation and a long‐lived B cell and antibody responses, suggesting that IFN‐γ production by Th1 cells is not sufficient per se to prevent robust humoral responses.
Another contribution of Th1 responses and IFN‐γ to the B‐cell response is their role in the generation of AMB cells. Indeed, T‐bet expression in B cells in malaria is regulated by IFN‐γ,92 triggered by a combination of CD40, IL‐12, and IL‐18 signaling. AMB cells are not activated by cross‐linking the BCR unlike classical memory B cells, which may explain why they are difficult to activate in vitro, but age‐associated CD11c+T‐bet+ B cells can be activated by TLR 7 and 9 signaling,87, 110 and antigen‐specific AMB‐like B cells in mice are transiently expanded after immunization with a TLR7/8 ligand together with Hen Egg Lysosome100 or a recombinant Plasmodium antigen.49 IFN‐γ or IL‐21 together with TLR signaling drives the generation of CD11c+T‐bet+ AMB‐like cells,142 which is particularly relevant in Plasmodium infections where there is a large co‐expression of IFN‐γ and IL‐21 by CD4+ T cells.10 Similarly, IL‐21 is required to activate AMB‐like cells in response to E. muris infection,143 and IL‐21 and SAP are required to activate AMB‐like cells during autoimmunity.144 These data support the view that the signals to activate AMB in a variety of situations including malaria where there is or has been chronic antigen presence, are partially but not entirely shared with those required for the generation of classical memory B cells.
We are left with apparently contradictory findings. The same signals that seem to impair GC responses during malaria promote generation of AMB‐like cells. Are AMB cells a GC subset or are they generated outside of the GC, and is T cell support required for their generation? Most AMB cells have surface IgG, and Immunoglobulin variable regions from AMB generated during malaria are heavily mutated, suggesting a GC origin.24, 97, 145 Signals able to drive AMB‐like cell activation, including IL‐21 and SAP, seem to also suggest a GC origin, and MHCII has been shown to be required to activate AMB like cells to E. muris. 143, 144 T‐bet+ AMB‐like cells have been suggested to be involved in the formation of spontaneous GC in a mouse model of lupus.146 Moreover, extrafollicular development of T‐bet‐expressing plasmablasts has been reported in response to E. muris infection,102, 143 suggesting that AMB‐like cells might also arise independently of the GC. To date, the origin of AMB during malaria remains uncertain. If, as some studies suggest, AMB and classical memory B cells share a common developmental origin,24, 145 then a key element might be the balance between IL‐21 and IFN‐γ, with excessive IFN‐γ favoring GC disruption and AMB accumulation, while IL‐21 supporting both AMB and classical memory B cell generation. However, it is important to highlight that the robust IFN‐γ responses during P. chabaudi and other intracellular viral and bacterial pathogens do not preclude protective long‐lasting B‐cell and antibody responses, nor disrupt the GC response. In addition, the role of IL‐21 and GC in the generation of AMB in response to Plasmodium infection remains unexplored. Figure 3 summarizes the main signals, either known or predicted, to be involved in the generation of classical and AMB.
Figure 3.
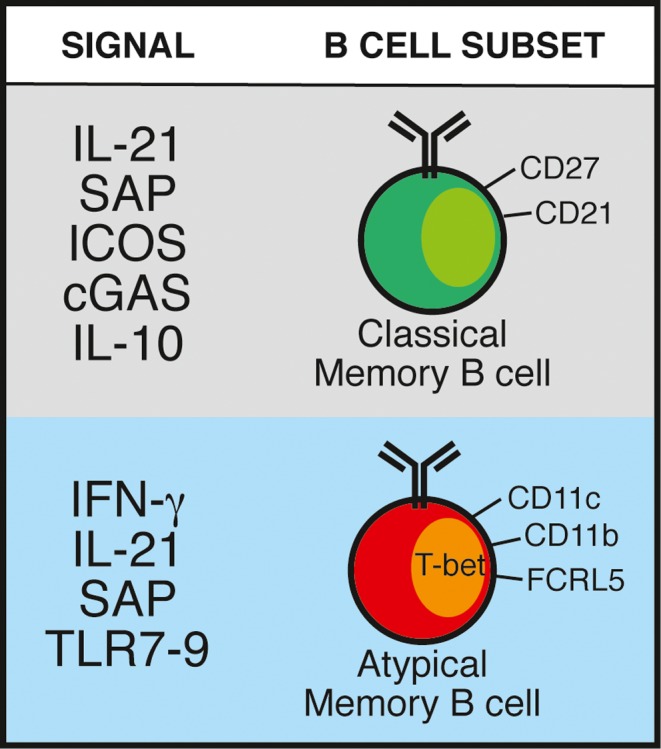
Signals known or suggested to support the generation of either classical or atypical memory B cells in response to Plasmodium spp. infection. SAP: signaling lymphocyte activation molecule (SLAM)‐associated protein; ICOS: inducible T cell co‐stimulator; cGAS: pattern recognition receptor cyclic GMP‐AMP synthase; TLR: toll‐like receptor
2. CONCLUDING REMARKS
Many studies show that B‐cell and antibody responses to malaria are short‐lived and often low level or absent, particularly in young children in endemic areas. Given the very varied methods of B cell or antibody measurement in a large variety of studies, the different transmission intensities of the study sites, and in some cases single time‐point measurements at unknown times after a detectable parasitemia, it is hard to put together a cohesive picture of what is happening. Low levels of antibodies and/or drops in antibody titer may be due in part to their production by short‐lived plasma cells in acute infection (Figure 1B), and we do not know when in the kinetics of the humoral response antibody measurements have been taken. Thus, the drop in antibody titer may well represent the contraction phase of a normal B‐cell response rather than evidence of a deficient B cell memory response to Plasmodium. However, some studies clearly show poor Plasmodium‐specific antibody responses in endemic areas and we need to understand the reasons for this. It is difficult to carry out a systematic study over time in children or adults in all field settings following a defined infection. Controlled human Plasmodium infections (CHMI) of adults in endemic countries, only recently made possible, may help us elucidate this important aspect of the humoral response in malaria.
There is, conversely, a large body of literature showing long‐lived B‐cell and antibody responses to Plasmodium parasites in humans and animal models, which challenge the concept of Plasmodium infection always driving the B‐cell response to exhaustion of dysfunction. Studies being performed in areas of different transmission intensity with consequently different reinfection rates might explain some of the apparent discrepancies. AMB, suggested as a reason for dysfunctional B‐cell responses to malaria, may not be the culprit. Whether they could be a source of protective antibodies and thus play a role in immunity to Plasmodium as has been shown in other infections in experimental models, remains to be conclusively demonstrated. The restimulation protocols used to demonstrate AMB anergy during malaria have not been exhaustive and might not be adequate to stimulate this subset of B cells. Moreover, even if AMB detected in the blood of malaria exposed individuals were indeed anergic, this would not rule out the possibility these might be terminally differentiated B‐cell subsets which have contributed to an antibody response in lymphoid organs during their past history. Thus, the true nature of AMB in malaria still remains elusive and intriguing, and animal models will very likely shed light on their origins, developmental history, and function during infection. Understanding the mechanisms that govern differential longevity of protective B‐cell responses among different individuals or to different Plasmodium antigens will greatly expand our capacity to improve antibody‐based malaria vaccines. In this regard, it is important to keep in mind that a single B cell target might not be sufficient to confer protection from complex protozoan parasites, including Plasmodium. Putative strategies might combine vaccines against different targets which provide protection by different mechanisms and target different stages of the parasite life cycle, while taking into account the occurrence of polymorphisms on the selected targets. Moreover, B‐cell responses might not be sufficient, and modulation of other immune arms might be required to confer full protection from Plasmodium infection while preventing pathology.
ACKNOWLEDGEMENTS
The authors thank Dr Matthew Lewis for his critical reading of the manuscript. JL and DPM are supported by the Francis Crick Institute (FC 10101), which receives its funding from the UK Medical Research Council, Cancer Research UK, and the Wellcome Trust, UK. JL is a Wellcome Trust Senior Investigator (WT104777/Z/14/Z). DPM has an Independent Research Fellowship in Immunology from Hull York Medical School, UK. FMN is a Senior Fellow with EDCTP, and is funded from an MRC/DFID African Research Leadership Award, and the Wellcome Trust (107499/Z/15/Z). RA receives a DELTAS Africa Initiative [DEL‐15‐003] fellowship. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA), and supported by the New Partnership for Africa's Development Planning, and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [107769/Z/10/Z], and the UK government. The views expressed in this publication are those of the authors and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government. All authors declare that they have no conflicts of interest.
Pérez‐Mazliah D, Ndungu FM, Aye R, Langhorne J. B‐cell memory in malaria: Myths and realities. Immunol Rev. 2020;293:57–69. 10.1111/imr.12822
This article is part of a series of reviews covering Immunity to Malaria appearing in Volume 293 of Immunological Reviews.
Contributor Information
Damián Pérez‐Mazliah, Email: damian.perezmazliah@york.ac.uk.
Jean Langhorne, Email: jean.langhorne@crick.ac.uk.
REFERENCES
- 1. World Health Organization . World malaria report 2018. Geneva, Switzerland: World Health Organization; 20181‐210. [Google Scholar]
- 2. Alonso PL, Sacarlal J, Aponte JJ, et al. Efficacy of the RTS, S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: Randomised controlled trial. Lancet. 2004;364:1411‐1420. [DOI] [PubMed] [Google Scholar]
- 3. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621‐663. [DOI] [PubMed] [Google Scholar]
- 4. Fowkes FJI, Richards JS, Simpson JA, Beeson JG. The relationship between anti‐merozoite antibodies and incidence of Plasmodium falciparum malaria: A systematic review and meta‐analysis. PLoS Medicine. 2010;7:e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen S, McGREGOR IA, Carrington S. Gamma‐globulin and acquired immunity to human malaria. Nature. 1961;192:733‐737. [DOI] [PubMed] [Google Scholar]
- 6. Sabchareon A, Burnouf T, Ouattara D, et al. Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am J Trop Med Hyg. 1991;45(3):297-308. [DOI] [PubMed] [Google Scholar]
- 7. Osier FHA, Fegan G, Polley SD, et al. Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect Immun. 2008;76:2240‐2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burns JM, Dunn PD, Russo DM. Protective immunity against Plasmodium yoelii malaria induced by immunization with particulate blood‐stage antigens. Infect Immun. 1997;65:3138‐3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von der Weid T, Langhorne J, Honarvar N. Gene‐targeted mice lacking B cells are unable to eliminate a blood stage malaria infection. J Immunol. 1996;156:2510‐2516. [PubMed] [Google Scholar]
- 10. Pérez‐Mazliah D, Ng DHL, Freitas do Rosário AP, et al. Disruption of IL‐21 signaling affects T cell‐B cell interactions and abrogates protective humoral immunity to malaria. PLoS Pathog. 2015;11:e1004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hill DL, Schofield L, Wilson DW. IgG opsonization of merozoites: Multiple immune mechanisms for malaria vaccine development. Int J Parasitol. 2017;47:585‐595. [DOI] [PubMed] [Google Scholar]
- 12. Teo A, Feng G, Brown GV, Beeson JG, Rogerson SJ. Functional antibodies and protection against blood‐stage malaria. Trends Parasitol. 2016;32:887‐898. [DOI] [PubMed] [Google Scholar]
- 13. Chan J‐A, Howell KB, Reiling L, et al. Targets of antibodies against Plasmodium falciparum–infected erythrocytes in malaria immunity. J Clin Invest. 2012;122:3227‐3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clark IA, Budd AC, Alleva LM, Cowden WB. Human malarial disease: A consequence of inflammatory cytokine release. Malar J. 2006;5:1‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donati D, Zhang LP, Chen Q, et al. Identification of a polyclonal B‐Cell activator in Plasmodium falciparum . Infect Immun. 2004;72:5412‐5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosenberg YJ. Autoimmume and polyclonal B cell responses during murine malaria. Nature. 1978;274:170‐172. [DOI] [PubMed] [Google Scholar]
- 17. Abele DC, Tobie JE, Hill GJ, Contacos PG, Evans CB. Alterations in serum proteins and 19S antibody production during the course of induced malarial infections in man. Am J Trop Med Hyg. 1965;14:191‐197. [DOI] [PubMed] [Google Scholar]
- 18. Cadman ET, Abdallah AY, Voisine C, et al. Alterations of splenic architecture in malaria are induced independently of Toll‐like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect Immun. 2008;76:3924‐3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bockstal V, Geurts N, Magez S. Acute disruption of bone marrow B Lymphopoiesis and apoptosis of transitional and marginal zone B Cells in the spleen following a blood‐stage Plasmodium chabaudi infection in mice. J Parasitol Res. 2011;2011:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Urban BC, Hien TT, Day NP, et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect Immun. 2005;73:1986‐1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belyaev NN, Brown DE, Diaz A‐IG, et al. Induction of an IL7‐R + c‐Kit hi myelolymphoid progenitor critically dependent on IFN‐γ signaling during acute malaria. Nat Immunol. 2010;11:477‐485. [DOI] [PubMed] [Google Scholar]
- 22. Belyaev NN, Biró J, Langhorne J, Potocnik AJ. Extramedullary myelopoiesis in Malaria depends on mobilization of myeloid‐restricted progenitors by IFN‐γ induced chemokines. PLoS Pathog. 2013;9:e1003406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weiss GE, Crompton PD, Li S, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria‐endemic area. J Immunol. 2009;183:2176‐2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Portugal S, Tipton CM, Sohn H, et al. Malaria‐associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. Elife. 2015;4:e07218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Illingworth J, Butler NS, Roetynck S, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. 2013;190:1038‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moir S, Ho J, Malaspina A, et al. Evidence for HIV‐associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV‐infected viremic individuals. J Exp Med. 2008;205(8):1797-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oliviero B, Mantovani S, Ludovisi S, et al. Skewed B cells in chronic hepatitis C virus infection maintain their ability to respond to virus‐induced activation. J Viral Hepat. 2015;22:391‐398. [DOI] [PubMed] [Google Scholar]
- 28. Joosten SA, van Meijgaarden KE, del Nonno F, et al. Patients with tuberculosis have a dysfunctional circulating B‐cell compartment, which normalizes following successful treatment. PLoS Pathog. 2016;12:e1005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rubtsov AV, Marrack P, Rubtsova K. T‐bet expressing B cells – Novel target for autoimmune therapies? Cell Immunol. 2017;321:35‐39. [DOI] [PubMed] [Google Scholar]
- 30. Rubtsova K, Rubtsov AV, Cancro MP, Marrack P. Age‐associated B cells: A T‐bet? dependent effector with roles in protective and pathogenic immunity. J Immunol. 2015;195:1933‐1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149‐159. [DOI] [PubMed] [Google Scholar]
- 32. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357:1903‐1915. [DOI] [PubMed] [Google Scholar]
- 33. Yu X, Tsibane T, McGraw PA, et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455:532‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Slifka MK, Ahmed R. Limiting dilution analysis of virus‐specific memory B cells by an ELISpot assay. J Immunol Methods. 1996;199:37‐46. [DOI] [PubMed] [Google Scholar]
- 35. Hammarlund E, Thomas A, Poore EA, et al. Durability of vaccine‐induced immunity against tetanus and diphtheria toxins: A cross‐sectional analysis. Clin Infect Dis. 2016;62:1111‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammarlund E, Lewis MW, Hansen SG, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131‐1137. [DOI] [PubMed] [Google Scholar]
- 37. Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: Long‐term B cell memory in humans after smallpox vaccination. J Immunol. 2003;171:4969‐4973. [DOI] [PubMed] [Google Scholar]
- 38. Maple PAC, Jones CS, Wall EC, et al. Immunity to diphtheria and tetanus in England and Wales. Vaccine. 2000;19:167‐173. [DOI] [PubMed] [Google Scholar]
- 39. Ly A, Hansen DS. Development of B cell memory in malaria. Front Immunol. 2019;10:111‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cockburn IA, Seder RA. Malaria prevention: from immunological concepts to effective vaccines and protective antibodies. Nat Immunol. 2018;19:1199‐1211. [DOI] [PubMed] [Google Scholar]
- 41. EL Silveira, V, Dominguez MR, Soares IS. To B or not to B: Understanding B Cell responses in the development of malaria infection. Front Immunol. 2018;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marsh K, Otoo L, Hayes RJ, Carson DC, Greenwood BM. Antibodies to blood stage antigens of Plasmodium falciparum in rural Gambians and their relation to protection against infection. Trans R Soc Trop Med Hyg. 1989;83:293‐303. [DOI] [PubMed] [Google Scholar]
- 43. Gupta S, Snow RW, Donnelly CA, Marsh K, Newbold C. Immunity to non‐cerebral severe malaria is acquired after one or two infections. Nat Med. 1999;5:340. [DOI] [PubMed] [Google Scholar]
- 44. Kinyanjui SM, Conway DJ, Lanar DE, Marsh K. IgG antibody responses to Plasmodium falciparum merozoite antigens in Kenyan children have a short half‐life. Malar J. 2007;6:1318‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akpogheneta OJ, Duah NO, Tetteh KKA, et al. Duration of naturally acquired antibody responses to blood‐stage Plasmodium falciparum is age dependent and antigen specific. Infect Immun. 2008;76:1748‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weiss GE, Traore B, Kayentao K, et al. The Plasmodium falciparum‐specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010;6:e1000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crompton PD, Kayala MA, Traore B, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci. 2010;107:6958‐6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ndungu FM, Cadman ET, Coulcher J, et al. Functional memory B cells and long‐lived plasma cells are generated after a single Plasmodium chabaudi infection in mice. PLoS Pathog. 2009;5:e1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pérez‐Mazliah D, Gardner PJ, Schweighoffer E, et al. Plasmodium‐specific atypical memory B cells are short‐lived activated B cells. Elife. 2018;7:e39800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Achtman AH, Stephens R, Cadman ET, Harrison V, Langhorne J. Malaria‐specific antibody responses and parasite persistence after infection of mice with Plasmodium chabaudi chabaudi . Parasite Immunol. 2007;29:435‐444. [DOI] [PubMed] [Google Scholar]
- 51. Chougnet C, Deloron P, Lepers JP, et al. Humoral and cell‐mediated immune responses to the Plasmodium falciparum Antigens PF155/RESA and CS protein: Seasonal variations in a population recently reexposed to endemic malaria. Am J Trop Med Hyg. 1990;43:234‐242. [DOI] [PubMed] [Google Scholar]
- 52. Migot F, Chougnet C, Raharimalala L, Astagneau P, Lepers JP, Deloron P. Human immune responses to the Plasmodium falciparum ring‐infected erythrocyte surface antigen (Pf155/RESA) after a decrease in malaria transmission in Madagascar. Am J Trop Med Hyg. 1993;48:432‐439. [DOI] [PubMed] [Google Scholar]
- 53. Migot F, Chougnet C, Henzel D. et al. Anti‐malaria antibody‐producing B cell frequencies in adults after a Plasmodium falciparum outbreak in Madagascar. Clin Exp Immunol. 1995;102:529‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Waa JAV, Jensen JB, Akood MA, Bayoumi R. Longitudinal study on the in vitro immune response to Plasmodium falciparum in Sudan. Infect Immun. 1984;45:505‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moncunill G, Mayor A, Jiménez A, et al. High antibody responses against Plasmodium falciparum in immigrants after extended periods of interrupted exposure to malaria. PLoS ONE. 2013;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yman V, White MT, Asghar M, et al. Antibody responses to merozoite antigens after natural Plasmodium falciparum infection: Kinetics and longevity in absence of re‐exposure. BMC Med. 2019;1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jelinek T, Schulte C, Infectious RBC. Imported falciparum malaria in Europe: Sentinel surveillance data from the European network on surveillance of imported infectious diseases. Clin Infect Dis. 2002;2002(34):572‐576. [DOI] [PubMed] [Google Scholar]
- 58. Matteelli A, Colombini P, Gulletta M, Castelli F, Carosi G. Epidemiological features and case management practices of imported malaria in northern Italy 1991–1995. Trop Med Int.Heal. 1999;4:653‐657. [DOI] [PubMed] [Google Scholar]
- 59. Perri GD, Solbiati M, Vento S,. West African immigrants and new patterns of malaria imported to North Eastern Italy. J Travel Med. 1994;1994(1):147‐151. [DOI] [PubMed] [Google Scholar]
- 60. do Rosário AP, Langhorne, J . T cell‐derived IL‐10 and its impact on the regulation of host responses during malaria. Int J Parasitol. 2012;42:549‐555. [DOI] [PubMed] [Google Scholar]
- 61. Ndungu FM, Lundblom K, Rono J, Illingworth J, Eriksson S, Färnert A. Long‐lived Plasmodium falciparum specific memory B cells in naturally exposed Swedish travelers. Eur J Immunol. 2013;43:2919‐2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chougnet C, Deloron P, Savel J. Persistence of cellular and humoral response to synthetic peptides from defined Plasmodium falciparum antigens. Ann Trop Med Parasitol. 2016;85:357‐363. [DOI] [PubMed] [Google Scholar]
- 63. Braga EM, Fontes CJF, Krettli AU. Persistence of humoral response against sporozoite and blood‐stage malaria antigens 7 years after a brief exposure to Plasmodium vivax . J Infect Dis. 1998; 177:1132‐1135. [DOI] [PubMed] [Google Scholar]
- 64. Wipasa J, Suphavilai C, Okell LC, et al. Long‐lived antibody and B cell memory responses to the human malaria parasites, Plasmodium falciparum and Plasmodium vivax . PLoS Pathog. 2010;6:e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nogaro SI, Hafalla JC, Walther B, et al. The breadth, but not the magnitude, of circulating memory B cell responses to Plasmodium falciparum increases with age/exposure in an area of low transmission. PLoS ONE. 2011;6(10):e25582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Triller G, Scally SW, Costa G, et al. Natural parasite exposure induces protective human anti‐malarial antibodies. Immunity. 2017;47:1197‐1209e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murugan R, Buchauer L, Triller G, et al. Clonal selection drives protective memory B cell responses in controlled human malaria infection. Sci Immunol. 2018;3:eaap8029. [DOI] [PubMed] [Google Scholar]
- 68. Changrob S, McHenry AM, Nyunt MH, et al. Persistence of long‐lived memory B cells specific to duffy binding protein in individuals exposed to Plasmodium vivax . Sci Rep. 2018;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Taylor RR, Egan A, McGuinness D,et al. Selective recognition of malaria antigens by human serum antibodies is not genetically determined but demonstrates some features of clonal imprinting. Int Immunol. 1996;8:905‐915. [DOI] [PubMed] [Google Scholar]
- 70. Drakeley CJ, Corran PH, Coleman PG, et al. Estimating medium‐ and long‐term trends in malaria transmission by using serological markers of malaria exposure. Proc Natl Acad Sci. 2005;102:5108‐5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Udhayakumar V, Kariuki S, Kolczack M, et al. Longitudinal study of natural immune responses to the Plasmodium falciparum apical membrane antigen (AMA‐1) in a holoendemic region of malaria in western Kenya: Asembo Bay Cohort Project VIII. Am J Trop Med Hyg. 2001;65:100‐107. [DOI] [PubMed] [Google Scholar]
- 72. Longley RJ, Reyes‐Sandoval A, Montoya‐Díaz E, et al. Acquisition and longevity of antibodies to Plasmodium vivax preerythrocytic antigens in Western Thailand. Clin Vaccine Immunol. 2016;23:117‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dorfman JR, Bejon P, Ndungu FM, et al. B cell memory to 3 Plasmodium falciparum blood‐stage antigens in a malaria‐endemic area. J Infect Dis. 2005;191:1623‐1630. [DOI] [PubMed] [Google Scholar]
- 74. Ayieko C, Maue AC, Jura WGZO, et al. Changes in B cell populations and merozoite surface protein‐1‐specific memory B cell responses after prolonged absence of detectable Plasmodium falciparum infection. PLoS ONE. 2013;8:e67230‐e67312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ndungu FM, Olotu A, Mwacharo J, et al. Memory B cells are a more reliable archive for historical antimalarial responses than plasma antibodies in no‐longer exposed children. Proc Natl Acad Sci. 2012;109:8247‐8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guan Y, Sajadi MM, Kamin‐Lewis R, et al. Discordant memory B cell and circulating anti‐env antibody responses in HIV‐1 infection. Proc Natl Acad Sci. 2009;106:3952‐3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: The challenge of becoming a long‐lived plasma cell. Nat Publ Gr. 2006;6:741‐750. [DOI] [PubMed] [Google Scholar]
- 78. Ng DHL, Skehel JJ, Kassiotis G, Langhorne J. Recovery of an antiviral antibody response following attrition caused by unrelated infection. PLoS Pathog. 2014;10:e1003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Matar CG, Anthony NR, O'Flaherty BM, et al. Gammaherpesvirus co‐infection with malaria suppresses anti‐parasitic humoral immunity. PLoS Pathog. 2015;11:e1004858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. White MT, Griffin JT, Akpogheneta O, et al. Dynamics of the antibody response to Plasmodium falciparum infection in African children. J Infect Dis. 2014;210:1115‐1122. [DOI] [PubMed] [Google Scholar]
- 81. Clark EH, Silva CJ, Weiss GE, et al. Plasmodium falciparum malaria in the Peruvian Amazon, a region of low transmission, is associated with immunologic memory. Infect Immun. 2012;80:1583‐1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Deloron P, Chougnet C. Is Immunity to malaria really short‐lived? Parasitol Today. 1992;8:375‐378. [DOI] [PubMed] [Google Scholar]
- 83. Bouchaud O, Cot M, Kony S, et al. Do African immigrants living in France have long‐term malarial immunity?. Am J Trop Med Hyg. 2005;72:21‐25. [PubMed] [Google Scholar]
- 84. Knox JJ, Buggert M, Kardava L, et al. T‐bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI insight. 2017;2:92943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Johrens K, Shimizu Y, Anagnostopoulos I, et al. T‐bet‐positive and IRTA1‐positive monocytoid B cells differ from marginal zone B cells and epithelial‐associated B cells in their antigen profile and topographical distribution. Haematologica. 2005;90:1070‐1077. [PubMed] [Google Scholar]
- 86. Rodrigues V, Laforge M, Campillo‐Gimenez L, et al. Abortive T follicular helper development is associated with a defective humoral response in Leishmania infantum‐infected macaques. PLoS Pathog. 2014;10:e1004096‐e1004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A B‐cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T‐bet drive lupus‐like autoimmunity. J Clin Invest. 2017;127:1392‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sullivan RT, Kim CC, Fontana MF, et al. FCRL5 delineates functionally impaired memory B cells associated with Plasmodium falciparum exposure. PLoS Pathog. 2015;11:e1004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ehrhardt GRA. Expression of the immunoregulatory molecule FcRH4 defines a distinctive tissue‐based population of memory B cells. J Exp Med. 2005;202:783‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Portugal S, Obeng‐Adjei N, Moir S, Crompton PD, Pierce SK. Atypical memory B cells in human chronic infectious diseases_ An interim report. Cell Immunol. 2017;321:18‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Obeng‐Adjei N, Portugal S, Holla P, et al. Malaria‐induced interferon‐γ drives the expansion of Tbethi atypical memory B cells. PLoS Pathog. 2017;13:e1006576‐e1006630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sundling C, Rönnberg C, Yman V, et al. B cell profiling in malaria reveals expansion and remodeling of CD11c+ B cell subsets. JCI Insight. 2019;4:126492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Scholzen A, Teirlinck AC, Bijker EM, et al. BAFF and BAFF receptor levels correlate with B cell subset activation and redistribution in controlled human malaria infection. J Immunol. 2014;192:3719‐3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Patgaonkar M, Herbert F, Powale K, et al. Vivax infection alters peripheral B‐cell profile and induces persistent serum IgM. Parasite Immunol. 2018;40:e12580‐e12614. [DOI] [PubMed] [Google Scholar]
- 96. Nogaro SI, Hafalla JC, Walther B, et al. The Breadth, but not the magnitude, of circulating memory B cell responses to Plasmodium falciparum increases with age/exposure in an area of low transmission. PLoS ONE. 2011;6:e25582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Muellenbeck MF, Ueberheide B, Amulic B, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J Exp Med. 2013;210:389‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kim CC, Baccarella AM, Bayat A, Pepper M, Fontana MF. FCRL5+ memory B cells exhibit robust recall responses. Cell Rep. 2019;27: 1446‐1460e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Barnett BE, Staupe RP, Odorizzi PM, et al. Cutting edge: B cell‐intrinsic T‐bet expression is required to control chronic viral infection. J Immunol. 2016;197:1017‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T‐box transcription factor T‐bet, a key player in a unique type of B‐cell activation essential for effective viral clearance. Proc Natl Acad Sci U. S. A. 2013;110:E3216‐E3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Winslow GM, Papillion AM, Kenderes KJ, Levack RC. CD11c+ T‐bet+ memory B cells: Immune maintenance during chronic infection and inflammation? Cell Immunol. 2017;321:8‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Racine R, Chatterjee M, Winslow GM. CD11c expression identifies a population of extrafollicular antigen‐specific splenic plasmablasts responsible for CD4 T‐independent antibody responses during intracellular bacterial infection. J Immunol. 2008;181:1375‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jones DD, DeIulio GA, Winslow GM. Antigen‐driven induction of polyreactive IgM during intracellular bacterial infection. J Immunol. 2012;189:1440‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rubtsova K, Rubtsov AV, Halemano K, et al. T cell production of IFNy in response to TLR7/IL‐12 stimulates optimal B cell responses to viruses. PLoS ONE. 2016;11:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wehr C, Eibel H, Masilamani M, et al. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. 2004;113:161‐171. [DOI] [PubMed] [Google Scholar]
- 106. Frisullo G, Nociti V, Iorio R, et al. Increased expression of T‐bet in circulating B cells from a patient with multiple sclerosis and celiac disease. Hum Immunol. 2008;69:837‐839. [DOI] [PubMed] [Google Scholar]
- 107. Wang Z, Wang Z, Wang J, Diao Y, Qian X, Zhu N. T‐bet‐expressing B cells are positively associated with Crohn's disease activity and support Th1 inflammation. DNA Cell Biol. 2016;35:628‐635. [DOI] [PubMed] [Google Scholar]
- 108. Claes N, Fraussen J, Vanheusden M, et al. Age‐associated B cells with proinflammatory characteristics are expanded in a proportion of multiple sclerosis patients. J Immunol. 2016;197:4576‐4583. [DOI] [PubMed] [Google Scholar]
- 109. Becker AM, Dao KH, Han BK, et al. SLE peripheral blood B cell, T cell and Myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One.8:e67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rubtsov AV, Rubtsova K, Fischer A, et al. Toll‐like receptor 7 (TLR7)‐driven accumulation of a novel CD11c+ B‐cell population is important for the development of autoimmunity. Blood. 2011;118:1305‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c‐expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J Immunol. 2015;195:71‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Isnardi I, Ng Y‐S, Menard L, et al. Complement receptor 2/CD21‐ human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026‐5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rakhmanov M, Keller B, Gutenberger S, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate‐like B cells. Proc Natl Acad Sci U. S. A. 2009;1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rivera‐Correa J, Guthmiller JJ, Vijay R, et al. Plasmodium DNA‐mediated TLR9 activation of T‐bet+ B cells contributes to autoimmune anaemia during malaria. Nat Commun. 2017;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kenderes KJ, Levack RC, Papillion AM, Cabrera‐Martinez B, Dishaw LM, Winslow GM. T‐Bet+ IgM memory cells generate multi‐lineage effector B cells. Cell Rep. 2018;24:824‐837e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Defrance T, Taillardet M, Genestier L. T cell‐independent B cell memory. Curr Opin Immunol. 2011;23:330‐336. [DOI] [PubMed] [Google Scholar]
- 117. Good‐Jacobson KL, Tarlinton DM. Multiple routes to B‐cell memory. Int Immunol. 2012;24:403‐408. [DOI] [PubMed] [Google Scholar]
- 118. Mewono L, Agnandji ST, Matondo Maya DW, et al. Malaria antigen‐mediated enhancement of interleukin‐21 responses of peripheral blood mononuclear cells in African adults. Exp Parasitol. 2009;122:37‐40. [DOI] [PubMed] [Google Scholar]
- 119. Obeng‐Adjei N, Portugal S, Tran T, et al. Circulating Th1‐cell‐type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. 2015;13:425‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Roetynck S, Olotu A, Simam J, et al. Phenotypic and functional profiling of CD4 T cell compartment in distinct populations of healthy adults with different antigenic exposure. PLoS ONE. 2013;8:e55195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mewono L, Matondo Maya DW, Matsiegui P‐B, et al. Interleukin‐21 is associated with IgG1 and IgG3 antibodies to erythrocyte‐binding antigen‐175 peptide 4 of Plasmodium falciparum in Gabonese children with acute falciparum malaria. Eur Cytokine Netw. 2008;19:30‐36. [DOI] [PubMed] [Google Scholar]
- 122. Pérez‐Mazliah D, Nguyen MP, Hosking C, et al. Follicular helper T cells are essential for the elimination of Plasmodium infection. EBioMedicine. 2017;24:216‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The costimulatory molecule ICOS regulates host Th1 and follicular Th cell differentiation in response to Plasmodium chabaudi chabaudi AS infection. J Immunol. 2016;196:778‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zander R, Obeng‐Adjei N, Guthmiller J, et al. PD‐1 co‐inhibitory and OX40 co‐stimulatory crosstalk regulates helper T cell differentiation and anti‐plasmodium humoral immunity. Cell Host Microbe. 2015;17:628‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Laidlaw BJ, Lu Y, Amezquita RA, et al. Interleukin‐10 from CD4+ follicular regulatory T cells promotes the germinal center response. Sci Immunol. 2017;2:eaan4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Guthmiller JJ, Graham AC, Zander RA, Pope RL, Butler NS. Cutting edge: IL‐10 is essential for the generation of germinal center B cell responses and anti‐ plasmodium humoral immunity. J Immunol. 2017;198:617‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hahn WO, Butler NS, Lindner SE, et al. cGAS‐mediated control of blood‐stage malaria promotes Plasmodium‐specific germinal center responses. JCI insight. 2018;3:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long‐term humoral immunity. Nature. 2003;421:282‐287. [DOI] [PubMed] [Google Scholar]
- 129. Qi H. From SAP‐less T cells to helpless B cells and back: dynamic T‐B cell interactions underlie germinal center development and function. Immunol Rev. 2012;247:24‐35. [DOI] [PubMed] [Google Scholar]
- 130. Butler NS, Moebius J, Pewe LL, et al. Therapeutic blockade of PD‐L1 and LAG‐3 rapidly clears established blood‐stage Plasmodium infection. Nat Immunol. 2011;13:188‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kurup SP, Obeng‐Adjei N, Anthony SM, et al. Regulatory T cells impede acute and long‐term immunity to blood‐stage malaria through CTLA‐4. Nat Med. 2017;23:1220‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell. 2000;100:655‐669. [DOI] [PubMed] [Google Scholar]
- 133. Knox JJ, Kaplan DE, Betts MR. T‐bet‐expressing B cells during HIV and HCV infections. Cell Immunol. 2017;321:26‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Peng SL, Szabo SJ, Glimcher LH. T‐bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U. S. A. 2002;99:5545‐5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Coutelier JP, van der Logt JT, Heessen FW, Warnier G, Snick JV. IgG2a restriction of murine antibodies elicited by viral infections. J Exp Med. 1987;165:64‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Markine‐Goriaynoff D, Coutelier J‐P. Increased efficacy of the immunoglobulin G2a Subclass in antibody‐mediated protection against lactate dehydrogenase‐elevating virus‐induced polioencephalomyelitis revealed with switch mutants. J Virol. 2002;76:432‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Dobaño C, Santano R, Vidal M, et al. Differential patterns of IgG subclass responses to Plasmodium falciparum antigens in relation to malaria protection and RTS. S Vaccination Front Immunol. 2019;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Roussilhon C, Oeuvray C, Müller‐Graf C, et al. Long‐term clinical protection from Falciparum Malaria is strongly associated with IgG3 antibodies to merozoite surface protein 3. PLoS Medicine. 2007;4:e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zander RA, Vijay R, Pack AD, et al. Th1‐like Plasmodium‐specific memory CD4+ T cells support humoral immunity. Cell Rep. 2017;21:1839‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ioannidis LJ, Nie CQ, Ly A, Ryg‐Cornejo V, Chiu CY, Hansen DS. Monocyte‐ and neutrophil‐derived CXCL10 impairs efficient control of blood‐stage malaria infection and promotes severe disease. J Immunol. 2016;196:1227‐1238. [DOI] [PubMed] [Google Scholar]
- 141. Ryg‐Cornejo V, Ioannidis L, Ly A, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. 2016;14:68‐81. [DOI] [PubMed] [Google Scholar]
- 142. Naradikian MS, Myles A, Beiting DP, et al. Cutting Edge: IL‐4, IL‐21, and IFN‐γ Interact To Govern T‐bet and CD11c Expression in TLR‐Activated B Cells. J. Immunol. 2016;197:1023‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yates JL, Racine R, McBride KM, Winslow GM. T cell‐dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J Immunol. 2013;191:1240‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Manni M, Gupta S, Ricker E, et al. Regulation of age‐associated B cells by IRF5 in systemic autoimmunity. Nat Immunol. 2018;19:407‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zinöcker S, Schindler CE, Skinner J, et al. The V gene repertoires of classical and atypical memory B cells in malaria‐susceptible West African children. J Immunol. 2015;194:929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Domeier PP, Chodisetti SB, Soni C, et al. IFN‐γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. 2016;213:715‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]