

Abstract
Chemo‐ and regioselectivity are often difficult to control during olefin hydrosilylation catalyzed by d‐ and f‐block metal complexes. The cationic hydride of calcium [CaH]+ stabilized by an NNNN macrocycle was found to catalyze the regioselective hydrosilylation of aliphatic olefins to give anti‐Markovnikov products, while aryl‐substituted olefins were hydrosilyated with Markovnikov regioselectivity. Ethylene was efficiently hydrosilylated by primary and secondary hydrosilanes to give di‐ and monoethylated silanes. Aliphatic hydrosilanes were preferred over other commonly employed hydrosilanes: Arylsilanes such as PhSiH3 underwent scrambling reactions promoted by the nucleophilic hydride, while alkoxy‐ and siloxy‐substituted hydrosilanes gave isolable alkoxy and siloxy calcium derivatives.
Keywords: alkaline earth metals, calcium hydride, hydrosilylation, Lewis acids, regioselective catalysis
The CaH effect: The calcium hydride cation [CaH]+ catalyzes the regioselective hydrosilylation of ethylene, α‐olefins, and styrene derivatives. Aliphatic hydrosilanes are preferred over commonly employed silanes such as PhSiH3 as the latter undergoes scrambling reactions.

Hydrosilylation of olefins is of considerable importance, since organosilicon intermediates and fine chemicals can be synthesized by addition of a Si–H function to unsaturated C=C double bonds.1 This reaction is efficiently catalyzed by transition metal catalysts,2 in particular platinum complexes (Speier and Karstedt catalysts).3 In the context of current quests for inexpensive, innocuous, and earth‐abundant alternatives, base metal catalysts containing Mn, Fe, Co, and Ni have been reported.2b, 4 Systems based on rare earth metals5 were also introduced as hydrosilylation catalysts, but operate through the combination of hydrometalation and σ‐bond metathesis steps instead of the sequence of oxidative addition/reductive elimination as commonly observed for late transition metal catalysts (Chalk–Harrod mechanism).6 Catalysts based on p‐block elements also promote this reaction.7 Harder et al. reported on the use of Group 1 and Group 2 metal benzyl complexes for the hydrosilylation of styrene derivatives with arylsilanes (Scheme 1);8 the molecular calcium hydride [(DIPPBDI)(thf)Ca(μ‐H)]2 (DIPPBDI=CH[C(Me)N‐DIPP]2, DIPP=2,6‐iPr2C6H3) was found to catalyze the hydrosilylation of 1,1‐diphenylethylene (1,1‐DPE) with phenylsilane.9 The triphenylsilyl complex [Ca(SiPh3)2(thf)4] hydrosilylated styrene derivatives with anti‐Markovnikov selectivity as did related alkali metal silanide and hydrosilicate complexes.10 Although simple Na[HBEt3] was reported to hydrosilylate styrene derivatives with aryl hydrosilanes, no activity was observed when aliphatic olefins like 1‐hexene or silanes such as Et3SiH were used.11
Scheme 1.

a) Previous reports of s‐block metal catalyzed hydrosilylation. b) Hydrosilylation catalyzed by calcium hydride cation [CaH]+.
In contrast to olefin hydrogenation by molecular alkaline earth metal hydrides,13 hydrosilylation of unactivated alkenes using aliphatic silanes by s‐block metal catalysts remains elusive.12 Herein we report on the hydrosilylation of ethylene, α‐olefins, and styrene derivatives using a molecular calcium hydride cation [CaH]+ stabilized by the macrocyclic polyamine ligand Me4TACD (1,4,7,10‐tetramethyl‐1,4,7,10‐tetraazacyclododecane).
Recently we isolated cationic calcium hydrides by hydrogenolysis of benzyl complexes [(L)Ca(η1‐CH2Ph)x(thf)(2−x)][BAr4](2−x) (L=Me4TACD; x=1,2; Ar=C6H3‐3,5‐Me2), which were limited by their solubility, stability, and scalability.13a When bis(allyl)calcium14 was treated with the conjugated Brønsted acid of the macrocyclic ligand [LH][BAr4] (1),15 the allyl calcium cation [(L)Ca(η3‐C3H5)(thf)][BAr4] (2) was obtained in high yield (Scheme 2 and Figure 1).16 Although 2 was inert towards H2 even at 70 °C, the stoichiometric reaction with RSiH3 (R=Ph, nOct) in THF gave the dimeric calcium hydride cation [(L)2Ca2(μ‐H)2(thf)][BAr4]2 (3) alongside the corresponding allylsilane (C3H5)RSiH2 after 5 min at 25 °C. A crystal structure analysis of 3 by X‐ray diffraction revealed a nonsymmetrical dimer with one coordinated THF as observed for the Me4TACD‐stabilized hydride of divalent ytterbium.17 The 1H NMR spectrum of 3 in [D8]THF shows a Ci‐symmetric structure even at −80 °C, indicating fast reversibility of the THF coordination (see the Supporting Information). Calculations on the DFT level suggest exothermic THF coordination (ca. 33 kJ mol−1, see the Supporting Information) to the solvent‐free dimer,13a in line with labile solvation of other molecular calcium hydrides.18
Scheme 2.

Improved synthesis of the cationic calcium hydride 3 starting from the allyl calcium cation 2.
Figure 1.

Structure of the molecular cations in 2 and 3. Displacement parameters are shown at the 50 % probability level. Anions, lattice solvents, and hydrogen atoms except for the hydrides are omitted for clarity.
The molecular cation in 3 was highly reactive toward hydrosilanes: Treating a solution of 3 with the commonly employed hydrosilylane PhSiH3 led to broad resonances for the calcium‐ and silicon‐bonded hydrides in the 1H NMR spectrum. Cross peaks appeared in an EXSY NMR experiment (see the Supporting Information), indicating their exchange on the NMR timescale. After 30 min, signals for SiH4 and Ph2SiH2 were observed as the result of reversible aryl exchange.19 Addition of D2 (1 bar) lowered the intensity of the hydride resonances and signals of H2 and HD appeared within 3 d. These exchange processes may be explained by reversible coordination of the metal hydride to the silicon center to form a hypervalent silicate.20 Although such a species was not observed by 1H and 29Si NMR spectroscopy even at −90 °C, NOESY signals indicate close proximity of the silicon hydrides to the methyl protons in the Me4TACD ligand (see the Supporting Information). Transfer of a hydride from [CaH]+ to the hydrosilane to give a solvent‐separated ion pair with [PhSiH4]− anion remains undetected but cannot be ruled out.8 An experiment performed with nOctSiH3 under identical conditions did not show any formation of nOct2SiH2 or SiH4 after 24 h; however, deuterolysis of nOctSiH3 in the presence of 3 (10 mol %) to give nOctSiH3−xDx (x=0–3) and HD/H2 was observed after 7 d at 25 °C under 1 bar of D2. When alkoxy hydrosilanes were used, irreversible hydride transfer from calcium to silicon occurred (Scheme 3). Reaction with MeSiH(OMe)2 gave the dimeric methoxy calcium complex 4, which was independently synthesized from 3 and methanol. Commonly used O(SiMe2H)2 also underwent Si–O cleavage to selectively give the dimethylsiloxy complex 5 and Me2SiH2 within 2 h at 70 °C. Coordination of the silyl ether to the Lewis acidic calcium center, as recently observed for a cationic magnesium complex, might facilitate the ether cleavage.21
Scheme 3.

Reaction of calcium hydride 3 with alkoxy‐ and siloxy‐substituted hydrosilanes.
To assess the suitability of different hydrosilanes, complex 3 was tested in the hydrosilylation of ethylene. At ambient conditions (25 °C, 1 bar ethylene), PhSiH3 gave Et2PhSiH within 6 h (Table 1, entry 1). Because of competing aryl exchange, ca. 5 % of Ph2EtSiH and Et3SiH were also formed at 70 °C (Table 1, entry 2). Higher selectivity was observed with electron‐rich (para‐R‐C6H4)SiH3 (Table 1, entry 3, R=MeO and 4, R=Me2N), while electron‐deficient (para‐F3C‐C6H4)SiH3 showed immediate scrambling followed by decomposition of the catalyst (Table 1, entry 5). Alkyl‐substituted primary hydrosilanes were selectively converted within 30–35 min (Table 1, entries 6–9). Monitoring the reaction by NMR spectroscopy revealed full consumption of the primary hydrosilane prior to the second addition of ethylene. While the selectivity with Ph2SiH2 was lower due to scrambling (Table 1, entry 10), alkyl‐substituted secondary silanes were fully converted within 15–20 min (Table 1, entries 11–14). Hydrosilylation of sterically more demanding iBu2SiH2 was incomplete after 60 min (Table 1, entry 15), while bulkier hydrosilanes Mes2SiH2 and tBu2SiH2 as well as tertiary hydrosilanes Et3SiH and Me2EtSiH did not react at all (Table 1, entries 16 and 17).
Table 1.
Hydrosilylation of ethylene by complex 3.[a]
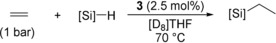
|
Entry |
Hydrosilane |
Product[b] |
t [min] |
Conv. [%][c] |
TOF [h−1][d] |
|---|---|---|---|---|---|
|
1[e] |
PhSiH3 |
Et2PhSiH |
360 |
90 |
12 |
|
2 |
PhSiH3 |
Et2PhSiH |
30 |
90 |
144 |
|
3 |
(para‐MeO‐ C6H4)SiH3 |
Et2(para‐MeO‐ C6H4)SiH |
30 |
99 |
160 |
|
4 |
(para‐Me2N‐ C6H4)SiH3 |
Et2(para‐Me2N‐ C6H4)SiH |
30 |
99 |
160 |
|
5[e] |
(para‐F3C‐ C6H4)SiH3 |
–[f] |
30 |
0 |
0 |
|
6 |
nBuSiH3 |
Et2 nBuSiH |
35 |
99 |
144 |
|
7 |
nHexSiH3 |
Et2 nHexSiH |
30 |
99 |
160 |
|
8 |
nOctSiH3 |
Et2 nOctSiH |
30 |
99 |
160 |
|
9 |
CySiH3 |
Et2CySiH |
30 |
90 |
144 |
|
10 |
Ph2SiH2 |
EtPh2SiH |
15 |
90 |
144 |
|
11 |
PhMeSiH2 |
EtPhMeSiH |
15 |
99 |
160 |
|
12 |
nOctMeSiH2 |
EtnOctMeSiH |
15 |
99 |
160 |
|
13 |
CyMeSiH2 |
EtCyMeSiH |
15 |
99 |
160 |
|
14 |
Et2SiH |
Et3SiH |
20 |
99 |
120 |
|
15 |
iBu2SiH2 |
EtiBu2SiH |
60 |
28 |
11 |
|
16 |
Mes2SiH2/ tBu2SiH2 |
– |
60 |
0 |
0 |
|
17 |
Et3SiH/ Me2EtSiH |
– |
60 |
0 |
0 |
[a] 0.1 mmol of substrate in 0.6 mL of [D8]THF, 1,4‐(SiMe3)2C6H4 (25 μmol) as internal standard. [b] Characterized by NMR spectroscopy and GC‐MS. [c] Determined by 1H NMR spectroscopy. [d] Calculated based on the amount of ethylene consumed. [e] 25 °C. [f] Decomposition of catalyst.
The Lewis acidity of the silicon center of the hydrosilane could promote the nucleophilic addition of the metal hydride to form a hypervalent silicate, but also facilitates the aryl exchange.19b Alkyl groups that lower the Lewis acidity increase the chemoselectivity and are favored for hydrosilylation catalyzed by 3. As the homologous magnesium hydride cation [MgH]+ did not show any reaction with PhSiH3 under comparable conditions,22 the combination of the Lewis acidic metal center with the nucleophilicity of the hydride ligand appears crucial.
While monitoring the catalysis by 1H NMR spectroscopy, we observed no calcium ethyl species. Only after the hydrosilane was fully consumed or when hydride 3 was dissolved in [D8]THF and pressurized with ethylene (1 bar), was formation of an ethyl calcium species detected by characteristic signals at δ −1.02 (q) and 1.26 (t) ppm for the methylene and methyl protons, respectively.24 After 10 min at 25 °C, higher n‐alkyl calcium species were also evident from their characteristic resonances, indicating additional insertion of ethylene into the calcium–n‐alkyl bond (see the Supporting Information). While oligomerization of ethylene was not observed for solid [CaH2]∞ 23 and [(DIPPBDI)Ca(μ‐H)]2,24 strontium hydride [(DIPePBDI)Sr(μ‐H)]2 (DIPeP=2,6‐(pent‐3‐yl)2‐phenyl) formed oligoethylene at room temperature.20a Unlike the BDI‐stabilized ethyl complexes, the highly reactive cationic n‐alkyl calcium derivative of 3 could not be isolated as it readily decomposed in THF solution (t 1/2<10 min at 25 °C, 20 min at −20 °C) to give short alkanes (C2–C6) and other undefined species. Only fully protonated alkanes were detected when the reaction was carried out in [D8]THF, indicating that the reaction with the ligand backbone as in related lanthanide complexes25 is favored over solvent deprotonation.
Higher α‐olefins such as 1‐octene and 1‐hexene were hydrosilylated at 70 °C to give the anti‐Markovnikov products with high regioselectivity. Depending on the stoichiometry and the hydrosilane, secondary (Table 2, entries 1 and 2) or tertiary (Table 2, entries 3 and 4) silanes formed, while no reaction occurred with the tertiary products or Et3SiH (Table 2, entry 5). The silanes could be readily isolated after the catalyst 3 was precipitated with n‐pentane and filtered off (see the Supporting Information). Hydrosilylation of 1,4‐hexadiene only gave the 4‐alkenylsilane, and as for 2‐hexene, no reaction of the internal double bond was detected even after prolonged reaction time or with an excess of hydrosilane (Table 2, entries 6 and 7). Likewise, the internal double bond in 4‐vinylcyclohexene, cyclohexene, and norbornene, as well as the 1,1‐disubstituted double bond in 2‐ethyl‐1‐butene (Table 2, entries 8–11) were not hydrosilylated. Allyl methyl ether was not hydrosilylated as the nucleophilic hydride was readily converted into the methoxy complex 4 under formation of propene (Table 2, entry 12).
Table 2.
Regioselective hydrosilylation of aliphatic olefins by complex 3.[a]

|
Entry |
Olefin |
Hydrosilane |
Product[b] |
t [h] |
Conv. [%][c] |
TOF [h−1] |
|---|---|---|---|---|---|---|
|
1 |
|
Et2SiH2 |
Et2 nHexSiH |
24 |
96 |
0.8 |
|
2 |
|
nOctSiH3 |
nOct2SiH2 |
24 |
95 |
0.8 |
|
3[d] |
nOctSiH3 |
nOct3SiH |
24 |
87 |
0.7 |
|
|
4 |
Et2SiH2 |
Et2 nOctSiH |
24 |
95 |
0.8 |
|
|
5 |
Et3SiH |
– |
24 |
0 |
0 |
|
|
6 |
|
Et2SiH2 |
|
24 |
95 |
0.8 |
|
7 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
8 |
|
nOctSiH3 |
|
24 |
70 |
0.6 |
|
9 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
10 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
11 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
12 |
|
nOctSiH3 |
–[e] |
0.1 |
0 |
0 |










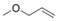
[a] Substrate (0.1 mmol) in [D8]THF (0.6 mL), 1,4‐(SiMe3)2C6H4 (25 μmol) as internal standard. [b] Characterized by NMR spectroscopy and GC‐MS. [c] Determined by 1H NMR spectroscopy. [d] 0.5 equiv of silane [e] Formation of methoxide complex 4 and propene.
Styrene and PhSiH3 reacted with moderate selectivity to give the Markovnikov‐silylated product, while by‐products included unreacted Ph2SiH2 and the dibenzylsilane (PhCHMe)2SiH2, which were formed from SiH4 through competing silane scrambling (Table 3, entry 1). Electron‐rich (para‐MeO‐C6H4)SiH3 gave a similar product mixture with higher selectivity for the expected monoalkylated arylsilane (Table 3, entry 2). Slower, but highly selective conversion was observed for nOctSiH3, as no silane scrambling was evident after 90 min (Table 3, entry 3). In contrast to the hydrosilylation of α‐alkenes, no hydrosilylation was detected with the secondary silane Et2SiH2, and only oligostyrene was obtained after 6 h (Table 3, entry 4). 1,1‐DPE was selectively hydrosilylated with primary nOctSiH3 after 4 h at 70 °C (Table 3, entry 5), which is slower than hydrosilylation by [(DMAT)2Ca(thf)2] (DMAT=α‐Me3Si‐2‐Me2N‐benzyl) using PhSiH3 in benzene.8 While this calcium catalyst gave the anti‐Markovnikov product when the reaction was carried out in THF, complex 3 gave the Markovnikov product exclusively. This indicates that the hydride‐insertion mechanism is operative for 3 even in THF.8, 10c Again, the sterically demanding tertiary carbanion [Ph2CMe]− readily formed by insertion remained unreacted after 6 h in the presence of secondary Et2SiH2 (Table 3, entry 6), in line with a metal‐centered σ‐bond metathesis. α‐Methylstyrene was also selectively hydrosilylated as were the internal double bonds in E‐ and Z‐stilbene, but longer reaction times were required (Table 3, entries 7–9). Higher substituted 1‐phenylcyclohexene as well as tri‐ and tetraphenylethylene did not show any conversion (Table 3, entries 10–12). Triphenyl(vinyl)silane gave a mixture of both regioisomers (Table 3, entries 13), formed through competing insertion to give either the sterically favored linear or the α‐silicon‐stabilized branched carbanion (Ph3SiCHCH3)−.11, 26
Table 3.
Regioselective hydrosilylation of activated olefins by complex 3.[a]

|
Entry |
Olefin |
Hydrosilane |
Product[b] |
t [h] |
Conv. [%][c] |
TOF [h−1] |
|---|---|---|---|---|---|---|
|
1 |
|
PhSiH3 |
|
0.5 |
60[d] |
24 |
|
2 |
(para‐MeO‐C6H4)SiH3 |
0.5 |
81[d] |
32 |
||
|
3 |
nOctSiH3 |
1.5 |
99 |
13.3 |
||
|
4 |
Et2SiH2 |
oligostyrene |
6 |
0 |
0 |
|
|
5 |
|
nOctSiH3 |
|
4 |
99 |
5 |
|
6 |
Et2SiH2 |
– |
6 |
0 |
0 |
|
|
7 |
|
nOctSiH3 |
|
48 |
94 |
0.6 |
|
8 |
|
nOctSiH3 |
|
22 |
99 |
0.9 |
|
9 |
|
nOctSiH3 |
22 |
99 |
0.9 |
|
|
10 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
11 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
12 |
|
nOctSiH3 |
– |
24 |
0 |
0 |
|
13 |
|
nOctSiH3 |
|
16 |
72/28 |
1.3 |














[a] Substrate (0.1 mmol) in [D8]THF (0.6 mL) 1,4‐(SiMe3)2C6H4 (25 μmol) as internal standard. [b] Characterized by NMR spectroscopy and GC‐MS. [c] Determined by 1H NMR spectroscopy. [d] Ar2SiH2 and (MeCHPh)2SiH2 (diastereomers) as by‐products.
In conclusion, the cationic calcium hydride 3 catalyzed the hydrosilylation of ethylene and α‐olefins with anti‐Markovnikov selectivity as well as styrene derivatives with Markovnikov selectivity. Selective catalysis was observed with (alkyl)hydrosilanes, whereas PhSiH3 as well as alkoxy‐ and siloxy‐substituted hydrosilanes underwent nucleophilic substitution of the hydride. Using nOctSiH3, the following order of reactivity for 3 was established: ethylene > styrene > 1‐octene. All experimental evidence suggests that [CaH]+ forms an alkyl calcium complex24 as the result of hydrometalation of olefin followed by σ‐bond metathesis with the hydrosilane to give the hydrosilylated product and [CaH]+.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft through the International Research Training Group “Selectivity in Chemo‐ and Biocatalysis” (GRK 1628) for financial support, F. Ritter and M. Paul for assistance with X‐ray crystallography, and Dr. G. Fink for NMR measurements.
D. Schuhknecht, T. P. Spaniol, L. Maron, J. Okuda, Angew. Chem. Int. Ed. 2020, 59, 310.
References
- 1.
- 1a. Marciniec B., Hydrosilylation, Springer, Berlin, 2009; [Google Scholar]
- 1b. Troegel D., Stohrer J., Coord. Chem. Rev. 2011, 255, 1440–1459; [Google Scholar]
- 1c. Nakajima Y., Shimada S., RSC Adv. 2015, 5, 20603–20616. [Google Scholar]
- 2.
- 2a. Meister T. K., Riener K., Gigler P., Stohrer J., Herrmann W. A., Kühn F. E., ACS Catal. 2016, 6, 1274–1284; [Google Scholar]
- 2b. Du X. Y., Huang Z., ACS Catal. 2017, 7, 1227–1243. [Google Scholar]
- 3.
- 3a. Speier J. L., Adv. Organomet. Chem., 1979, 17 407–447; [Google Scholar]
- 3b. Lewis L. N., Stein J., Gao Y., Colborn R. E., Hutchins G., Platinum Met. Rev. 1997, 41, 66. [Google Scholar]
- 4.
- 4a. Hofmann R., Vlatković M., Wiesbrock F., Polymers 2017, 9, 534; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Obligacion J. V., Chirik P. J., Nat. Rev. Chem. 2018, 2, 15–34; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Zaranek M., Pawluc P., ACS Catal. 2018, 8, 9865–9876. [Google Scholar]
- 5. Liu D. S., Liu B. Y., Pan Z. X., Li J. F., Cui C. M., Sci. China Chem. 2019, 62, 571–582. [Google Scholar]
- 6. Chalk A. J., Harrod J. F., J. Am. Chem. Soc. 1965, 87, 16–21. [Google Scholar]
- 7.
- 7a. Oertle K., Wetter H., Tetrahedron Lett. 1985, 26, 5511–5514; [Google Scholar]
- 7b. Lambert J. B., Zhao Y., Wu H., J. Org. Chem. 1999, 64, 2729–2736; [DOI] [PubMed] [Google Scholar]
- 7c. Rubin M., Schwier T., Gevorgyan V., J. Org. Chem. 2002, 67, 1936–1940; [DOI] [PubMed] [Google Scholar]
- 7d. Simonneau A., Oestreich M., Angew. Chem. Int. Ed. 2013, 52, 11905–11907; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12121–12124; [Google Scholar]
- 7e. Holthausen M. H., Mehta M., Stephan D. W., Angew. Chem. Int. Ed. 2014, 53, 6538–6541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6656–6659; [Google Scholar]
- 7f. Revunova K., Nikonov G. I., Dalton Trans. 2015, 44, 840–866; [DOI] [PubMed] [Google Scholar]
- 7g. Jakobsson K., Chu T., Nikonov G. I., ACS Catal. 2016, 6, 7350–7356; [Google Scholar]
- 7h. Lipke M. C., Liberman-Martin A. L., Tilley T. D., Angew. Chem. Int. Ed. 2017, 56, 2260–2294; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2298–2335. [Google Scholar]
- 8. Buch F., Brettar J., Harder S., Angew. Chem. Int. Ed. 2006, 45, 2741–2745; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2807–2811. [Google Scholar]
- 9.
- 9a. Ruspic C., Spielmann J., Harder S., Inorg. Chem. 2007, 46, 5320–5326; [DOI] [PubMed] [Google Scholar]
- 9b. Harder S., Chem. Rev. 2010, 110, 3852–3876. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Leich V., Spaniol T. P., Maron L., Okuda J., Chem. Commun. 2014, 50, 2311–2314; [DOI] [PubMed] [Google Scholar]
- 10b. Leich V., Lamberts K., Spaniol T. P., Okuda J., Dalton Trans. 2014, 43, 14315–14321; [DOI] [PubMed] [Google Scholar]
- 10c. Leich V., Spaniol T. P., Okuda J., Organometallics 2016, 35, 1179–1182; [Google Scholar]
- 10d. Schuhknecht D., Leich V., Spaniol T. P., Okuda J., Chem. Eur. J. 2018, 24, 13424–13427. [DOI] [PubMed] [Google Scholar]
- 11. Zaranek M., Witomska S., Patroniak V., Pawluc P., Chem. Commun. 2017, 53, 5404–5407. [DOI] [PubMed] [Google Scholar]
- 12.After submission of this manuscript, Hill et al. reported hydrosilylation of unactivated olefins with PhSiH3 using molecular Mg and Ca hydrides.
- 12a. Garcia L., Dinoi C., Mahon M. F., Maron L., Hill M. S., Chem. Sci. 2019, 10, 8108–8118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Garcia L., Mahon M. F., Hill M. S., Organometallics 2019, 38, 3778–3785. [Google Scholar]
- 13.
- 13a. Schuhknecht D., Lhotzky C., Spaniol T. P., Maron L., Okuda J., Angew. Chem. Int. Ed. 2017, 56, 12367–12371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12539–12543; [Google Scholar]
- 13b. Bauer H., Alonso M., Fischer C., Rosch B., Elsen H., Harder S., Angew. Chem. Int. Ed. 2018, 57, 15177–15182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15397–15402; [Google Scholar]
- 13c. Wilson A. S. S., Dinoi C., Hill M. S., Mahon M. F., Maron L., Angew. Chem. Int. Ed. 2018, 57, 15500–15504; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15726–15730; [Google Scholar]
- 13d. Shi X., Qin G., Wang Y., Zhao L., Liu Z., Cheng J., Angew. Chem. Int. Ed. 2019, 58, 4356–4360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4400–4404. [Google Scholar]
- 14. Jochmann P., Dols T. S., Spaniol T. P., Perrin L., Maron L., Okuda J., Angew. Chem. Int. Ed. 2009, 48, 5715–5719; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5825–5829. [Google Scholar]
- 15.
- 15a. Dyke J., Levason W., Light M. E., Pugh D., Reid G., Bhakhoa H., Ramasami P., Rhyman L., Dalton Trans. 2015, 44, 13853–13866; [DOI] [PubMed] [Google Scholar]
- 15b. Bhakhoa H., Rhyman L., Lee E. P., Mok D. K. W., Ramasami P., Dyke J. M., Dalton Trans. 2017, 46, 15301–15310. [DOI] [PubMed] [Google Scholar]
- 16. Lichtenberg C., Jochmann P., Spaniol T. P., Okuda J., Angew. Chem. Int. Ed. 2011, 50, 5753–5756; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5872–5875. [Google Scholar]
- 17. Schuhknecht D., Truong K. N., Spaniol T. P., Maron L., Okuda J., Chem. Commun. 2018, 54, 11280–11283. [DOI] [PubMed] [Google Scholar]
- 18. Causero A., Ballmann G., Pahl J., Farber C., Intemann J., Harder S., Dalton Trans. 2017, 46, 1822–1831. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Castillo I., Tilley T. D., J. Am. Chem. Soc. 2001, 123, 10526–10534; [DOI] [PubMed] [Google Scholar]
- 19b. Liu X., Xiang L., Louyriac E., Maron L., Leng X., Chen Y., J. Am. Chem. Soc. 2019, 141, 138–142. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Rösch B., Gentner T. X., Elsen H., Fischer C. A., Langer J., Wiesinger M., Harder S., Angew. Chem. Int. Ed. 2019, 58, 5396–5401; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5450–5455; [Google Scholar]
- 20b. Mukherjee D., Höllerhage T., Leich V., Spaniol T. P., Englert U., Maron L., Okuda J., J. Am. Chem. Soc. 2018, 140, 3403–3411. [DOI] [PubMed] [Google Scholar]
- 21. Pahl J., Elsen H., Friedrich A., Harder S., Chem. Commun. 2018, 54, 7846–7849. [DOI] [PubMed] [Google Scholar]
- 22. Lemmerz L. E., Mukherjee D., Spaniol T. P., Wong A., Menard G., Maron L., Okuda J., Chem. Commun. 2019, 55, 3199–3202. [DOI] [PubMed] [Google Scholar]
- 23. Wright L., Weller S., J. Am. Chem. Soc. 1954, 76, 5305–5308. [Google Scholar]
- 24.
- 24a. Wilson A. S. S., Hill M. S., Mahon M. F., Dinoi C., Maron L., Science 2017, 358, 1168–1171; [DOI] [PubMed] [Google Scholar]
- 24b. Wilson A. S. S., Hill M. S., Mahon M. F., Organometallics 2019, 38, 351–360. [Google Scholar]
- 25.
- 25a. Venugopal A., Fegler W., Spaniol T. P., Maron L., Okuda J., J. Am. Chem. Soc. 2011, 133, 17574–17577; [DOI] [PubMed] [Google Scholar]
- 25b. Fegler W., Venugopal A., Spaniol T. P., Maron L., Okuda J., Angew. Chem. Int. Ed. 2013, 52, 7976–7980; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8134–8138. [Google Scholar]
- 26. Wiesinger M., Maitland B., Farber C., Ballmann G., Fischer C., Elsen H., Harder S., Angew. Chem. Int. Ed. 2017, 56, 16654–16659; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16881–16886. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary