

Abstract
Herein, we have synthesized an enantiomerically pure propeller‐shaped PAH, C90H48, possessing three [7]helicene and three [5]helicene subunits. This compound can be obtained in gram quantities in a straightforward manner. The photophysical and chiroptical properties were investigated using UV/Vis absorption and emission, optical rotation and circular dichroism spectroscopy, supported by DFT calculations. The nonlinear optical properties were investigated by two‐photon absorption measurements using linearly and circularly polarized light. The extremely twisted structure and packing of the homochiral compound were investigated by single‐crystal X‐ray diffraction analysis.
Keywords: chirality, enantiomers, helicenes, polycyclic aromatic hydrocarbons, stereospecific synthesis
Chirality: The challenge and cumbersome process of resolving enantiomers of a large optically active PAH has been overcome by stereospecific synthesis, taking advantage of the thermodynamic control of the reaction and kinetic (un)stability of [n]helicenes (see figure).
Introduction
Polycyclic aromatic hydrocarbons1 with multiple helicene substructures are of special interest for synthetic and materials chemists owing to their unique chiroptical and nonlinear optical properties, structural beauty, and synthetic challenge.2 These interests have been fueled by their potential applications3 as circularly polarized light (CPL) emitters in OLEDs,4 CPL responsive OFETs,5 spin filters,6 and photoswitches.7 In the past few years, there has been a great advancement in the synthesis of these class of compounds.8 PAHs with double,9 triple,10 quadruple,11 quintuple,12 and sextuple13 helicene subunits ([4],10a [5],9d, 10c, 13f, 13g, 14 [6],9e, 11b, 12b [7],9a, 9c, 13b, 13c, 13e or [9]13a helicenes) have been achieved. Despite this enormous synthetic success, however, the studies of this class of compounds are still limited by separation of their enantiomerically pure forms in minute quantities and investigation of their chiroptical properties by circular dichroism (CD) and circularly polarized luminescence spectroscopy, and these compounds have rarely been used for any application purposes.
The nonstereoselective synthesis of PAHs with m, number of helicene substructures, can possibly yield ≤2m stereoisomers depending on the symmetry and stereodynamics of the molecule. The asymmetric synthesis of PAHs with multiple helices is yet to be developed.2b By employing the nonasymmetric synthesis, the obtained mixture of enantiomers is usually separated with the help of HPLC employing chiral stationary phase columns. These are very expensive and their cost increases dramatically on moving from the analytical to semipreparative and preparative scale. Additionally, there is no universal column that could be used for the same class of molecules. Often even for the compounds with a very similar structure, the baseline separation cannot be achieved with the same column. Moreover, the large structure and limited solubility makes the separation of such compounds into enantiomers cumbersome. This issue can be considered a great hurdle in transforming these structural beauties into applications.
Herein, we describe the bottom‐up stereospecific synthesis of a novel enantiomerically pure propeller‐shaped molecule possessing six helicene substructures comprising three [7]helicene and three [5]helicene subunits. Our synthetic design takes advantage of thermodynamic control of reaction and kinetic (un)stability of [n]helicenes. The synthesized compound was fully characterized by 1D and 2D NMR, UV/Vis absorption and emission, optical rotation (OR) and circular dichroism (CD) spectroscopy, MALDI‐TOF mass spectrometry, and single‐crystal X‐ray diffraction. The nonlinear optical properties were investigated by two‐photon absorption experiments using linearly and circularly polarized light.
One of the best studied examples of multihelicene compounds is sextuplet helicene 1 reported by group of Kamikawa and Gingras independently13f, 13g at the same time (Figure 1). The six helicene subunits in 1 give rise to 20 possible stereoisomers, namely, 10 pairs of enantiomers. Interestingly, Kamikawa's palladium‐catalyzed room‐temperature synthesis afforded exclusively diastereomer 1 a, which upon heating for only three hours at 100 °C transformed into diastereomer 1 b. Gingras’ nickel‐mediated Yamamoto‐type reaction at 120 °C resulted in a mixture of diastereomers, which were also transformed exclusively into diastereomer 1 b when heated at the same temperature for 30 min. The sole formation of diastereomer 1 b at higher temperature can be attributed to its much higher thermodynamic stability in comparison to the other stereoisomers and labile configuration of [5]helicenes15 (ΔG ≠ (298 K)=24.1 kcal mol−1). Preferred formation of the thermodynamically most stable stereoisomers was also observed in the course of the synthesis of other multihelicene compounds at higher temperatures.13c Despite the formation of a single diastereomer, the challenge to resolve its enantiomers by HPLC with chiral stationary phase columns remains. This challenge has already been faced by Kamikawa, who resolved isomer 1 b into enantiomers. The attempts to resolve 1 a were not successful.13g
Figure 1.
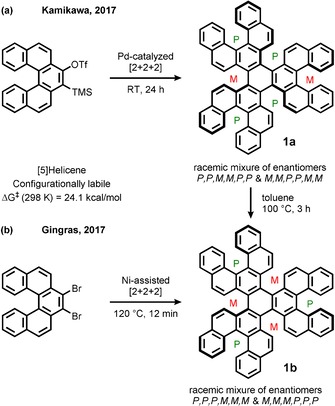
(a) Kamikawa's and (b) Gingras’ synthesis of sextuple helicenes obtained as the thermodynamically most stable single pair of enantiomers out of 20 possible stereoisomers.
Recently, we investigated the trend of configurational stabilities among [n]helicenes (n=4–11) and found an exponential increase in configurational stability when increasing the number of ortho‐fused rings (n).16 [n]Helicenes with n>6 are configurationally stable even at elevated temperatures. If in Gingras’ synthesis of 1 b the 7,8‐dibromo[5]helicene is replaced by configurationally stable and enantiomerically pure 9,10‐dibromo[7]helicene (M or P), one should obtain sextuple helicene as a single enantiomer benefiting from the configurational stability of [7]helicene and flexibility of [5]helicene.
The enantiopure 9,10‐dibromo[7]helicene was synthesized17 from the optically pure 1,1′‐bi‐(2‐phenanthrol) in four steps with retention of configuration and optical purity. The latter compound can be accessed in an enantiopure form by asymmetric catalysis or with the help of chiral auxiliaries.18 To our delight, the nickel‐mediated Yamamoto‐type [2+2+2] cyclotrimerization of (M)‐9,10‐dibromo[7]helicene (M ‐2) at 110 °C formed exclusively diastereomer (M,M,M,P,P,P)‐3 in enantiomerically pure form (Scheme 1 a) in 57 % yield. At this temperature, the [7]helicene subunit has a half‐life for enantiomerization of 1.8×109 min;15, 19 therefore, [7]helicene subunits do not undergo enantiomerization under these conditions. On the other hand, the newly formed [5]helicene subunits are still configurationally flexible. Restricting the configurational resilience to only [5]helicenes limits the number of theoretically possible stereoisomers to four diastereomers, C‐2 symmetric (M,M,M,P,P,M) and (M,M,M,P,M,M), and d‐3 symmetric (M,M,M,P,P,P) and (M,M,M,M,M,M), unlike the 20 stereoisomers of 1. The exclusive formation of diastereomer (M,M,M,P,P,P)‐3 can be rationalized when considering the relative thermodynamic stability of each stereoisomer (Scheme 1 b). (M,M,M,P,P,P)‐3 is 15 kcal mol−1 lower in energy than the energetically adjacent stereoisomer (M,M,M,P,P,M)‐3. The energy difference between the thermodynamically most and least stable stereoisomer is 23 kcal mol−1. Compound (M,M,M,P,P,P)‐3 can be easily synthesized in gram quantities and purified by routine silica gel column chromatography using petrol ether/toluene (1:1) as an eluent. The structure and purity of (M,M,M,P,P,P)‐3 could be unambiguously confirmed by 1D and 2D NMR spectroscopy and MALDI‐TOF mass spectrometry.
Scheme 1.
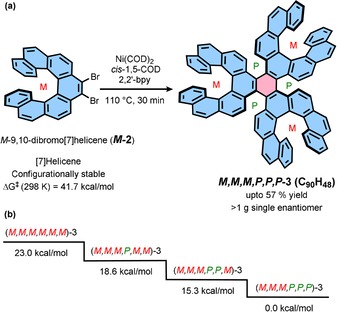
(a) Stereospecific synthesis of enantiopure PAH 3 and (b) relative energies of the four possible diastereomers.
Each proton and carbon signal could be assigned with the aid of 2D NMR (COSY, NOESY, HMBC, and HSQC) experiments, which confirmed the structure of compound 3 (Figures S5–S8). The assignment of proton resonances is shown in Figure 2. The downfield shift of proton h is due to the steric interaction in the fjord region of the helicene subunits, which is in consistent with that of [5]helicenes. Moreover, the presence of only eight signals indicates the higher d‐3 symmetry of the molecule. At this point, the formation of C‐2 symmetric isomers could be completely ruled out. Later, the single‐crystal X‐ray analysis, OR, and CD spectroscopic data supported by (TD)‐DFT calculations (vide‐infra) conferred the structure to that of diastereomer (M,M,M,P,P,P)‐3. The other enantiomer (P,P,P,M,M,M)‐3 is obtained when the synthesis is started with enantiopure (P)‐9,10‐dibromo[7]helicene instead of the (M)‐isomer.
Figure 2.
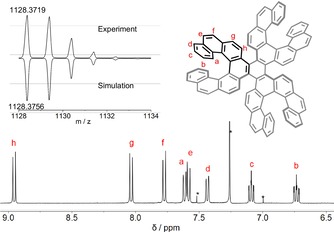
1H NMR spectrum of (M,M,M,P,P,P)‐3 in CDCl3 at 298 K and assigned corresponding proton signals (residual solvent CHCl3 peak and satellite peaks are marked with an asterisk). Top left: experimental and simulated high‐resolution MALDI mass isotopic pattern.
Single crystals of homochiral molecule (M,M,M,P,P,P)‐3 were grown by a slow diffusion of hexane into a solution of 3 in toluene in an NMR tube. Compound 3 crystallizes in the chiral orthorhombic P212 121 space group (Figure 3). Similar to 1 b, compound 3 also exhibits an extremely twisted structure and highly deformed benzene ring (B), which adopts twisted chair‐like conformation. The crystal packing analysis revealed the slipped stacking arrangement of molecules held together by π–π interactions (Figure S9, Supporting Information).
Figure 3.
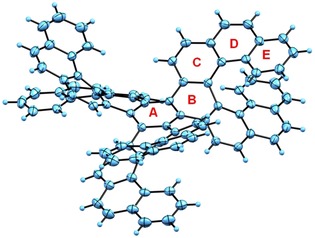
Solid‐state structure of (M,M,M,P,P,P)‐3. Thermal ellipsoids are shown at the 50 % probability level.
The UV/Vis absorption and emission spectra of (M,M,M,P,P,P)‐3 in chloroform, THF, and toluene are nearly identical, indicating no solvatochromism (Figure S1, Supporting Information). Compound 3 exhibited similar photophysical properties as 1 b. The calculated optical energy gap of 3, 2.33 eV, from the onset of absorption spectrum (535 nm) is marginally smaller than that of 1 b (2.38 eV). The absorption maximum in the visible range is, however, redshifted by 50 eV with nearly double molar absorptivity (Figure 4, λ max=418 nm, ϵ=77,276 m −1 cm−1). To get an insight into electronic transitions, the absorption UV/Vis spectrum was analyzed with the aid of TD‐DFT at B3LYP/6‐31G(d,p) level. The calculated absorption spectrum is red‐shifted by 41.3 eV compared to experimental spectrum. The significant extension of π‐conjugation upon replacing three [5]helicene units with three [7]helicene units resulted in only a small decrease of the calculated HOMO–LUMO energy gap (3.14 eV for 3 and 3.33 eV for 1 b). This trend is also observed in naked carbo[n]helicenes.20 Interestingly, the HOMO and LUMO frontier molecular orbitals are energetically degenerate with the HOMO−1 and LUMO+1 orbitals, respectively. While the calculated maximum absorption in the visible range (447 nm, oscillator strength (f)=1.05) is mainly stemming from the HOMO−1→LUMO, HOMO→LUMO+1, HOMO−1→LUMO+1, and HOMO→LUMO transitions, the lowest energy absorption (473 nm, f=0.10) is mainly from the HOMO−1→LUMO, HOMO−1→LUMO+1, and HOMO→LUMO transitions. The emission spectrum of 3 is redshifted by 46 eV compared to 1 b with similar fluorescence lifetime (5.22 ns) and slightly enhanced quantum yield (7.3 %).
Figure 4.
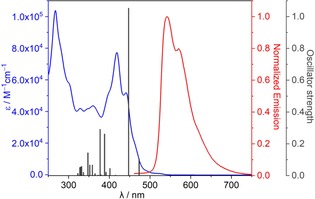
Measured UV/Vis absorption (blue line, c=1.4×10−5 m) and emission (red line, c=1.4×10−5 m, λ ex=410 nm) spectra of (M,M,M,P,P,P)‐3 in chloroform. Calculated (vertical gray lines) UV/Vis spectrum (TD‐DFT/B3LYP/6‐31G(d,p)).
Another interesting feature of PAHs with multiple [n]helicene units is the OR value. (M,M,M,P,P,P)‐1 b exhibited a specific OR ( ) of +495°, which is much smaller than that of (M)‐[5]helicene21 (−1670°) and could be elucidated from the presence of three P and three M helicene units. Moreover, the observed net positive value of optical rotation was attributed to the more pronounced helical twist of internal P helicene units. These inferences are further confirmed by the observed specific OR ( ) value of −222° (c=0.047, CHCl3) for (M,M,M,P,P,P)‐3. (M)‐[7]Helicene21 has a much larger specific OR of −6200°. Despite the more pronounced helical twist of internal (P)‐[5]helicene, the much larger OR value of external (M)‐[7]helicene resulted in the net negative value. The assigned absolute configuration of (M,M,M,P,P,P)‐3 is further confirmed by the reproduced experimental ECD spectrum with TD‐DFT calculations (Figure 5). Similar to OR, the negative cotton effect in the low energy region (460 nm, Δϵ=−72 m −1 cm−1, g=Δϵ/ϵ=3.3×10−3) stems from the external (M)‐[7]helicene units. The anisotropic factor g is in the order of 10−3 through the complete spectral range.
Figure 5.
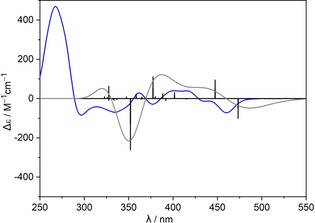
Experimental (blue line, c=1.4×10−5 m) and calculated (black vertical lines and gray line, TD‐DFT/B3LYP/6‐31G(d,p)) ECD spectrum of (M,M,M,P,P,P)‐3 in chloroform.
Previously, it was observed that the solvent polarity has a large influence on the two photon cross‐section value.22 Therefore, the two‐photon absorption (TPA) cross‐section of (M,M,M,P,P,P)‐3 was measured in two solvents with a large difference in polarity, THF, and toluene, via two‐photon induced fluorescence relative spectroscopic technique (Figure 6). The observed overlap between one‐ and two‐photon absorption spectra is, on one hand, attributed to the noncentrosymmetric molecule, in which the parity selection rule does not hold. On the other hand, it is caused by the high density of states, see Figure 4. The maximum non‐linear absorption occurs at higher energy transitions when compared with the linear absorption maximum. This is in consistent with previously published TPA data.23 The maximum TPA cross‐section of (M,M,M,P,P,P)‐3 is 436 GM in toluene, which increased by a factor of 1.2 to 517 GM in high polarity solvent THF. Two‐photon excitation was verified by log–log plots of fluorescence intensities vs. excitation power at various wavelengths, all gave slopes of 2 (Figure S3, Supporting Information). The higher two‐photon induced fluorescence signal is recorded upon excitation with CPL rather than linearly polarized light (Figure S3, Supporting Information). This indicates that the ratio of the TPA cross‐section for circularly and linearly polarized excitation is greater than 1 (Ω>1), which can be attributed to opposite symmetry of ground and excited states.24 This is in concomitance with the observed overlap of one‐ and two‐photon absorption spectra.
Figure 6.
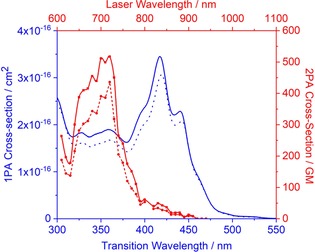
One‐photon (blue lines) and two‐photon (red lines) absorption spectra of (M,M,M,P,P,P)‐3 in THF (solid line) and toluene (dotted line).
In summary, we have successfully achieved the stereospecific synthesis of a large propeller‐shaped PAH, (M,M,M,P,P,P)‐3, in gram quantities and in an enantiomerically pure form, benefiting from the configurational stability of [7]helicene units and flexibility of [5]helicene units at elevated temperatures. The exclusive formation of (M,M,M,P,P,P)‐3 out of the twenty possible stereoisomer is rationalized by the thermodynamic control of the reaction and kinetic flexibility of [5]helicene units under the given reaction conditions. The highly twisted structure of (M,M,M,P,P,P)‐3 offers unique photophysical and nonlinear optical properties. Additionally, our findings provide an insight into how the OR values are changed in propeller shaped PAHs with multiple helicene subunits, this will be a guide in designing the molecules with larger OR. We believe that access to optically active compounds, such as 3, in large quantities will push their applications in organic electronic devices.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the Julius‐Maximilians‐Universität Würzburg within the “Excellent Ideas” programme. We sincerely thank Prof. C. Lambert (University of Würzburg) for generous support of our research. We are very grateful to Prof. C. Lambert and Prof. M. Juríček (University of Zurich) for their suggestions and proofreading our manuscript. We thank Prof. F. Würthner (University of Würzburg) for giving an access to a CD spectrometer and single‐crystal X‐ray diffraction facilities in his group.
F. Zhang, E. Michail, F. Saal, A.-M. Krause, P. Ravat, Chem. Eur. J. 2019, 25, 16241.
References
- 1.
- 1a. Wang X.-Y., Narita A., Müllen K., Nat. Chem. Rev. 2017, 2, 0100; [Google Scholar]
- 1b. Segawa Y., Ito H., Itami K., Nat. Rev. Mater. 2016, 1, 15002. [Google Scholar]
- 2.
- 2a. Rickhaus M., Mayor M., Juríček M., Chem. Soc. Rev. 2016, 45, 1542–1556; [DOI] [PubMed] [Google Scholar]
- 2b. Gingras M., Chem. Soc. Rev. 2013, 42, 968–1006; [DOI] [PubMed] [Google Scholar]
- 2c. Shen Y., Chen C.-F., Chem. Rev. 2011, 111, 1463–1535. [DOI] [PubMed] [Google Scholar]
- 3. Brandt J. R., Salerno F., Fuchter M. J., Nat. Chem. Rev. 2017, 1, 0045. [Google Scholar]
- 4. Jhulki S., Mishra A. K., Chow T. J., Moorthy J. N., Chem. Eur. J. 2016, 22, 9375–9386. [DOI] [PubMed] [Google Scholar]
- 5. Yang Y., da Costa R. C., Fuchter M. J., Campbell A. J., Nat. Photonics 2013, 7, 634–638. [Google Scholar]
- 6.
- 6a. Naaman R., Paltiel Y., Waldeck D. H., Nat. Chem. Rev. 2019, 3, 250–260; [Google Scholar]
- 6b. Mondal P. C., Fontanesi C., Waldeck D. H., Naaman R., Acc. Chem. Res. 2016, 49, 2560–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Ravat P., Šolomek T., Juríček M., ChemPhotoChem 2019, 3, 180–186; [Google Scholar]
- 7b. Ravat P., Šolomek T., Häussinger D., Blacque O., Juríček M., J. Am. Chem. Soc. 2018, 140, 10839–10847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Isla H., Crassous J., Comptes Rendus Chimie 2016, 19, 39–49. [Google Scholar]
- 8.
- 8a. Fernández-García J. M., Evans P. J., Filippone S., Herranz M. Á., Martín N., Acc. Chem. Res. 2019, 52, 1565–1574; [DOI] [PubMed] [Google Scholar]
- 8b. Cruz C. M., Márquez I. R., Castro-Fernández S., Cuerva J. M., Maçôas E., Campaña A. G., Angew. Chem. Int. Ed. 2019, 58, 8068–8072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8152–8156; [Google Scholar]
- 8c. Reger D., Haines P., Heinemann W., Frank M., Guldi D., Jux N., Angew. Chem. Int. Ed. 2018, 57, 5938–5942; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6044–6049; [Google Scholar]
- 8d. Nakakuki Y., Hirose T., Sotome H., Miyasaka H., Matsuda K., J. Am. Chem. Soc. 2018, 140, 4317–4326; [DOI] [PubMed] [Google Scholar]
- 8e. Li C., Yang Y., Miao Q., Chem. Asian J. 2018, 13, 884–894; [DOI] [PubMed] [Google Scholar]
- 8f. Schaller G. R., Topić F., Rissanen K., Okamoto Y., Shen J., Herges R., Nat. Chem. 2014, 6, 608. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hu Y., Paternò G. M., Wang X.-Y., Wang X.-C., Guizzardi M., Chen Q., Schollmeyer D., Cao X.-Y., Cerullo G., Scotognella F., Müllen K., Narita A., J. Am. Chem. Soc. 2019, 141, 12797–12803; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Yang W., Longhi G., Abbate S., Lucotti A., Tommasini M., Villani C., Catalano V. J., Lykhin A. O., Varganov S. A., Chalifoux W. A., J. Am. Chem. Soc. 2017, 139, 13102–13109; [DOI] [PubMed] [Google Scholar]
- 9c. Ferreira M., Naulet G., Gallardo H., Dechambenoit P., Bock H., Durola F., Angew. Chem. Int. Ed. 2017, 56, 3379–3382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3428–3431; [Google Scholar]
- 9d. Katayama T., Nakatsuka S., Hirai H., Yasuda N., Kumar J., Kawai T., Hatakeyama T., J. Am. Chem. Soc. 2016, 138, 5210–5213; [DOI] [PubMed] [Google Scholar]
- 9e. Fujikawa T., Segawa Y., Itami K., J. Am. Chem. Soc. 2015, 137, 7763–7768. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kawai K., Kato K., Peng L., Segawa Y., Scott L. T., Itami K., Org. Lett. 2018, 20, 1932–1935; [DOI] [PubMed] [Google Scholar]
- 10b. Hahn U., Maisonhaute E., Nierengarten J.-F., Angew. Chem. Int. Ed. 2018, 57, 10635–10639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10795–10799; [Google Scholar]
- 10c. Saito H., Uchida A., Watanabe S., J. Org. Chem. 2017, 82, 5663–5668. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Liu G., Koch T., Li Y., Doltsinis N. L., Wang Z., Angew. Chem. Int. Ed. 2019, 58, 178–183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 184–189; [Google Scholar]
- 11b. Fujikawa T., Segawa Y., Itami K., J. Am. Chem. Soc. 2016, 138, 3587–3595. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Meng D., Liu G., Xiao C., Shi Y., Zhang L., Jiang L., Baldridge K. K., Li Y., Siegel J. S., Wang Z., J. Am. Chem. Soc. 2019, 141, 5402–5408; [DOI] [PubMed] [Google Scholar]
- 12b. Kato K., Segawa Y., Scott L. T., Itami K., Angew. Chem. Int. Ed. 2018, 57, 1337–1341; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1351–1355. [Google Scholar]
- 13.
- 13a. Wang Y., Yin Z., Zhu Y., Gu J., Li Y., Wang J., Angew. Chem. Int. Ed. 2019, 58, 587–591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 597–601; [Google Scholar]
- 13b. Zhu Y., Guo X., Li Y., Wang J., J. Am. Chem. Soc. 2019, 141, 5511–5517; [DOI] [PubMed] [Google Scholar]
- 13c. Navakouski M., Zhylitskaya H., Chmielewski P. J., Lis T., Cybińska J., Stępień M., Angew. Chem. Int. Ed. 2019, 58, 4929–4933; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4983–4987; [Google Scholar]
- 13d. Zuzak R., Castro-Esteban J., Brandimarte P., Engelund M., Cobas A., Piątkowski P., Kolmer M., Pérez D., Guitián E., Szymonski M., Sánchez-Portal D., Godlewski S., Peña D., Chem. Commun. 2018, 54, 10256–10259; [DOI] [PubMed] [Google Scholar]
- 13e. Zhu Y., Xia Z., Cai Z., Yuan Z., Jiang N., Li T., Wang Y., Guo X., Li Z., Ma S., Zhong D., Li Y., Wang J., J. Am. Chem. Soc. 2018, 140, 4222–4226; [DOI] [PubMed] [Google Scholar]
- 13f. Berezhnaia V., Roy M., Vanthuyne N., Villa M., Naubron J.-V., Rodriguez J., Coquerel Y., Gingras M., J. Am. Chem. Soc. 2017, 139, 18508–18511; [DOI] [PubMed] [Google Scholar]
- 13g. Hosokawa T., Takahashi Y., Matsushima T., Watanabe S., Kikkawa S., Azumaya I., Tsurusaki A., Kamikawa K., J. Am. Chem. Soc. 2017, 139, 18512–18521. [DOI] [PubMed] [Google Scholar]
- 14. Naulet G., Sturm L., Robert A., Dechambenoit P., Röhricht F., Herges R., Bock H., Durola F., Chem. Sci. 2018, 9, 8930–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goedicke C., Stegemeyer H., Tetrahedron Lett. 1970, 11, 937–940. [Google Scholar]
- 16. Ravat P., Hinkelmann R., Steinebrunner D., Prescimone A., Bodoky I., Juríček M., Org. Lett. 2017, 19, 3707–3710. [DOI] [PubMed] [Google Scholar]
- 17. Terrasson V., Roy M., Moutard S., Lafontaine M.-P., Pepe G., Felix G., Gingras M., RSC Adv. 2014, 4, 32412–32414. [Google Scholar]
- 18.
- 18a. Link A., Sparr C., Chem. Soc. Rev. 2018, 47, 3804–3815; [DOI] [PubMed] [Google Scholar]
- 18b. Nakano K., Hidehira Y., Takahashi K., Hiyama T., Nozaki K., Angew. Chem. Int. Ed. 2005, 44, 7136–7138; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7298–7300. [Google Scholar]
- 19. Martin R. H., Marchant M.-J., Tetrahedron Lett. 1972, 13, 3707–3708. [Google Scholar]
- 20. Chen C.-F., Shen Y., in Helicene Chemistry: From Synthesis to Applications, Springer, Heidelberg, 2017, pp. 19–40. [Google Scholar]
- 21. Nakai Y., Mori T., Inoue Y., J. Phys. Chem. A 2012, 116, 7372–7385. [DOI] [PubMed] [Google Scholar]
- 22. Griesbeck S., Michail E., Wang C., Ogasawara H., Lorenzen S., Gerstner L., Zang T., Nitsch J., Sato Y., Bertermann R., Taki M., Lambert C., Yamaguchi S., Marder T. B., Chem. Sci. 2019, 10, 5405–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruz C. M., Castro-Fernández S., Maçôas E., Cuerva J. M., Campaña A. G., Angew. Chem. Int. Ed. 2018, 57, 14782–14786; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14998–15002. [Google Scholar]
- 24. Vivas M. G., De Boni L., Bretonniere Y., Andraud C., Mendonca C. R., Opt. Express 2012, 20, 18600–18608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary