

Abstract
Intestine is a major target of vitamin D and several studies indicate an association between vitamin D deficiency and inflammatory bowel diseases (IBD), but also increased colorectal cancer (CRC) risk and mortality. However, the putative effects of 1α,25‐dihydroxyvitamin D3 (calcitriol), the active vitamin D metabolite, on human colonic stem cells are unknown. Here we show by immunohistochemistry and RNAscope in situ hybridization that vitamin D receptor (VDR) is unexpectedly expressed in LGR5 + colon stem cells in human tissue and in normal and tumor organoid cultures generated from patient biopsies. Interestingly, normal and tumor organoids respond differentially to calcitriol with profound and contrasting changes in their transcriptomic profiles. In normal organoids, calcitriol upregulates stemness‐related genes, such as LGR5,SMOC2,LRIG1,MSI1,PTK7, and MEX3A, and inhibits cell proliferation. In contrast, in tumor organoids calcitriol has little effect on stemness‐related genes while it induces a differentiated phenotype, and variably reduces cell proliferation. Concordantly, electron microscopy showed that calcitriol does not affect the blastic undifferentiated cell phenotype in normal organoids but it induces a series of differentiated features in tumor organoids. Our results constitute the first demonstration of a regulatory role of vitamin D on human colon stem cells, indicating a homeostatic effect on colon epithelium with relevant implications in IBD and CRC.
Keywords: colon cancer, colon stem cells, organoids, stemness genes, vitamin D
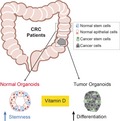
Vitamin D deficiency has been linked to enhanced colorectal cancer (CRC) risk and mortality. Human colonic stem cells, known as crypt stem cells, are essential in gut homeostasis, and their alteration is a feature of CRC. Here, Alberto Muñoz and colleagues generated a living‐biobank of stem‐cell‐derived normal and tumor colon organoids from CRC patients. Vitamin D upregulates stemness‐related genes and the undifferentiated phenotype in normal organoids, while it induces differentiation in tumor organoids. These findings uncover a regulatory role of vitamin D on crypt stem cells, with relevance to CRC development.
Abbreviations
- ChIP
chromatin immunoprecipitation
- CRC
colorectal cancer
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- GSEA
gene set enrichment analysis
- IBD
inflammatory bowel diseases
- IGV
integrative genome visualization
- PBS
phosphate‐buffered saline
- PGE2
prostaglandin E2
- RSPO1
R‐spondin 1
- VDR
vitamin D receptor
Introduction
Calcitriol (1α,25‐dihydroxyvitamin D3) is the active vitamin D metabolite and a main regulator of the physiology and homeostasis of the intestine. It induces Ca2+ and phosphate absorption, and contributes to epithelial integrity/barrier function, detoxification, and protection against infection 1. These actions of calcitriol are the consequence of it binding to and activating vitamin D receptor (VDR), a member of the nuclear receptor superfamily that acts predominantly as a transcription factor modulating the expression of a high number of genes in a tissue‐ and cell‐dependent fashion 2, 3. Epidemiological and preclinical studies suggest an association between vitamin D deficiency and inflammatory bowel diseases (IBD), various extraskeletal disorders, and several neoplasias, particularly colorectal cancer (CRC) 1, 3, 4. Supporting this, experimental results in immortal carcinoma cell lines and animal models of CRC demonstrate a multilevel protective action of calcitriol and other VDR agonists 3, 5.
High level of Vdr RNA has been found in the small and large intestine/colon as compared to other mouse tissues 6. VDR was reported to be preferentially expressed in top differentiated enterocytes, which supports its role in Ca2+ and phosphate absorption and barrier function, and to a lesser extent in cells at the crypt base 7, 8. However, a detailed analysis of VDR protein expression and function in human colon tissue is lacking. Intestinal epithelium is the highest renewal body tissue, which has hampered its long‐term cell culture in vitro. However, the identification of LGR5 as a marker of colon crypt bottom stem cells 9 made possible their isolation and unlimited growth as three‐dimensional culture, forming structures called organoids or mini‐guts 10, 11. Organoid technology is a powerful tool for multiple applications (diagnosis, personalized medicine…) and has now allowed us to study VDR expression and calcitriol action in human colon stem cells using CRC patient‐derived organoids.
Here, we combined RNAscope in situ hybridization and immunohistochemistry to study the expression of LGR5 stem cell marker and VDR protein in human colon mucosa. Interestingly, our results show for the first time that a high proportion of crypt bottom stem cells co‐express LGR5 and VDR. This led us to investigate the role of calcitriol in colon stem cells. To this end, we generated a unique living biobank of CRC patient‐derived organoids from matched healthy (normal) and tumor colon biopsies. Remarkably, matched normal and tumor organoids responded distinctly to calcitriol, which has contrasting effects on cell phenotype and gene expression. Our results strongly support a major regulatory role of vitamin D on human colon normal and cancer stem cells that has potential implications in important medical conditions such as IBD and CRC.
Results
VDR is expressed in human colon crypt stem cells
To analyze the expression of VDR protein in human colon mucosa, we performed immunohistochemistry assays in healthy tissue from CRC patients. In the same sections, to identify the colon stem cells, we carried out RNAscope in situ hybridization for detection of the stem marker LGR5 9 as all available anti‐LGR5 antibodies showed a lack of specificity in test experiments (data not shown). We observed a strong predominantly nuclear VDR signal in both top differentiated enterocytes/cells and a population of cells located at the crypt bottom, while those in the mid‐crypt displayed a lower signal (Figs 1A and S1). Quantification of data at cell level in 100 crypts from four CRC patients (25 crypts/patient) revealed that VDR is expressed in 64% of crypt bottom cells and that 87% of LGR5 + cells were positive for VDR expression (Fig. 1B). We additionally performed double immunostaining of tissue microarrays of healthy colon from 25 CRC patients with antibodies against VDR and PTK7, a stemness‐related cell marker 12. In line with LGR5 data, VDR (nuclear) and PTK7 (extranuclear) proteins were largely co‐expressed in crypt bottom cells (Fig. S1). These findings provide evidence, for the first time, that VDR is expressed in human colon stem cells in vivo.
Figure 1.
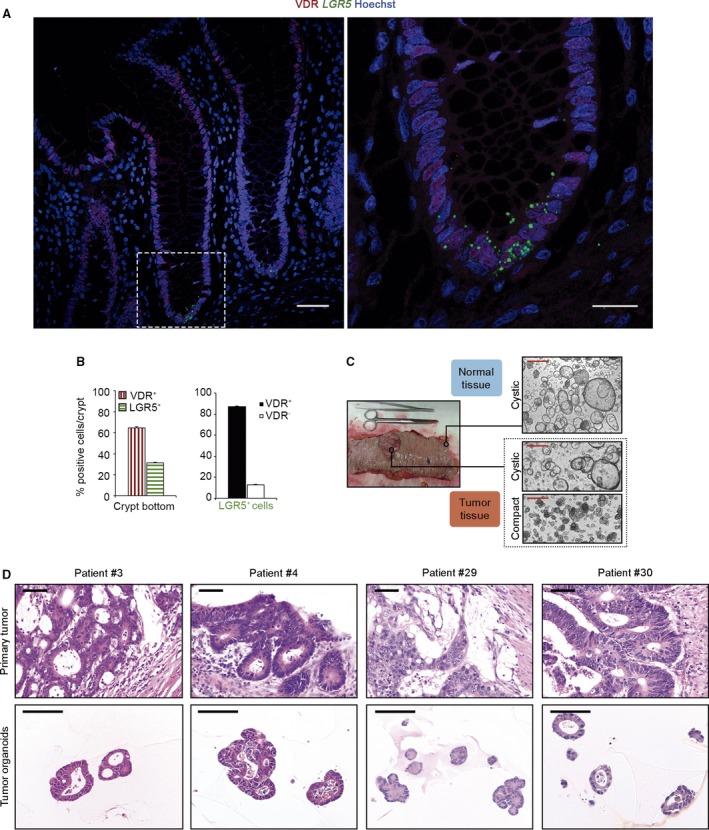
Human colon stem cells express VDR. (A) Images of VDR immunofluorescence (red) and LGR5 RNAscope in situ hybridization (green) in human colon crypts. Scale bars: 50 μm left and 20 μm right. (B) Quantification of VDR and LGR5‐positive cells in 25 crypts/patient (n = 4 patients). Bars represent mean ± standard error of the mean (SEM). (C) Representative phase‐contrast images of normal and tumor colon organoids. Scale bars: 500 μm. (D) Hematoxylin/eosin images showing the primary tumor architecture and tumor organoid phenotype. Scale bars: 100 μm.
Human normal and tumor colon organoids express VDR and respond to calcitriol
To investigate the effect of vitamin D on human colon stem cells, we successfully generated a living biobank of long‐term normal and tumor organoid cultures from 39 CRC patients who had undergone surgery for curative purposes (Table S1). Normal organoids had relatively homogeneous cystic morphology formed by one or two cell layers, whereas tumor organoids were frequently multilayered and showed two alternative morphologies, cystic or compact (Fig. 1C) and resembled the primary tumor morphology (Fig. 1D). The analysis by RT‐qPCR of VDR RNA expression revealed slightly higher levels in normal than in tumor organoids (Fig. 2A). RNA expression of CYP24A1, the most responsive calcitriol‐target gene that encodes the enzyme responsible for its degradation, was higher in tumor than in normal organoids, as occurs in tumor vs. healthy colon tissue 13. Calcitriol treatment increased CYP24A1 RNA to comparable levels in the two types of organoids (Fig. 2B). In addition, calcitriol increased both VDR and CYP24A1 proteins to a variable extent in matched normal and tumor organoids from a subset of patients (Fig. 2C) and CA2, a protein typically expressed in normal colon organoids 14 (Fig. 2C). We also performed RNAscope in situ hybridization for LGR5 detection in organoids and, as in colon tissue, VDR protein and LGR5 RNA were mostly co‐expressed in normal and tumor organoids. VDR signal was increased by calcitriol in both types of organoids whereas that of LGR5 seemed to increase apparently only in normal organoids (Fig. 2D). Altogether, these results showed that both types of organoids, derived respectively from human normal and cancer stem cells, express VDR and respond to calcitriol.
Figure 2.
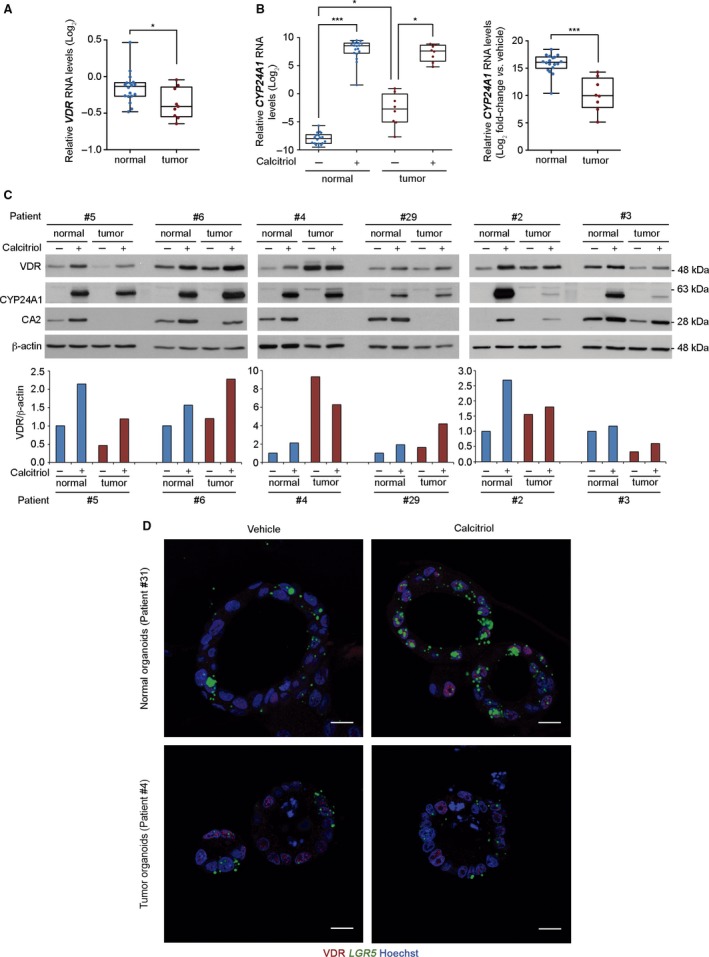
Human colon patient‐derived normal and tumor organoids express VDR and respond to calcitriol. (A) Box‐plot of VDR RNA level in 18 normal and 9 tumor organoid cultures in relation to that of SW480‐ADH colon cancer cells. Box plots represent median ± max/min. Statistical analysis was performed by nonparametric Mann–Whitney test, *P < 0.05. (B) Box‐plot of CYP24A1 RNA level in relation to that of SW480‐ADH colon cancer cells (left) and its fold‐change (right) in the organoid cultures used in (A) treated for 96 h with 100 nm calcitriol or vehicle. Box plots represent median ± max/min. Statistical analysis was performed by nonparametric Kruskal–Wallis test using the Benjamini, Krieger and Yekutieli post test (left) and Mann–Whitney test (right), *P < 0.05, ***P < 0.001. (C) Western blot analysis of VDR, CYP24A1, and CA2 protein levels in whole‐cell extracts from six matched normal and tumor organoid cultures treated for 96 h with 100 nm calcitriol or vehicle. β‐actin was used as a loading control. The graphics below show the VDR/β‐actin ratio. (D) Images of VDR immunofluorescence (red) and LGR5 RNAscope in situ hybridization (green) in human normal and tumor organoids treated for 96 h with 100 nm calcitriol or vehicle. Scale bars: 15 μm.
Calcitriol differentially modulates cell phenotype and proliferation in normal and tumor colon organoids
Based on the induction of differentiation by calcitriol on carcinoma cell lines 15, we next explored the effect of this hormonal agent on the cell phenotype in both types of organoids. Ultrastructural analysis of untreated normal organoids from three patients consistently showed an undifferentiated cell phenotype with decondensed euchromatin and large nucleoli typical of high transcriptional activity, absence of intercellular adhesion structures and microvilli, many free ribosomes, and few Golgi complexes and intermediate filament bundles. Unexpectedly, calcitriol treatment did not change the undifferentiated cell phenotype in normal organoids (Fig. 3A, upper panels). On the contrary, cells of three tumor organoid cultures that showed a similar undifferentiated phenotype responded to calcitriol with changes compatible with differentiation: heterochromatin patches, prominent microvilli and adhesion structures (desmosomes) (Fig. 3A, lower panels), bundles of intermediate filaments, abundant, well‐developed, rough endoplasmic reticulum cisternae, autophagic vacuoles, and Golgi complexes (Fig. 3B).
Figure 3.
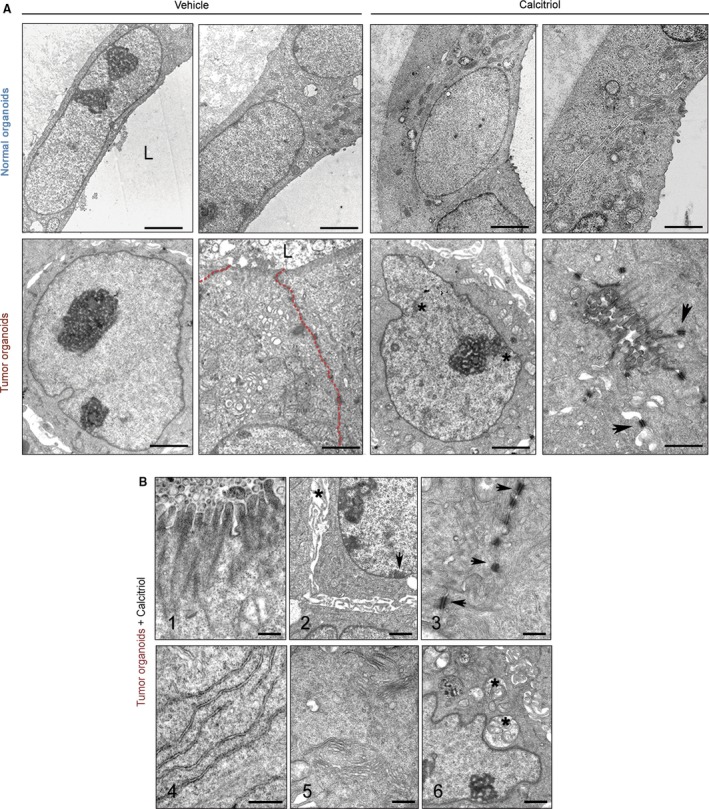
Calcitriol induces cell differentiation in human tumor organoids. (A) Representative ultrastructural images of normal (patient #11) and tumor organoids (patient #38) treated with 100 nm calcitriol or vehicle for 96 h. Upper panel scale bars (from left to right): 2 μm, 2 μm, 2 μm, and 1 μm. Lower panel scale bars: 2 μm, 1 μm, 2 μm, and 1 μm. L, lumen; asterisks, heterochromatin aggregates; arrowheads, desmosomes; red‐dotted line, intercellular region lacking mature adhesion structures. (B) Epithelial differentiation features induced by calcitriol in tumor organoids from patients #4 and #29. (1) Microvilli. (2) Heterochromatin clumps (arrows) and dilated intercellular spaces (asterisks). (3) Desmosomes (arrows). (4) Rough endoplasmic reticulum. (5) Golgi complexes. (6) Autophagic vacuoles (asterisk). Scale bars (from 1 to 6): 0.4 μm, 1 μm, 0.5 μm, 0.5 μm, 0.5 μm and 1 μm.
We then explored whether the antiproliferative effect of calcitriol reported in carcinoma cell lines 2, 3 was reproduced in colon organoids. Calcitriol consistently diminished cell proliferation in all seven patient‐derived normal organoids under study and, to a variable extent, in tumor organoids (Fig. 4A,B). No differences were found in the number of organoids, which suggests that calcitriol does not affect the clonogenicity of either normal or cancer stem cells (Fig. 4C). Taken together, the VDR RNA level in normal and tumor organoids show a statistically significant correlation with the antiproliferative response to calcitriol (Fig. 4D,E), which may also depend on other calcitriol‐related features and/or the tumor‐specific mutational status. Together, these results indicate that calcitriol contributes to maintain the undifferentiated phenotype and restricts the proliferation of normal colon stem cells and their progeny, while it induces differentiation and variably inhibits the proliferation of cancer stem cells.
Figure 4.
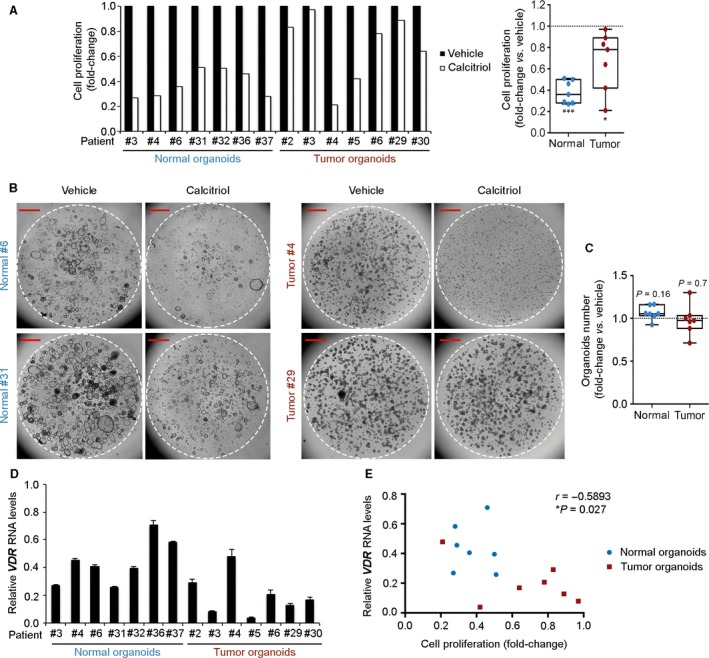
Antiproliferative action of calcitriol in human normal and tumor organoids. (A) Proliferation assay of normal and tumor organoid cultures from seven patients treated with 100 nm calcitriol or vehicle for 10 days. Box‐plot represents median ± max/min. Statistical analysis was performed by nonparametric Wilcoxon signed‐rank test, *P < 0.05, ***P < 0.001. (B) Phase‐contrast images of human normal and tumor organoid cultures from (A). Broken line indicates the border of Matrigel. Scale bars: 1 mm. (C) Graph showing the number of normal and tumor organoids used in (A) after 10 days of calcitriol treatment. Box‐plot represents median ± max/min. Statistical analysis was performed by Wilcoxon signed‐rank test. The exact P‐values are indicated for nonsignificant results. (D) RT‐qPCR analysis of VDR RNA level in normal and tumor organoids from (A) in relation to that of SW480‐ADH colon carcinoma cells. Data are represented as mean ± standard deviation (SD). (E) Scattegram showing the inverse correlation between VDR RNA levels and cell proliferation in organoids used in (A). Statistical significance was determined by Pearson test.
Calcitriol differentially regulates the expression of stemness, differentiation, proliferation and tumorigenesis genes in colon cancer patient‐derived normal and tumor organoids
Next we studied the long‐term effect of calcitriol on the transcriptomic profiles of six matched normal and tumor organoid cultures by RNA‐seq analysis upon 96 h of treatment. Given the differences in function and mutational profiles between colon segments (right‐side, left‐side) 16, 17, to attenuate interindividual variability 18, we selected patients with left‐side colon (descending plus sigma) tumors (Table S1). Mutational analysis of tumor organoids confirmed that they harbored patterns of genetic alterations that are typical of sporadic CRC, with APC (5/6) and KRAS mutations (4/6) (Fig. 5A). Accordingly, nuclear staining of MLH1, PMS2, MSH2, and MSH6 proteins in all cases (data not shown) and lack of POLE mutation ruled out a hypermutated MMR phenotype.
Figure 5.
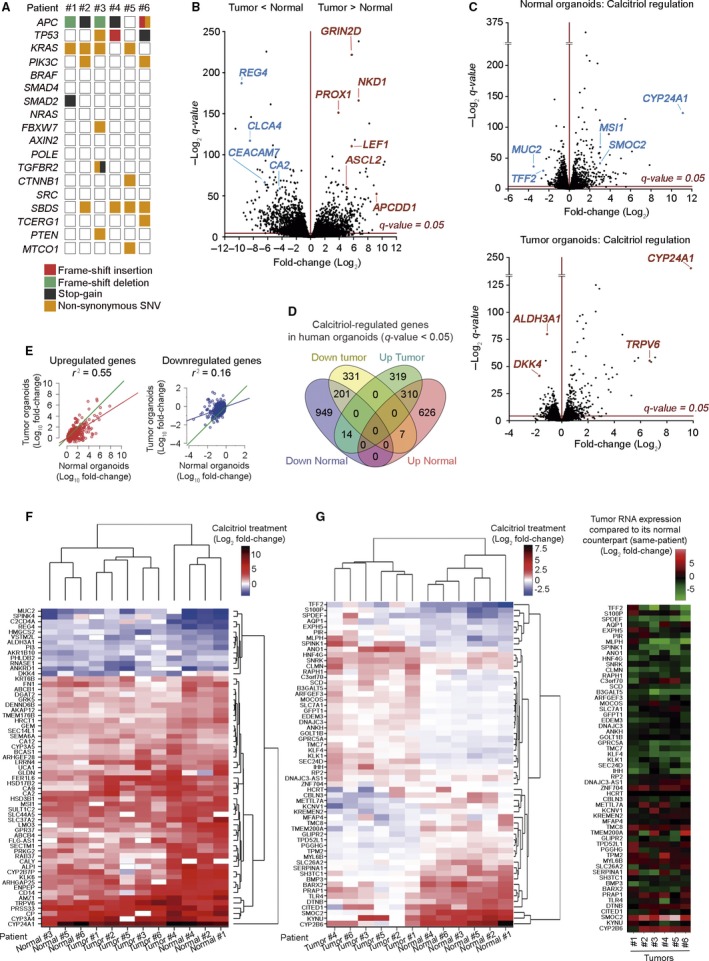
Transcriptomic profile changes by calcitriol in human normal and tumor organoids. (A) Overview of the mutations found in the tumor organoid cultures of six selected patients. (B) Volcano plot comparing human normal and tumor RNA‐seq signatures from the six matched organoid cultures analyzed in (A). The x‐axis shows the fold‐change (Log2) and the y‐axis shows the q‐value (−Log2). Each dot represents a gene. Blue/red dots represent genes that are down‐ or upregulated (respectively) in tumor vs. normal organoids. Dots above the line are significant. (C) Volcano plot comparing normal (upper blot) and tumor (lower blot) organoid signatures from (B) treated with 100 nm calcitriol or vehicle for 96 h. (D) Venn diagram showing the overlap between calcitriol‐significant regulated genes in normal and tumor organoids. The number of genes included in each group is depicted and the complete list of genes is in Table S2. (E) Graphs representing the linear correlation between the effect of calcitriol on gene expression (left graph, induction; right graph, repression), computed as the ratio (Log10 fold‐change) of the RNA‐seq counts, in treated organoids over controls. Only genes significantly (FDR<0.05) regulated by the treatment in normal organoids are represented. Green line, theoretical perfect correlation (r 2 = 1). Statistical analysis was performed by Multiple r‐squared regression test. (F) Heatmap showing significant genes commonly regulated by calcitriol in matched normal and tumor organoids with an average Log2 fold‐change > 1 and expression > 4 cpm. (G) Heatmaps showing the genes with the greatest variance between sample groups of Log2 fold‐change upon calcitriol treatment in matched normal and tumor organoids (left) and tumor RNA expression of those genes compared to its normal counterpart (right).
Transcriptomic profiles of untreated normal and tumor organoids showed a high number of differentially expressed genes, which mostly coincide with those previously reported (Fig. 5B; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64392) 14. Paired analysis of data from normal and tumor organoids of each patient followed by joint analysis of all patients’ data revealed that calcitriol significantly changed the expression of 2,107 genes (943 upregulated, 1,164 downregulated) in normal organoids and 1,182 genes (643 upregulated, 539 downregulated) in tumor organoids, which were partially coincident (532, 19.3%) (Fig. 5C,D and Table S2; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100785). A strong correlation was found between genes induced by calcitriol in normal and tumor organoids, whereas the correlation was much weaker for repressed genes (Fig. 5E). Common calcitriol‐target genes in both types of organoids included known VDR‐regulated genes such as CYP24A1 and TRPV6 (Fig. 5F and Table S2). Notably, some clusters of genes showed different quantitative or even inverse regulation by calcitriol in normal and tumor organoids (Fig. 5G, left). The expression level of these genes in untreated normal and tumor organoids is shown (Fig. 5G, right). Calcitriol targets also included genes involved in a variety of metabolic, detoxification, nuclear receptor‐related, and other pathways (Table S3). Supporting post‐translational regulation by vitamin D, as described in several systems 19, 20, VDR was not identified as a significant regulated gene in the RNA‐seq analysis.
Among the top up/downregulated genes by calcitriol in normal and tumor organoids, there were some involved in cell stemness (MSI1, SMOC2), differentiation (CEACAM7, HMGCS2, and MUC2), proliferation (GRK5, RARRES1), and tumorigenesis (ALDH3A1, S100P) (Table S4). So, next we used an independent cohort of 18 normal and 9 tumor organoids to a) confirm the effect of calcitriol on the expression of several genes that were found to be significantly regulated in the RNA‐seq assay and, b) study the effect of calcitriol on genes involved in those processes or pathways relevant for colon biology. As expected, differentiation genes were expressed at higher levels in untreated normal than in tumor organoids (Fig. S2). Interestingly, the opposite was found for stemness‐related genes, which showed lower expression in untreated normal than in tumor organoids (Fig. S2). In line with the RNA‐seq data, RT‐qPCR analyses confirmed the upregulation by calcitriol of multiple stemness‐related genes including LGR5, MSI1, PTK7, SMOC2, LRIG1, and MEX3A, a marker of slow‐dividing subpopulation of LGR5 + stem cells, in normal organoids (Fig. 6A) 9, 12, 21, 22. In tumor organoids, the effects of calcitriol on stemness‐related genes were weaker as only MSI1 and LRIG1 were upregulated (Fig. 6A).
Figure 6.
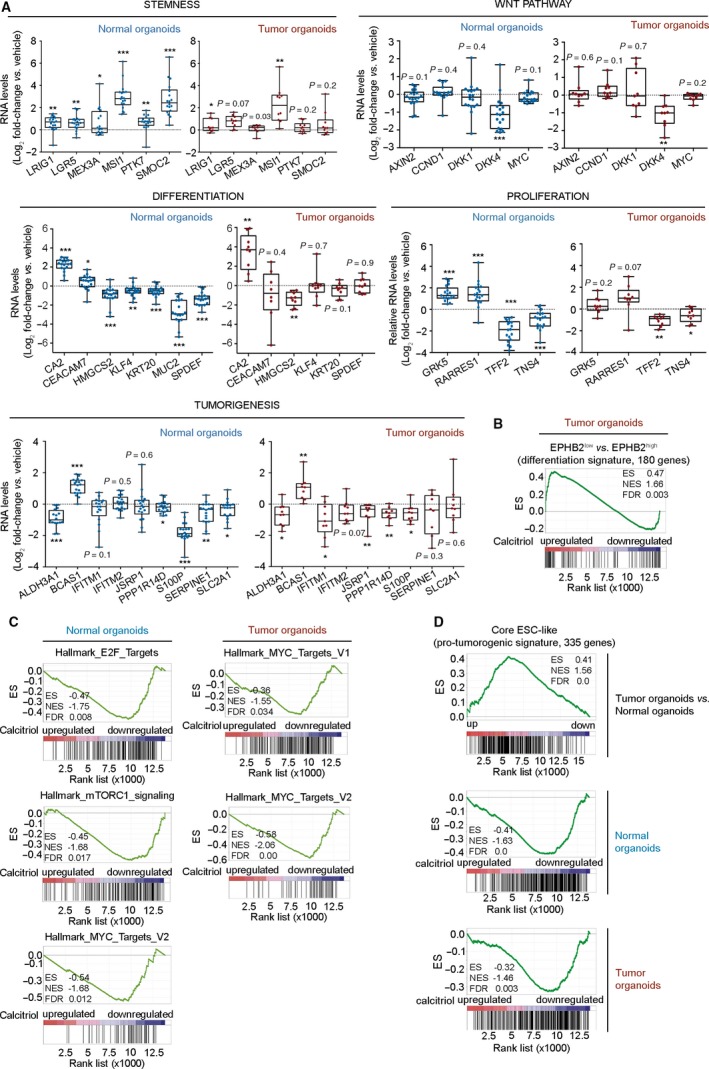
Calcitriol distinctly changes gene expression profile of human normal and tumor organoids. (A) RT‐qPCR analysis of the RNA level of selected genes in an independent series of 18 normal and nine tumor organoid cultures treated with 100 nm calcitriol or vehicle for 96 h. Box plots represent median ± max/min. Statistical analysis was performed by One‐sample t‐test (normal organoids) and nonparametric Wilcoxon signed‐rank test (tumor organoids), *P < 0.05, **P < 0.01, ***P < 0.001. The exact P‐values are indicated for nonsignificant results. (B) GSEA comparing the reported human healthy colon EPHB2‐based differentiation gene signature and the RNA‐seq analysis of human tumor colon organoids treated with calcitriol. (C) GSEA comparing the proliferation‐related molecular signatures databases and the RNA‐seq analyses of human normal and tumor organoids treated with calcitriol. (D) GSEA comparing the pro‐tumorigenic signature core ESC‐like with the RNA‐seq analysis of genes up/downregulated in tumor vs. normal organoids (upper graph), and with that of normal or tumor organoids treated with calcitriol (lower graphs).
Consistent with the ultrastructural analysis, calcitriol induced desmosomal (DSC2), microvilli (PLS1), and adhesion (CDH1) genes in tumor organoids (Table S2). We also confirmed by RT‐qPCR that several genes expressed in differentiated enterocytes (KRT20) or in goblet/secretory cells (KLF4, SPDEF, and MUC2) were downregulated by calcitriol in normal but not in tumor organoids (Fig. 6A). Additionally, Gene Set Enrichment Analysis (GSEA) showed that the differentiation signature EPHB2low vs. EPHB2high of human colon cells 12 was significantly enriched in calcitriol‐treated tumor, but not normal, organoids (Fig. 6B).
We also observed that a series of genes involved in cell proliferation (LRIG1, GRK5, RARRES1, TFF2, and TNS4) and tumorigenesis (ALDH3A1, BCAS1, S100P, PPP1R14D, and SERPINE1) were regulated by calcitriol, preferentially in normal organoids, in a way compatible with the previously described antiproliferative action of calcitriol and a potential antitumoral effect (Fig. 6A). Concordant with its effects on cell proliferation, GSEA revealed an inverse association of the calcitriol profile (RNA‐seq) with E2F, mTOR, and c‐MYC proliferative signatures (Fig. 6C). GSEA also showed that untreated tumor organoids were enriched in the ‘Core transcriptional embryonic stem cell (ESC)‐like’ pro‐tumorigenic signature that, as described by Wong and cols 23 is frequently activated in aggressive human epithelial cancers. Remarkably, calcitriol was able to abolish this association in both types of organoids (Fig. 6D). In contrast to data on carcinoma cell lines cultured in 2D conditions 15, calcitriol did not repress classical genes of the Wnt pathway except DKK4 (Fig. 6A), a gene that increases invasiveness, angiogenesis, and chemoresistance 24, 25 (Table S5).
To confirm the specificity of the RNA‐seq results, we silenced VDR expression in normal organoids through lentiviral transduction of a shRNAmir against VDR, which inhibited the regulation of several calcitriol‐target genes (Fig. S3). Overall, these results show that calcitriol has different/opposite regulatory effects on stemness and differentiation genes in human normal and tumor organoids.
In addition, we generated colon organoids from wild‐type (wt) and, as control, Vdr‐deficient (Vdr −/− ) mice and analyzed the transcriptomic profile induced by calcitriol treatment using RNA microarrays. As expected, no genes were regulated by calcitriol in organoids from Vdr −/− mice, while 143 target genes were identified in wt organoids. Remarkably, only 32 genes overlapped between those regulated by calcitriol in human (RNA‐seq analysis) and mouse (RNA microarrays, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE105117) colon organoids (Fig. S4A), while 111 genes were exclusively regulated in mouse organoids (Table S6). This result agrees with the reported lack of conservation of VDR response elements between primates and rodents 26. We also show that the calcitriol expression profile in wt mouse organoids was related with the Mex3ahigh/Lgr5high mouse signature of slow‐dividing intestinal stem cells 22 (Fig. S4B).
Identification of direct calcitriol‐target genes in human colon organoids
To further elucidate the action of calcitriol in colon homeostasis, we searched for direct transcriptional target genes in normal organoids. To this end, three independent chromatin immunoprecipitation‐sequencing (ChIP‐seq) assays were performed using six additional normal patient‐derived organoids (2 per ChIP‐seq) treated with calcitriol for 2 h. A variable number of DNA binding sites for VDR were found after calcitriol treatment (Fig. 7A). As in other systems 27, a large number of VDR‐binding sites were located in intronic or intergenic regions (Fig. S5A). Identified peaks were assigned to the nearest gene at a maximum distance of 10 kb. As many as 559 genes were coincident between two ChIP‐seq assays and 182 genes were coincident in all three, including CYP24A1 and TRPV6 (Table S7; http://www.ncbi.nlm.nih.gov/protein/GEO107283). Moreover, GSEA revealed that the calcitriol‐direct target gene signature, defined as the 182 genes commonly identified in all ChIP‐seq assays, was strongly associated with calcitriol‐upregulated genes in the RNA‐seq analysis (Fig. 7B). This result agrees with the current model of VDR/nuclear receptor action: direct binding to upregulated genes and predominant indirect action by signaling interference, protein–protein interaction or via microRNAs on downregulated genes 28. Remarkably, 68 genes of the 182‐gene signature overlapped with those regulated (62 up‐ and 6 downregulated) by calcitriol in the RNA‐seq assay (Table S7). The stemness‐related MSI1 and SMOC2 genes, several genes with antiproliferative and/or tumor suppressive function (KCNIP3, PPP1R1C, PRKG2, PRR5L, GRK5, and RARRES1, the latter with a VDR‐binding site 37 kb away from 5′UTR), and CYP24A1 and TRPV6 (Fig. 7C) were among the calcitriol‐direct transcriptional targets (Table S7). ChIP‐seq assays also showed enrichment in the consensus direct repeat (DR)3 VDR motif (Fig. 7D). The comparison of our RNA‐seq data analysis with public transcription factor (TF) ChIP‐seq datasets confirmed VDR as the most significant TF involved in gene upregulation (Fig. S5B).
Figure 7.
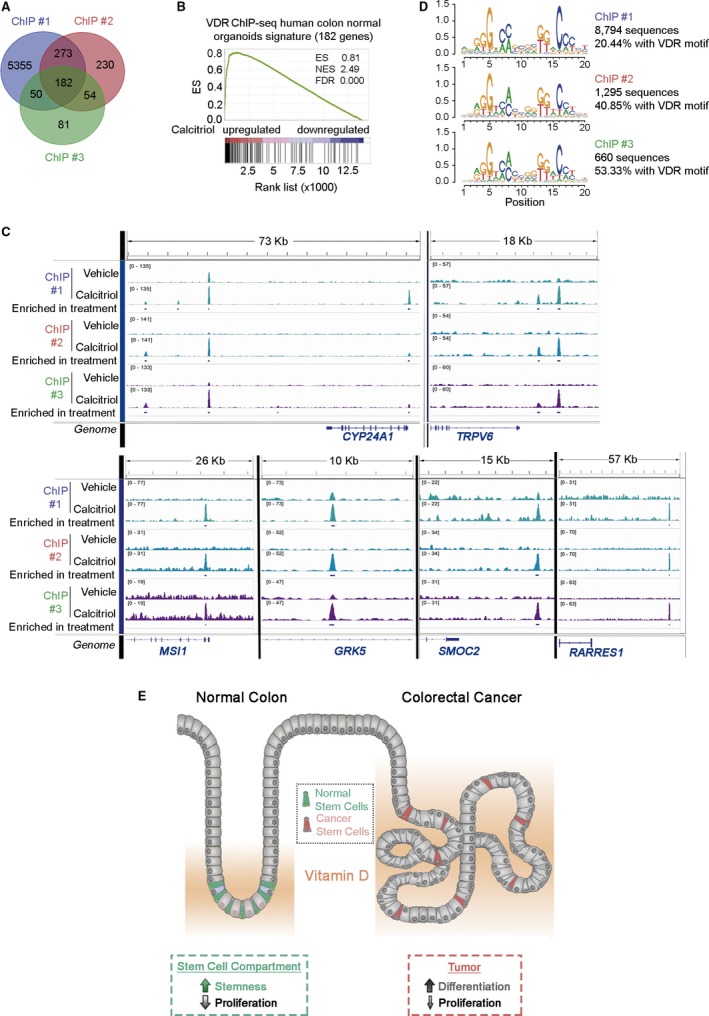
Direct transcriptional calcitriol‐target genes in human colon normal organoids. (A) Venn diagram showing the overlap between genes with VDR binding sites identified by ChIP‐seq in three independent experiments using six human normal organoid cultures treated with 100 nm calcitriol for 2 h. (B) GSEA comparing the genes containing VDR‐binding sites identified in the ChIP‐seq assays and those regulated by calcitriol in normal organoids in the RNA‐seq analysis. (C) Integrative Genome Visualization (IGV) representation of VDR‐binding sites identified in the ChIP‐seq assays in CYP24A1 and TRPV6 (controls), MSI1,GRK5,SMOC2, and RARRES1 genes. (D) Homer de novo motif analysis of VDR‐binding sites in ChIP‐seq studies. (E) Scheme showing the proposed action of vitamin D on normal and cancer colon stem cells.
Discussion
The association of vitamin D deficiency with the risk and severity of clinically relevant gut diseases such as IBD and CRC, together with the observation that calcitriol induces a more differentiated phenotype of enterocytes in vivo and of colon carcinoma cells in 2D culture conditions 1, 3, 15, led us to analyze the effects of calcitriol on human organoids. These 3D structures constitute the state‐of‐the‐art system to study normal and tumoral epithelial stem cells in vitro. We report unanticipated differential effects of calcitriol on both types of organoids that are relevant for understanding the relevance of vitamin D in normal and pathological intestinal biology.
Vitamin D action in human colon has classically been investigated in immortalized carcinoma cell lines, and recently in ex vivo short‐lived colon biopsy samples and primary fibroblasts from patient‐derived normal and tumor tissue 29, 30. We report for the first time that human colon stem cells express VDR in vivo. VDR is a major determinant of tissue responsiveness to vitamin D. Others are the circulating level of the liver‐synthesized calcitriol precursor 25‐hydroxyvitamin D/calcidiol, the activity of CYP27B1 and CYP24A1 enzymes that synthesize and degrade calcitriol, respectively, and the nuclear expression of VDR co‐repressors and co‐activators 2, 3. This finding points to a potential effect of calcitriol on the behavior of normal and cancer stem cells.
To directly address these issues, we established a unique long‐term culture collection of matched patient‐derived normal and tumor organoids and tested the effects of calcitriol. First, we validated that VDR is co‐expressed in vivo with LGR5 and PTK7, markers of colon crypt base columnar stem cells, and furthermore, it mediates in normal organoids the induction of LGR5 by calcitriol. The direct binding of VDR to, and transcriptional regulation of, other stemness‐related genes such as MSI1 and SMOC2 in normal organoids further supports the stemness‐inducing action of calcitriol in colon tissue. Together, these results strengthen the association of the vitamin D‐induced gene expression program with the maintenance of stemness of, perhaps, several colon stem cell populations. Homeostasis of intestinal mucosa, one of the tissues with the highest cell renewal, depends on an adequate balance between proliferation and differentiation of crypt bottom stem cells and their progeny in response to a number of intrinsic and stromal signals such as Wnt, EGF, Notch, Hippo, BMP, and Hedgehog. The new actions of vitamin D described in this study may contribute to the homeostasis of healthy intestinal mucosa, and to the repair and regeneration of damaged intestine upon injury. In line with this, epidemiological studies indicate an association of vitamin D deficiency with IBD, and experiments with animal models suggest a beneficial effect of VDR agonists in these conditions 31.
We also determined that calcitriol declines, but does not abolish, cell proliferation in normal organoids. Although Wnt and EGF pathways are essential for colon stem cell proliferation in vivo and, concordantly, for the generation of organoids in vitro 32, 33, we show that calcitriol reduces cell proliferation in normal organoids, yet classical Wnt pathway genes were unaffected in our transcriptomic analyses. Instead, calcitriol upregulates LRIG1, marker of quiescent + 4 stem cells 21, 31 and also master regulator of epithelial stem cells and tumor suppressor that encodes a multilevel tyrosine kinase receptor (EGFR and others) inhibitor 34, 35. Moreover, we observed in mouse, due the lack of the equivalent human signature, that calcitriol induces a gene expression profile that is related to the Mex3ahigh/Lgr5high signature of slow‐dividing intestinal stem cells 22. Additionally, calcitriol inhibits the activator of EGF pathway TFF2 gene 36 and regulates the expression of the pro‐tumorigenic gene TNS4 and of the negative regulators of colon carcinoma cell proliferation RARRES1 (also known as TIG1) and its target GRK5 37. Conceivably, these effects may contribute to the antiproliferative action of calcitriol in crypt stem cells.
Our results show that the gene regulation effect of calcitriol is much stronger in human than in mouse colon organoids, and that little coincidence exists between target genes in the two species. However, Peregrina and collaborators reported the presence of Vdr and a role of dietary vitamin D in the growth and maturation of mouse intestinal Lgr5 + cells 38.
The finding that calcitriol has pro‐differentiation and variable antiproliferative effects lacking stemness induction in tumor organoids adds to the reported actions on carcinoma cell lines and cancer‐associated fibroblasts, which indicates that calcitriol is a multifaceted protective agent against CRC. In line with the cancer stem cell model stating that tumors originate from the mutational alteration of resident tissue stem cells, human CRC is mainly supported by mutated LGR5 + stem cells 39. The pro‐differentiation effect of calcitriol on tumor organoid cells is in line, albeit less apparent, with that observed in colon carcinoma cell lines grown on plastic dishes 15. Notably, VDR agonists have been proposed to promote terminal differentiation of normal upper crypt absorptive enterocytes 1, which however is not observed in cells of normal colon organoids. This discrepancy may rely on differences in the microenvironment (location and other signals) and the distinct phenotype of these two cell populations (top crypt enterocytes/bottom crypt stem cells) in human tissue and organoid culture, respectively. In this scenario, our results favor a protective action of VDR agonists against CRC. Further supporting this we describe that calcitriol downregulates a gene signature associated to aggressiveness and poor prognosis of breast and lung carcinoma patients 23.
In summary, the newly identified functions in patient‐derived organoids reveal an unanticipated homeostatic action of vitamin D in the human intestinal mucosa. This strongly supports a role of calcitriol in the maintenance of colon crypt cell stemness and in the differentiation of colon cancer stem cells (Fig. 7E).
Materials and methods
Human samples
Fresh human tissues were provided by IdiPAZ and Fundación Jiménez Díaz Biobanks, integrated into the Spanish Biobank Network ( http://www.redbiobancos.es), from individuals diagnosed with colorectal cancer and subjected to surgery between 2013 and 2018. Normal tissue samples were obtained from the area distal to the tumor and the histology of the biopsies was evaluated by the pathology services of La Paz University Hospital and Fundación Jiménez Díaz. All human subjects gave informed consent. The study complied with ethical regulations and was approved by the Ethics Committee of La Paz University Hospital (HULP‐PI‐1425) and the Fundación Jiménez Díaz (PIC‐15/2014).
Establishment of 3D colon and tumor organoid cultures
To establish human normal colon organoids, colon crypts were isolated from human biopsies as previously described 10, 11. Briefly, colon mucosa biopsies were incubated with a mixture of antibiotics [Primocin (Invivogen, San Diego, CA, USA), Gentamycin and Fungizone (Thermo Fisher Scientific, MA, USA)] for 30 min in rotation at room temperature (RT). Next, tissue was cut into small pieces and incubated twice with 10 mm dithiothreitol (DTT) for 5 min at RT. Samples were then transferred to 8 mm EDTA solution for 5 min at RT and 60 min in slow rotation at 4 °C. Samples were washed in PBS until complete EDTA removal and transferred to a 50 mL conical tube in fresh PBS. Colon crypts were separated from the mucosa after shaking and supernatant was centrifuged at 115 g for 5 min at RT. Crypts were washed twice in washing buffer [Advanced DMEM/F12, 10 mm HEPES, and 10 mm Glutamax (Thermo Fisher Scientific)] and finally pelleted crypts were embedded in Matrigel (Corning, MA, USA). Drops were seeded on pre‐warmed, 48‐well culture dishes. After Matrigel solidification at 37 °C complete ‘normal’ culture medium was added [50% Advanced DMEM/F12, 50% Wnt3a‐conditioned medium, 10 mm HEPES, 10 mm Glutamax, 10 mm Nicotinamide (Sigma‐Aldrich, St. Louis, MD, USA), 1x N2 (Thermo Fisher Scientific), 1x B27 (Thermo Fisher Scientific), 1 mm N‐acetyl‐l‐cysteine (Sigma‐Aldrich), 1 : 500 Primocin, 0.1 μg·mL−1 Noggin ( Peprotech, New Jersey, NJ, USA), 1 μg·mL−1 Gastrin (Tocris, Bristol, UK), 1 μg·mL−1 RSPO1 (Sinobiological, Beijing, China), 50 ng·mL−1 EGF (Peprotech), 0.02 μm PGE2 (Sigma‐Aldrich), 1 μm LY‐2157299 (Axon‐Medchem, Groningen, The Netherlands), and 10 μm SB‐202190 (Sigma‐Aldrich)].
To establish mouse normal colon organoids, colon crypts were basically isolated as described for human crypts with minor modifications. Mouse colon was washed in PBS, incubated for 30 min with antibiotics (Primocin) and cut into small pieces. Next, tissue was incubated for 5 min with 8 mm EDTA at RT and 30 min at 4 °C in slow rotation. Then, it was washed in PBS to remove EDTA. Crypts were isolated after shaking and collected in a 50 mL conical tube after passing the solution through a 70‐μm mesh filter. The crypts solution was centrifuged at 260 g for 5 min at 4 °C. The pellet was washed in washing buffer and pelleted crypts were embedded in Matrigel on a prewarmed 48‐well culture dish. After Matrigel solidification, ‘mouse’ culture medium was added (‘normal’ culture medium minus Nicotinamide, Gastrin, PGE2, and SB‐202190).
Human tumor organoid cultures were generated as follows. Human tumor biopsies were washed in PBS several times and incubated in a mixture of antibiotics (Primocin, Gentamycin, and Fungizone) for 30 min in rotation at RT. To obtain single cells, tissue was cut into small pieces and digested enzymatically in a suspension of 1 mg·mL−1 collagenase type IV (Sigma‐Aldrich) (in PBS) for 30 min at 37 °C with continuous shaking in a waterbath. We forced cell disaggregation by passing the suspension through a 18G syringe. Next, we added 5% FBS and single cells were collected in a conical 50 mL tube after passing the solution through a 70‐μm mesh filter and centrifugation at 250 g for 5 min at 4 °C. To lyse erythrocytes, the pellet obtained after centrifugation was incubated in 157 mm NH4Cl for 5 min, washed in PBS and centrifuged again. Cells were then washed in washing buffer and finally pelleted cells were embedded in Matrigel and seeded on prewarmed 48‐well culture dishes. After Matrigel solidification, ‘tumor’ culture medium was added (‘normal’ culture medium minus Wnt3a‐conditioned medium, Nicotinamide and RSPO1).
Growth and expansion of organoid cultures
Normal and tumor culture medium was changed every other day. For passaging, we followed the protocol described 10, 11, with modifications. Briefly, Matrigel‐embedded organoids were collected using a scraper in a 15 mL conical tube and incubated with Cell Recovery Solution (Corning) for 30 min on ice with continuous shaking to remove Matrigel. After centrifugation at 260 g 5 min at 4 °C, organoids were washed in washing buffer and centrifuged again. Next, organoids were incubated with disaggregation buffer [washing buffer solution containing 1 mg·mL−1 dispase (Thermo Fisher Scientific)] for 10 min at RT in orbital rotation. Immediately afterwards, 2 mm EDTA was added and the mixture was incubated for an additional 5 min. Following this, the solution was homogenized by passing it through a 21G syringe, collected in a 15 mL conical tube, centrifuged at 250 g for 5 min at 4 °C and washed twice in washing buffer. Pelleted cells were embedded in Matrigel and seeded on culture dishes.
RNAscope in situ hybridization
In situ hybridization for LGR5 on formalin‐fixed paraffin‐embedded samples (colon mucosa and normal/tumor organoids) was performed using the RNAscope® 2.5 HD Assay‐RED (Advanced Cell Diagnostics; External Service, Dr. Ilse Rooman's laboratory at Vrije Universiteit Brussel) according to manufacturer's instructions and with standard sample pretreatment (15 min Target Retrieval and 30 min Protease Plus). Subsequently, sections were stained for VDR using a rabbit monoclonal antibody (D2K6W #12550, Cell Signaling, Danvers, MA, USA) diluted 1:50 in Antibody Diluent (Agilent, Santa Clara, CA, USA) and applied for 20 min at RT. For detection, a donkey anti‐rabbit Alexa Fluor 488‐labeled F(ab’)2 secondary antibody (Jackson ImmunoResearch Europe, Cambridgeshire, UK) was used, diluted 1:500 in PBS and applied for 30 min at RT. Nuclei were counterstained with Hoechst 33342. Images were taken with Zeiss LSM710 confocal microscope (Zeiss, Oberkochen, Germany). VDR and LGR5 colors were changed using imagej software for a better visualization (VDR in red and LGR5 in green). Images of at least 25 crypts/patient from four patients were taken in a confocal microscope. Quantification of VDR and LGR5 staining was performed manually by two people, considering the percentage of positive cells from a total of 46 cells of the crypt bottom per crypt.
Western blot
Normal (passages 1–5) and tumor (passages 3–8) organoids were pelleted after removing Matrigel with Cell Recovery Solution and whole‐cell extracts were prepared using RIPA buffer (0.05 m Tris/HCl pH 7.5, 0.1% SDS, 0.15 m NaCl, 1% Triton X‐100 (Sigma‐Aldrich) and 1% sodium deoxycholate) containing protease and phosphatase inhibitors (10 μg·mL−1 leupeptin, 10 μg·mL−1 aprotinin, 1 mm PMSF, 1 mm orthovanadate, and 1 mm NaF, all from Sigma‐Aldrich). Cell extracts were separated by SDS/PAGE, transferred to PVDF membranes and incubated with the following primary antibodies: rabbit monoclonal‐VDR (D2K6W) (Cell Signaling, #12550), rabbit polyclonal‐CA2 (CUSABIO, #PA004370HA01HU, TX, USA), rabbit polyclonal‐CYP24A1 (Santa Cruz Biotechnology, #sc‐66851, Dallas, TX, USA) and goat polyclonal‐β‐actin (Santa Cruz Biotechnologies, #sc‐1616). Different exposure times of the films were used to ensure that bands were not saturated. ImageJ was used for the semi‐quantification of VDR protein level.
Electron microscopy
For electron microscopy analysis, samples from three human normal (patients #11, #12, and #37; passages 2–11) and tumor organoid cultures (patients #4, #29, and #38; passages 10–21) treated with 100 nm calcitriol or vehicle for 96 h (n = 3 per group) were fixed with 3% glutaraldehyde (Merck‐Millipore, Burlington, MA, USA) in 0.12 m phosphate buffer, pH 7.4. Samples were then rinsed in 0.12 M phosphate buffer, postfixed in 2% osmium tetroxide (Sigma‐Aldrich), dehydrated in acetone and embedded in araldite (Durcupan, Sigma‐Aldrich, St. Louis, MO, USA). Ultrathin sections stained with uranyl acetate and lead citrate were examined with a JEOL 201 electron microscope.
Proliferation and clonogenicity assays
Normal (passages 2–7) and tumor (passages 8–27) organoids were disaggregated with 1 mg·mL−1 dispase and around 6000–7000 single cells were seeded on 20 μL Matrigel drops in 48‐well culture dishes. Organoids were incubated with normal or tumor complete medium plus Y‐27632 (Tocris) in the presence of 100 nm calcitriol or vehicle for 10 days. Medium and calcitriol/vehicle were replaced every other day. At day 10, normal and tumor organoids were used to study RNA expression and cell viability by estimating the amount of cellular ATP using the CellTiter‐Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) following the manufacturer's instructions. To analyze the clonogenicity, pictures of drops were taken at days 0 and 10 of the cell viability assay and the number of organoids in each drop was counted using mipar software 40. Phase‐contrast images of cultured organoids were captured with a DFC550 digital camera (Leica, Wetzlar, Germany) mounted on an inverted TS100 microscope (Nikon, Tokio, Japan).
Mutational status
Mutations present in six tumor organoid cultures from CRC patients (#1–6; passages 1–3) were analysed. Briefly, Matrigel was removed using ice‐cold Cell Recovery following manufacturer's instructions and DNA was extracted from pelleted organoids by incubation overnight (O/N) at 56 °C with lysis buffer [50 mm Tris/HCl pH 8.0, 100 mm EDTA pH 8, 100 mm NaCl, 1% SDS, and 20 mg·mL−1 proteinase K (Merck‐Millipore, MA, USA)]. Saturated NaCl buffer was added for 5 min and DNA was precipitated with isopropanol and washed twice with 70% ethanol. DNA was genotyped by sequencing the amplified product of a multiplexed‐PCR reaction (Amplicon sequencing) using a proof‐reading polymerase. Indexed libraries were pooled and loaded onto a MiSeq instrument (Illumina). Initial alignment was performed with BWA after primer sequence clipping and variant calling was done with the GATK Unified Genotyper and VarScan2 followed by ANNOVAR annotation. Mutations were called at a minimum 3% allele frequency. SNPs were filtered out with dbSNP and 1000 genome datasets. All detected variants were manually revised and confirmed.
Real‐time quantitative PCR
Matrigel‐embedded normal and tumor organoids (passages 1–6) were washed twice in PBS, lysed with TRIZOL (Thermo Fisher Scientific) and total RNA was purified using the NucleoSpin miRNA extraction kit (Machery‐Nagel, Düren, Germany). For cDNA retrotranscription iScript cDNA Synthesis kit, (Bio‐Rad, Hercules, CA, USA) was used. RT‐qPCR analyses were performed with the Taqman® Universal PCR Master Mix (Applied Biosystems, Waltham, CA, USA) using the following FAM‐labeled TaqMan probes (Applied Biosystems): ALDH3A1 (Hs00964880_m1), AXIN2 (Hs00610344_m1), BCAS1 (Hs00180227_m1), CA2 (Hs01070108_m1), CCND1 (Hs00765553_m1), CEACAM7 (Hs00185152_m1), CYP24A1 (Hs00167999_m1), DKK1 (Hs00183740_m1), DKK4 (Hs00205290_m1), GRK5 (Hs00992173), HMGCS2 (Hs00985427_m1), IFITM1 (Hs01652522_g1), IFITM2 (Hs00829485_sH), JSRP1 (Hs00376079_m1), KLF4 (Hs00358836_m1), KRT20 (Hs00300643_m1), LGR5 (Hs00173664_m1), LRIG1 (Hs00394267_m1), MSI1 (Hs01045894_m1), MUC2 (Hs03005103_g1), MYC (Hs00153408_m1), PPP1R14D (Hs00214613_m1), RARRES1 (Hs00161204_m1), S100P (Hs00195584_m1), SERPINE1 (Hs01126606_m1), SLC2A1 (Hs00892681_m1), SMOC2 (Hs00405777_m1), SPDEF (Hs01026050_m1), TFF2 (Hs00193719_m1), and TNS4 (Hs00262662_m1). RNA expression values were normalized vs. the VIC‐labeled TaqMan probe for housekeeping gene large ribosomal protein (RPLPO) (H99999902_m1) using the comparative CT method. Additionally, we used Power SYBR® Green PCR Master Mix (Applied Biosystems) and the following primers to VDR (forward 5′ AACGCTGTGTGGACATCGGC‐3′; reverse 5′‐GTCATGGCTTTCGTTGGACT‐3′), MEX3A (forward 5′‐CAAGCTCTGCGCTCTCTACA‐3′; reverse 5′‐ATGAACACTGGTTCCTCGCC‐3′) and PTK7 (forward 5′‐TCTGGGAGACCTCAAGCAGT‐3′; reverse 5′‐ATGCACAAAGCGGTTGTTGG‐3′) analysis, and their values were normalized vs. the housekeeping gene Succinate Dehydrogenase Complex subunit A (SDHA) (forward 5′‐TGGGAACAAGAGGGCATCTG‐3′; reverse 5′‐CCACCACTGCATCAAATTCATG‐3′) using the comparative CT method. All RT‐qPCR were performed in a CFX384 Touch Real‐Time PCR Detection System (Bio‐Rad). SW480‐ADH cells were used as relative value to compare VDR and CYP24A1 expression in Figs 2A,B and 4D. To study the mRNA levels of other genes, organoids treated with vehicle were used as control.
RNA‐sequencing
Matched normal and tumor organoids from six patients (#1–6; passages 1–3) were seeded in 48‐well culture dishes and 48 h later were treated with 100 nm calcitriol (Sigma‐Aldrich) or vehicle for 96 h. Medium and calcitriol/vehicle was replaced every other day. Total RNA was extracted and RNA integrity was confirmed as optimal for all samples on an Agilent 2100 Bioanalyzer (RIN range 9.1–9.9). Six hundred nanograms of RNA samples was used for library construction. PolyA+ fraction was purified and randomly fragmented, converted to double stranded cDNA and processed through subsequent enzymatic treatments of end‐repair, dA‐tailing and ligation to adapters as in Illumina's TruSeq Stranded mRNA Sample Preparation kit (Illumina, CA, USA). Adapter‐ligated library was completed by PCR with Illumina PE primers (eight cycles). The resulting purified cDNA library was applied to an Illumina flow cell for cluster generation and sequenced on an Ilumina HiSeq2000 by following manufacturer's protocols. Eight samples were combined per sequencing lane and a minimum of 25 million 50 base single‐reads were generated for each sample.
Sequencing reads were aligned to the transcriptome with TopHat2 41. Novel transcript discovery was not attempted. TopHat was provided with known gene annotations and other transcript data obtained from Gencode (Version 26 – Ensembl 75) basic gene set for the GRCh37/hg19 human genome assembly 42. Gene expression level was calculated from TopHat alignments as the number of reads per gene computed using HTSeq 43 using default settings and gene features as defined in the GRCh37.75 release of the human genome (gtf file). Differential gene expression analysis was performed with the Bioconductor 44 DEseq2 package for the r statistical Software 45. Single enrichment analysis was performed with differentially expressed genes between calcitriol vs. vehicle treatment. The analysis was carried out with the functional annotation tool included in the PathVisio bioinformatic resources 46, 47.
Chromatin immunoprecipitation‐sequencing
We performed three independent ChIP‐seq experiments with human normal organoids (two culture organoids/experiment: ChIP #1, patients #34&35 at passages 12 and 6; ChIP #2, patients #18&19 at passages 3 and 4; and ChIP #3, patients #38&39 at passages 2 and 4) following the ChIP‐seq Millipore protocol with modifications. Briefly, normal organoids from two patients cultured on 6‐well dishes were treated with 100 nm calcitriol or vehicle for 2 h. DNA‐protein crosslinking was done by incubation of Matrigel‐embedded organoids with 1% formaldehyde (methanol free) for 8 min shaking at 4 °C. Crosslinking was quenched with 1.25 mm glycine in PBS for 5 min. We transferred the organoid solution to a 50 mL conical tube for centrifugation at 700 g for 5 min at 4 °C. Supernatant was removed and the pellet was washed twice in cold PBS, transferred to a 1.5 mL tube and lysed in 1 mL ChIP‐lysis buffer [1% SDS, 10 mm EDTA, 50 mm Tris/HCl pH 8.1 and 1x protease cocktail inhibitors (Complete, EDTA‐free, Sigma‐Aldrich)] for 1 h. We used a 21G syringe to homogenize the solution and force the lysis. Samples were sonicated for 20 min at maximum intensity at 4 °C in a S220 Focused‐ultrasonicator (Covaris). We took 40 μL of calcitriol and vehicle‐samples as input control, and diluted 10‐fold the remaining sonicated cell supernatant in ChIP‐dilution buffer (0.01% SDS, 1.1% Triton X‐100, 1.2 mm EDTA, 167 mm NaCl, Tris/HCl pH 8.1 and 1x protease cocktail inhibitor). After 45 min precleaning incubation using magnetic beads (Magna ChIP™ Protein A+G Magnetic Beads, Merck‐Millipore), the supernatant was incubated with 10 μg rabbit monoclonal‐VDR (D2K6W) antibody (Cell Signaling #12550, 2 mg·mL−1, made to order) O/N in orbital rotation at 4 °C. Immunocomplexes were recovered using magnetic beads for 2 h in orbital rotation at 4 °C. Immunoprecipitated material was then washed in the following buffers in the indicated order: ChIP‐dilution buffer, ChIP‐low salt buffer (0.1% SDS, 1% Triton X‐100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris/HCl pH 8.0 and 1x protease cocktail inhibitor), ChIP‐high salt buffer (0.1% SDS, 1% Triton X‐100, 2 mm EDTA, 500 mm NaCl, 20 mm Tris/HCl pH 8 and 1x protease cocktail inhibitor), ChIP‐LiCl buffer (0.25 M LiCl, 1% NP‐40, 1% sodium deoxycholate, 1 mm EDTA, 10 mm Tris/HCl pH 8.0 and 1x protease cocktail inhibitor), ChIP‐TE buffer (two washes) and finally eluted in freshly prepared ChIP‐elution buffer (1% SDS, 0.1 M NaHCO3). The eluted solution and input controls were incubated with 0.2 M NaCl at 65 °C ON in order to de‐crosslink the DNA complexes, and subsequently we added 9.5 mm EDTA, 36.5 mm Tris/HCl pH 6.5 and 70 μg·mL−1 proteinase K for 1 h at 45 °C. DNA extraction was performed using the QIAquick® PCR purification kit (Qiagen, Venlo, The Netherlands).
Fragmented DNA were processed through subsequent enzymatic treatments of end‐repair, dA‐tailing, and ligation to adapters with NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs, Ipswich, MA, USA). Adapter‐ligated libraries were completed by limited‐cycle PCR and extracted with a (single) double‐sided SPRI size selection. Median fragment size is 340 bp from which 120 bp correspond to adaptor sequences. Libraries were applied to an Illumina flow cell for cluster generation and sequenced on an Illumina instrument by following manufacturer's protocols. Sequencing reads were aligned to the GRCh37/hg19 genome assembly with Bowtie2 48. Enriched peak calling was performed with MACS2 49 callpeak function, with –q‐value 0.15, –nomodel and –extsize 200 options using as control concatenated same‐patient alignment files from input treated (calcitriol) and untreated (vehicle). MACS2 bdgdiff function was used to obtain statistically enriched DNA regions using the pileup data obtained from the peak calling. Peaks annotation was performed using custom parameters in order to assign the peaks to the nearest gene in a range of 10 kb upstream/downstream from the gene body. Manual inspection of data and RNA‐seq identified genes of interest was performed using integrative genome visualization (igv) Tool v.2.3.79 50, 51.
Functional enrichment analysis
We used Gene Set Enrichment Analysis (GSEA) 52 to assess the degree of association between our RNA‐seq profile (genes regulated by calcitriol in human colon normal/tumor organoids), or microarray expression (genes regulated by calcitriol in mouse colon normal organoids), with other signatures defined with genes enriched (differentiation gene set: EPHB2low vs. EPHB2high human normal colon epithelial cells 12; proliferation gene sets: E2F, MYC and mTORC1 hallmark gene sets from MSigDB; pro‐tumorigenic gene set: Core ESC‐like from MSigDB; slow‐dividing mouse intestinal stem cells gen set: Mex3ahigh/Lgr5high mouse colon cells 22 and VDR ChIP‐seq in human colon normal organoids, Table S7). The over‐representation of transcription factor binding sites in genes regulated by calcitriol was computed with the Bioconductor TFEA.ChIP package.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad, CA, USA). P‐values < 0.05 were considered significant and * indicates P < 0.05, **P < 0.01, and ***P < 0.001. The exact P‐values are indicated for nonsignificant results. Box plots shown in Figs 2A,B, 4A,C and 6A represent median ± max/min. Graphs showing the quantification of VDR and LGR5 positive cells in human crypts (Fig. 1B) represent mean ± standard error of the mean (SEM). Graphs showing the RNA levels in VDR interference (Fig. S3), and relative VDR RNA levels of normal and tumor organoids (Fig. 4D) represent mean ± standard deviation (SD).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AM, EB, FXR, AF‐B, and AB designed the research studies; AF‐B, AB, and AC‐C conducted most experiments with the help of PB‐M, PJ, GF‐M and MJL; RC, LG‐P, and DGO obtaining the surgical samples; LdP, OD, AC‐C, and AF‐B performed the bioinformatics analyses of the data; ML conducted the electron microscopy analysis, and FR and SPB conducted the immunohistochemical analyses; AM, FXR, and AB wrote the manuscript.
Supporting information
Fig. S1. Human colon crypt bottom cells express PTK7 and VDR.
Fig. S2. Gene expression profile of untreated human normal and tumor orgnaoids.
Fig. S3. VDR silencing in human colon normal organoids.
Fig. S4. Gene expression profile of mouse colon organoids by calcitriol treatment.
Fig. S5. Genomic distribution of VDR‐binding sites and their association to calcitriol regulated genes.
Table S1. Clinicopathological characteristics of CRC patients from whom biopsies were obtained to generate normal and/or tumor organoid cultures.
Table S2. List of genes regulated by calcitriol (q < 0.05) in matched normal and tumor organoids identified by RNA‐seq analyses.
Table S3. GO molecular pathways regulated by calcitriol in human normal and tumor organoids according to significant target genes identified in the RNA‐seq analyses.
Table S4. RNA‐seq data of most regulated genes by calcitriol in human normal and tumor organoids.
Table S5. RNA‐seq data of calcitriol regulation of selected genes in human normal and tumor organoids.
Table S6. Humanized genes regulated by calcitriol only in colon organoids from wild‐type mice (Microarray, q‐value < 0.05).
Table S7. Assigned genes with VDR‐binding sites coincident in three ChIP‐seq assays performed in human normal organoid cultures (MACS2 significantly enriched with calcitriol treatment).
Appendix S1. Supplementary methods.
Acknowledgements
We thank our patients for their generosity. We are grateful to the Instituto de Salud Carlos III (ISCIII), RETICS Biobanks Network with the European Regional Development Fund (FEDER); the Fundación Jiménez Díaz Biobank PT17/0015/006 and La Paz University Hospital‐IdiPAZ Biobank PT13/0010/0003. We also thank Drs. E Gutiérrez, E Álvarez, and L Asensio (La Paz University Hospital) for assisting with patient recruitment and consent, Prof. MB Demay for the donation of Vdr +/− mice, Dr. HG Pálmer for the mutational analysis, Dreamgenics for helping with the bioinformatics analyses, MT Berciano for her help with the electron microscopy studies, MG González‐Bueno for her assistance with animal maintenance and genotyping, Dr. Y Heremans and Dr. I Rooman for RNAscope in situ hybridization and L Banham for manuscript editing. The studies were funded by Networks of Excellence from Spanish Ministry of Science, Innovation and Universities (MICINN) SAF2016‐76377‐R, ‘Nuclear Receptors in Cancer, Metabolism and Inflammation’ (NuRCaMeIn) SAF2017‐90604‐REDT to A.M., ISCIII‐Biomedical Research Networking Centres‐Oncology (CIBERONC) CB16/12/00273 to A.M. and A.B.; CB16/12/00453 to F.X.R.; CB16/12/00342 to E.B. and CB16/12/00241 to F.R. ISCIII‐Biomedical Research Networking Centres‐Respiratory Diseases (CIBERES) CB15/00037 to L.dP, and ISCIII‐FEDER PI15/00934 to F.R.
Alberto Muñoz and Antonio Barbáchano share co‐senior authorship
Contributor Information
Alberto Muñoz, Email: amunoz@iib.uam.es.
Antonio Barbáchano, Email: abarbachano@iib.uam.es.
Data availability
Data from the RNA‐seq analysis of patient‐derived colon normal and tumor organoids, the VDR ChIP‐seq analysis of human colon normal organoids, and microarrays of mice colon normal organoids that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the following accession codes: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100785 (RNA‐seq), http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107283 (ChIP‐seq), and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE105117 (microarray).
References
- 1. Barbachano A, Fernandez‐Barral A, Ferrer‐Mayorga G, Costales‐Carrera A, Larriba MJ & Munoz A (2017) The endocrine vitamin D system in the gut. Mol Cell Endocrinol 453, 79–87. [DOI] [PubMed] [Google Scholar]
- 2. Deeb KK, Trump DL & Johnson CS (2007) Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 7, 684–700. [DOI] [PubMed] [Google Scholar]
- 3. Feldman D, Krishnan AV, Swami S, Giovannucci E & Feldman BJ (2014) The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer 14, 342–357. [DOI] [PubMed] [Google Scholar]
- 4. Mondul AM, Weinstein SJ, Layne TM & Albanes D (2017) Vitamin D and cancer risk and mortality: state of the science, gaps, and challenges. Epidemiol Rev 39, 28–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferrer‐Mayorga G, Larriba MJ, Crespo P & Munoz A (2019) Mechanisms of action of vitamin D in colon cancer. J Steroid Biochem Mol Biol 185, 1–6. [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto H, Miyamoto K, Li B, Taketani Y, Kitano M, Inoue Y, Morita K, Pike JW & Takeda E (1999) The caudal‐related homeodomain protein Cdx‐2 regulates vitamin D receptor gene expression in the small intestine. J Bone Miner Res 14, 240–247. [DOI] [PubMed] [Google Scholar]
- 7. Modica S, Gofflot F, Murzilli S, D'Orazio A, Salvatore L, Pellegrini F, Nicolucci A, Tognoni G, Copetti M, Valanzano R et al (2010) The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. Gastroenterology 138, 636–48, 648 e1–12. [DOI] [PubMed] [Google Scholar]
- 8. Ahearn TU, McCullough ML, Flanders WD, Long Q, Sidelnikov E, Fedirko V, Daniel CR, Rutherford RE, Shaukat A & Bostick RM (2011) A randomized clinical trial of the effects of supplemental calcium and vitamin D3 on markers of their metabolism in normal mucosa of colorectal adenoma patients. Cancer Res 71, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ et al (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 10. Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD et al (2011) Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- 11. Jung P, Sato T, Merlos‐Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho E et al (2011) Isolation and in vitro expansion of human colonic stem cells. Nat Med 17, 1225–1227. [DOI] [PubMed] [Google Scholar]
- 12. Jung P, Sommer C, Barriga FM, Buczacki SJ, Hernando‐Momblona X, Sevillano M, Duran‐Frigola M, Aloy P, Selbach M, Winton DJ et al (2015) Isolation of human colon stem cells using surface expression of PTK7. Stem Cell Reports 5, 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun H, Wang C, Hao M, Sun R, Wang Y, Liu T, Cong X & Liu Y (2016) CYP24A1 is a potential biomarker for the progression and prognosis of human colorectal cancer. Hum Pathol 50, 101–108. [DOI] [PubMed] [Google Scholar]
- 14. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor‐Weiner A, Kester L et al (2015) Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palmer HG, Gonzalez‐Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de Herreros AG, Lafarga M et al (2001) Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E‐cadherin and the inhibition of beta‐catenin signaling. J Cell Biol 154, 369–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bauer KM, Hummon AB & Buechler S (2012) Right‐side and left‐side colon cancer follow different pathways to relapse. Mol Carcinog 51, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower C et al (2012) Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 61, 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cristobal A, van den Toorn HWP, van de Wetering M, Clevers H, Heck AJR & Mohammed S (2017) Personalized proteome profiles of healthy and tumor human colon organoids reveal both individual diversity and basic features of colorectal cancer. Cell Rep 18, 263–274. [DOI] [PubMed] [Google Scholar]
- 19. Peleg S & Nguyen CV (2010) The importance of nuclear import in protection of the vitamin D receptor from polyubiquitination and proteasome‐mediated degradation. J Cell Biochem 110, 926–934. [DOI] [PubMed] [Google Scholar]
- 20. Kongsbak M, von Essen MR, Boding L, Levring TB, Schjerling P, Lauritsen JP, Woetmann A, Odum N, Bonefeld CM & Geisler C (2014) Vitamin D up‐regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4 + T cells. PLoS One 9, e96695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S et al (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J 31, 3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barriga FM, Montagni E, Mana M, Mendez‐Lago M, Hernando‐Momblona X, Sevillano M, Guillaumet‐Adkins A, Rodriguez‐Esteban G, Buczacki SJA, Gut M et al (2017) Mex3a marks a slowly dividing subpopulation of Lgr5+ intestinal stem cells. Cell Stem Cell 20 (801–816), e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E & Chang HY (2008) Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pendas‐Franco N, Garcia JM, Pena C, Valle N, Palmer HG, Heinaniemi M, Carlberg C, Jimenez B, Bonilla F, Munoz A et al (2008) DICKKOPF‐4 is induced by TCF/beta‐catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25‐dihydroxyvitamin D3. Oncogene 27, 4467–4477. [DOI] [PubMed] [Google Scholar]
- 25. Ebert MP, Tanzer M, Balluff B, Burgermeister E, Kretzschmar AK, Hughes DJ, Tetzner R, Lofton‐Day C, Rosenberg R, Reinacher‐Schick AC et al (2012) TFAP2E‐DKK4 and chemoresistance in colorectal cancer. N Engl J Med 366, 44–53. [DOI] [PubMed] [Google Scholar]
- 26. Dimitrov V & White JH (2016) Species‐specific regulation of innate immunity by vitamin D signaling. J Steroid Biochem Mol Biol 164, 246–253. [DOI] [PubMed] [Google Scholar]
- 27. Pike JW & Christakos S (2017) Biology and mechanisms of action of the vitamin D hormone. Endocrinol Metab Clin North Am 46, 815–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weikum ER, Knuesel MT, Ortlund EA & Yamamoto KR (2017) Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 18, 159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mapes B, Chase M, Hong E, Ludvik A, Ceryes K, Huang Y & Kupfer SS (2014) Ex vivo culture of primary human colonic tissue for studying transcriptional responses to 1alpha,25(OH)2 and 25(OH) vitamin D. Physiol Genomics 46, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrer‐Mayorga G, Gomez‐Lopez G, Barbachano A, Fernandez‐Barral A, Pena C, Pisano DG, Cantero R, Rojo F, Munoz A & Larriba MJ (2017) Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 66, 1449–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Limketkai BN, Mullin GE, Limsui D & Parian AM (2017) Role of vitamin D in inflammatory bowel disease. Nutr Clin Pract 32, 337–345. [DOI] [PubMed] [Google Scholar]
- 32. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ & Clevers H (2009) Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 457, 608–611. [DOI] [PubMed] [Google Scholar]
- 33. Fumagalli A, Drost J, Suijkerbuijk SJ, van Boxtel R, de Ligt J, Offerhaus GJ, Begthel H, Beerling E, Tan EH, Sansom OJ et al (2017) Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci USA 114, E2357–E2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ordonez‐Moran P & Huelsken J (2012) Lrig1: a new master regulator of epithelial stem cells. EMBO J 31, 2064–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neirinckx V, Hedman H & Niclou SP (2017) Harnessing LRIG1‐mediated inhibition of receptor tyrosine kinases for cancer therapy. Biochim Biophys Acta 1868, 109–116. [DOI] [PubMed] [Google Scholar]
- 36. Engevik KA, Hanyu H, Matthis AL, Zhang T, Frey MR, Oshima Y, Aihara E & Montrose MH (2019) Trefoil Factor 2 activation of CXCR36 requires calcium mobilization to drive epithelial repair in gastric organoids. J Physiol 597, 2673–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu CC, Tsai FM, Shyu RY, Tsai YM, Wang CH & Jiang SY (2011) G protein‐coupled receptor kinase 5 mediates Tazarotene‐induced gene 1‐induced growth suppression of human colon cancer cells. BMC Cancer 11, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peregrina K, Houston M, Daroqui C, Dhima E, Sellers RS & Augenlicht LH (2015) Vitamin D is a determinant of mouse intestinal Lgr5 stem cell functions. Carcinogenesis 36, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujii M & Sato T (2017) Defining the role of Lgr5(+) stem cells in colorectal cancer: from basic research to clinical applications. Genome Med 9, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sosa JM, Huber DE, Welk B & Fraser HL (2014) Development and application of MIPAR™: a novel software package for two‐ and three‐dimensional microstructural characterization. Int Mat Manuf Innovation 3, 123–140. [Google Scholar]
- 41. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R & Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S et al (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anders S, Pyl PT & Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Love MI, Huber W & Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kutmon M, van Iersel MP, Bohler A, Kelder T, Nunes N, Pico AR & Evelo CT (2015) PathVisio 3: an extendable pathway analysis toolbox. PLoS Comput Biol 11, e1004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Iersel MP, Kelder T, Pico AR, Hanspers K, Coort S, Conklin BR & Evelo C (2008) Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 9, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Langmead B & Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thorvaldsdottir H, Robinson JT & Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G & Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Human colon crypt bottom cells express PTK7 and VDR.
Fig. S2. Gene expression profile of untreated human normal and tumor orgnaoids.
Fig. S3. VDR silencing in human colon normal organoids.
Fig. S4. Gene expression profile of mouse colon organoids by calcitriol treatment.
Fig. S5. Genomic distribution of VDR‐binding sites and their association to calcitriol regulated genes.
Table S1. Clinicopathological characteristics of CRC patients from whom biopsies were obtained to generate normal and/or tumor organoid cultures.
Table S2. List of genes regulated by calcitriol (q < 0.05) in matched normal and tumor organoids identified by RNA‐seq analyses.
Table S3. GO molecular pathways regulated by calcitriol in human normal and tumor organoids according to significant target genes identified in the RNA‐seq analyses.
Table S4. RNA‐seq data of most regulated genes by calcitriol in human normal and tumor organoids.
Table S5. RNA‐seq data of calcitriol regulation of selected genes in human normal and tumor organoids.
Table S6. Humanized genes regulated by calcitriol only in colon organoids from wild‐type mice (Microarray, q‐value < 0.05).
Table S7. Assigned genes with VDR‐binding sites coincident in three ChIP‐seq assays performed in human normal organoid cultures (MACS2 significantly enriched with calcitriol treatment).
Appendix S1. Supplementary methods.
Data Availability Statement
Data from the RNA‐seq analysis of patient‐derived colon normal and tumor organoids, the VDR ChIP‐seq analysis of human colon normal organoids, and microarrays of mice colon normal organoids that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the following accession codes: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100785 (RNA‐seq), http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107283 (ChIP‐seq), and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE105117 (microarray).