

Summary
Phylogenomics is increasingly used to infer deep‐branching relationships while revealing the complexity of evolutionary processes such as incomplete lineage sorting, hybridization/introgression and polyploidization. We investigate the deep‐branching relationships among subfamilies of the Leguminosae (or Fabaceae), the third largest angiosperm family. Despite their ecological and economic importance, a robust phylogenetic framework for legumes based on genome‐scale sequence data is lacking.
We generated alignments of 72 chloroplast genes and 7621 homologous nuclear‐encoded proteins, for 157 and 76 taxa, respectively. We analysed these with maximum likelihood, Bayesian inference, and a multispecies coalescent summary method, and evaluated support for alternative topologies across gene trees.
We resolve the deepest divergences in the legume phylogeny despite lack of phylogenetic signal across all chloroplast genes and the majority of nuclear genes. Strongly supported conflict in the remainder of nuclear genes is suggestive of incomplete lineage sorting.
All six subfamilies originated nearly simultaneously, suggesting that the prevailing view of some subfamilies as ‘basal’ or ‘early‐diverging’ with respect to others should be abandoned, which has important implications for understanding the evolution of legume diversity and traits. Our study highlights the limits of phylogenetic resolution in relation to rapid successive speciation.
Keywords: Fabaceae, gene tree conflict, incomplete lineage sorting, lack of phylogenetic signal, Leguminosae, phylogenomics
Introduction
Phylogenomic studies often focus on difficult‐to‐resolve, deep relationships in the Tree of Life (e.g. in land plants; Wickett et al., 2014), the deep‐branching relationships of animals (Simion et al., 2017), the root of Placentalia (Morgan et al., 2013; Romiguier et al., 2013) and the initial radiation of Neoaves (Suh, 2016). These studies have shown that many of these relationships remain unresolved even when deploying large genome‐scale molecular sequence data, owing to lack of phylogenetic signal and/or conflicting signals between different genomic regions (Rokas et al., 2003; Salichos & Rokas, 2013), such that the inferred relationships are often only implied by a small fraction of genes or characters (Shen et al., 2017). Therefore, fully resolved phylogenies will probably remain elusive, but phylogenomic analysis can provide important insights into the evolutionary processes that shape phylogeny and the underlying causes of lack of phylogenetic resolution. For instance, incomplete lineage sorting (ILS) or deep coalescence is widely recognized as a process causing phylogenetic discordance among gene trees and is routinely invoked to explain conflicting genealogies, even though few studies have provided compelling evidence for it (Suh et al., 2015). Lack of phylogenetic signal and gene tree estimation errors may be equally or more important (Scornavacca & Galtier, 2017; Richards et al., 2018), and, together with gene tree conflict as a result of ILS, can cause polytomies in species trees, especially associated with episodes of rapid divergence. It can be difficult to determine whether such polytomies should be viewed as ‘soft’ in the case of insufficient data, or ‘hard’ in the case of (nearly) simultaneous speciation (Suh, 2016), since the latter is often implied by absence of evidence for resolved relationships, rather than convincing evidence in favour of simultaneous speciation. For deep divergences, in particular, polytomies and reticulate patterns are expected to be difficult to analyse owing to the erosion of phylogenetic signal over time by saturation of substitutions.
The legume family (Leguminosae or Fabaceae) is one of the most prominent angiosperm families across global ecosystems and, with c. 20 000 spp. (Lewis et al., 2005), it ranks third in size after the orchids (Orchidaceae) and daisies (Compositae or Asteraceae). More than three decades since the first molecular phylogenies of the family were inferred (Doyle, 1995; Doyle et al., 1997), sustained phylogenetic research (reviewed in LPWG, 2013a) culminated in the recent reclassification of the Leguminosae into six subfamilies with diverse floral morphologies (Fig. 1a–f; LPWG, 2017). The defining feature of the family is the typical unicarpellate and unilocular superior fruit, which is referred to as the ‘legume’ or ‘pod’ (Fig. 1g). Legumes are the second most cultivated plant family after the Poaceae, and the species serve many purposes for humans, including timber, ornamentals, fodder crops, hallucinogens, medicines and, most notably, a large set of globally important pulse crops (Fig. 1i). A key trait of many legumes is the ability to fix atmospheric nitrogen (N) via symbiosis with ‘rhizobia’‐bacteria in root nodules (Fig. 1h), which leads to enriched soils, high leaf N content and protein‐rich seeds (McKey, 1994). Furthermore, legume species are omnipresent and often abundant in nearly all vegetation types across the planet, ranging from large rainforest trees to small temperate herbs, representing one of the most spectacular examples of evolutionary and ecological radiation of any angiosperm family (Fig. 1j–l).
Figure 1.

Diversity, ecology and economic importance of legumes. (a–f) The family is subdivided into six subfamilies: (a) Cercidoideae (Bauhinia madagascariensis); (b) Detarioideae (Macrolobium angustifolium); (c) Duparquetioideae (Duparquetia orchidacea); (d) Dialioideae (Baudouinia sp.); (e) Caesalpinioideae (Mimosa pectinatipinna); and (f) Papilionoideae (Medicago marina). (g) While the family has a very diverse floral morphology, the fruit (Brodriguesia santosii), which comes in many shapes and is most often referred to as a 'pod' or 'legume', is the defining feature of the family. (h) A large fraction of legume species is known to fix atmospheric nitrogen symbiotically with 'rhizobia', bacteria that are incorporated in root nodules, for example in Lupinus nubigenus. (i) Economically, the family is the second most important of flowering plants after the grasses, with a wide array of uses, including timber, ornamentals, fodder crops, and, notably, pulse crops such as peanuts (Arachis hypogaea), beans (Phaseolus vulgaris), chickpeas (Cicer arietinum) and lentils (Lens culinaris). (j–l) Ecologically, legumes are also extremely diverse and important, occurring and often dominating globally across disparate ecosystems, including: wet tropical forest, for example, Albizia grandibracteata in the East African Albertine Rift (j); savannas, seasonally dry tropical forests, and semi‐arid thorn‐scrub, for example. Mimosa delicatula in Madagascar (k); and temperate woodlands and grasslands, for example, Vicia sylvatica in the European Alps (l). Photographs: (a, b, d, i–l) Erik Koenen; (c) Jan Wieringa; (e–h) Colin Hughes.
Despite this prominence, a well‐resolved phylogenetic framework for the family, based on genome‐scale data, is lacking and the origin and early evolution, including deep‐branching relationships among the six legume subfamilies, are poorly understood, hampering research in comparative legume biology. Sister‐group relationships between subfamilies Papilionoideae and Caesalpinioideae (sensu LPWG, 2017), and of the clade combining these two subfamilies with the newly recognized subfamily Dialioideae, have been recovered previously (Lavin et al., 2005; Bruneau et al., 2008; LPWG, 2017). However, the relationships between the clade comprising those three subfamilies and the other subfamilies Cercidoideae, Detarioideae and Duparquetioideae have not been confidently resolved (cf. Bruneau et al., 2008; LPWG, 2017). Moreover, previous legume phylogenies have been exclusively inferred from a handful of chloroplast markers (Doyle et al., 1997; Wojciechowski et al., 2004; Lavin et al., 2005; Bruneau et al., 2008; Simon et al., 2009; Cardoso et al., 2012, 2013; LPWG, 2017), even though it is preferable to infer species trees based on analysis of unlinked nuclear loci to account for different evolutionary histories of individual genes (Maddison, 1997).
Alongside improving resolution in the legume phylogeny, our main objective is to investigate the causes of the lack of resolution surrounding the initial divergence and deep‐branching relationships of legumes. First, we ask whether lack of phylogenetic signal is causing lack of resolution, or whether previous studies simply did not analyse sufficiently large datasets. Second, considering gene tree discordance, can we choose among alternative topologies to reject a hard polytomy and find support for a fully bifurcating topology? In addition to analysing sequences of nearly all protein‐coding genes from the chloroplast genome, we analyse thousands of nuclear gene alignments harvested from transcriptomes and completely sequenced genomes, with a total aligned length several orders of magnitude longer than those previously used in legume phylogenetics. This means we can dissect and analyse phylogenetic signal and conflict across unlinked loci, and most likely rule out data deficiency as causing lack of resolution. We analyse these new datasets with maximum likelihood (ML) analysis, Bayesian inference (BI) and a multispecies coalescent summary method to infer the most likely relationships among the legume subfamilies. Having inferred the most likely species‐tree topology, we evaluate numbers of supporting and conflicting bipartitions across gene trees for critical nodes, and discuss the implications for understanding the early evolution of legumes.
Materials and Methods
DNA/RNA extraction and sequencing
For the newly generated chloroplast gene data, DNA was extracted from fresh leaves, silica‐dried leaf tissue or herbarium specimens, using the DNeasy Plant Mini Kit (Qiagen). Sequencing libraries were prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina (San Diego, CA, USA), and sequenced on the Illumina HiSeq 2000 sequencing platform, at low coverage (‘genome‐skimming’), or as part of hybrid capture experiments for a separate study on mimosoid legumes (E. J. M. Koenen et al., unpublished). For the newly generated nuclear gene data, we used transcriptome sequencing, using RNA extracted from fresh leaves using the RNeasy Plant Mini Kit (Qiagen). RNA sequencing libraries were prepared using the TruSeq RNA Library Prep Kit (Illumina) and sequenced on the Illumina HiSeq 2000 platform. All laboratory procedures were performed according to the specifications and protocols provided by kit manufacturers.
Sequence assembly
Raw reads for the chloroplast DNA data were cleaned and filtered as follows: Illumina adapter sequence artifacts were trimmed using trimmomatic v.0.32 (Bolger et al., 2014); overlapping read pairs were merged with pear v.0.9.8 (Zhang et al., 2014); low‐quality reads were discarded and low‐quality bases at read ends were trimmed with trimmomatic v.0.32 (using settings MAXINFO:40:0.1 LEADING:20 TRAILING:20). Quality‐filtered reads were assembled into contigs using spades v.3.6.2 (Bankevich et al., 2012). For RNA data, we used the Fastx‐toolkit v.0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) to remove low‐quality reads (< 80% of bases with a quality score of 20 or higher), tagdust v.1.12 (Lassmann et al., 2009) to remove adapter sequences, and Prinseq‐lite v.0.20.4 (Schmieder & Edwards, 2011) to trim low‐quality bases off the ends of reads. Transcriptome assembly was performed on the quality‐filtered reads using trinity (Grabherr et al., 2011; release 2012‐06‐08), with default settings.
Chloroplast proteome alignment
DNA sequences of protein‐coding chloroplast genes were newly generated for 49 accessions, or extracted from data sources (Supporting Information Table S1). Sequence data were extracted directly from annotated plastomes in GenBank or by Blast searches from de novo assembled contigs and transcriptomes against Medicago truncatula plastid coding reference sequences using custom Python scripts. Sequences for some outgroup taxa (data from Moore et al., 2010) were downloaded separately per gene from GenBank. For each gene, a codon alignment was inferred using macse v.1.01b (Ranwez et al., 2011) with default settings. Phylogenetic trees were inferred with raxml v.8.2 (Stamatakis, 2014) for each gene separately to screen for erroneously aligned sequences. For some species, individual gene sequences that led to anomalously long terminal branches (> 10 times longer than it's sister group) were removed, as these are likely to be poorly aligned and may produce spurious results. The genes accD and clpP were removed, because they have been lost, pseudogenized or relocated to the nuclear genome in several legume lineages (Magee et al., 2010; Williams et al., 2015; Dugas et al., 2015), leading to poor‐quality alignments. Gene alignments were concatenated, the full alignment visually checked, and obvious misalignments resolved. Furthermore, sequence errors (single A/T indels) that caused frameshift mutations were corrected and the accuracy of the alignment at codon level was assessed and corrected if necessary. For the genes ndhF, ndhI, rpl20 and rps18, where the ends of coding sequences had varying lengths, all sites between the first and last stop codons in the alignment were excluded, as they were poorly aligned. Finally, using bmge v.1.12 (Criscuolo & Gribaldo, 2010) the codon alignment was translated to amino acid sequences.
Nuclear gene data and matrix assembly
Whole‐genome and transcriptome data were downloaded from various sources and augmented with newly generated transcriptome sequence data for six Caesalpinioideae and Detarioideae taxa (Table S2). Peptide sequences were downloaded from annotated genomes, or extracted from transcriptome assemblies using transdecoder (http://transdecoder.github.io/). To assemble the nuclear peptide sequence data into aligned gene matrices, we performed mcl clustering using the pipeline of Yang & Smith (2014), with a hit fraction cut‐off of 0.75, inflation value of 2 and a minimum log‐transformed e‐value of 30. These settings yield clusters with good overlap between sequences and good alignability (omitting genes that are too variable), with loss of only a few short gene clusters. The resulting homologue gene clusters were subjected to two rounds of alignment with mafft v.7.187 (Katoh & Standley, 2013), gene tree inference with raxml v.8.2 (Stamatakis, 2014), and pruning and masking of tips and cutting deep paralogues as described in Yang & Smith (2014). In the first round we used 0.3 and 1.0 as relative and absolute cut‐offs for trimming tips, and 0.5 as the minimum cut‐off for cutting deep paralogues, and keeping all clusters with a minimum of 25 taxa for the second round. In the second round we used more stringent cut‐off values (0.2 and 0.5 for trimming tips and 0.4 for cutting deep paralogs) (see Yang & Smith (2014) for more information on these parameter settings). One‐to‐one orthologues (i.e. homologue gene clusters in which each taxon is represented by a single gene copy) and rooted ingroup (RT) homologues were extracted from the homologue cluster trees, with a minimum aligned length of 100 amino acids per homologue. RT homologues are extracted by rooting homologue cluster trees on the outgroup (here Aquilegia coerulea and Papaver somniferum), detecting gene duplications and pruning the paralogue copies with fewer taxa present until each taxon is represented by a single copy. The outgroup is also pruned, and clusters in which each taxon is only present once are also included, meaning that all one‐to‐one orthologues are also in the RT homologue set (see Yang & Smith, 2014 for a more details). Sequences with > 50% gaps and all sites with > 5% missing data were removed from the homologue alignments using bmge. For the one‐to‐one orthologues, alignments with < 50 taxa were discarded. For the larger set of RT homologues, alignments with < 25 taxa were discarded. These cut‐offs may appear overly strict, but are probably important to reduce negative influences of missing data, particularly to avoid fragmentary sequences acting as ‘rogue’ taxa in gene tree estimation. Furthermore, the large dataset size means that even after discarding alignments with < 50 or 25 taxa present, a very large number of gene trees and long concatenated alignments still remain for analysis (see Results).
Phylogenetic inferences
Gene tree inferences were made with ML analysis in raxml v.8.2 (Stamatakis, 2014). Species tree analyses were performed with ML in RAxML, using BI in phylobayes‐MPI 1.7 (Lartillot et al., 2013) and the multispecies coalescent summary method in astral v.5.6.3 (Mirarab et al., 2014a).
Gene trees of one‐to‐one orthologues and RT homologues were estimated with RAxML using the WAG + G model, with 100 rapid bootstrap replicates. We calculated 80% majority‐rule consensus trees for each orthologue or homologue and used these to calculate internode certainty all (ICA) values using RAxML, in order to include only nodes that received ≥ 80% bootstrap support (BS) in the individual gene trees. We also used the concordance analysis in phyparts (Smith et al., 2015), with a BS cut‐off of 50% and used the output to extract numbers of supporting and conflicting bipartitions for plotting pie charts on the species tree.
We used partitionfinder2 (Lanfear et al., 2017) to estimate partitions for the ML analysis on nucleotide sequences of the chloroplast alignment, with a minimum length per partition set to 500 nucleotides, and allowing different codon positions per gene in different partitions. The resulting 16 partitions were run with the GTR + GAMMA model, with 1000 rapid bootstrap replicates. For the amino acid sequences, the ML analyses of both the chloroplast and concatenated nuclear one‐to‐one orthologue alignments were run with the LG4X model, without partitioning, as this model accounts for substitution rate heterogeneity across the alignment by estimating four different LG substitution matrices (Le et al., 2012). For the chloroplast alignment, 1000 rapid bootstrap replicates were carried out.
Bayesian species tree analyses were performed in phylobayes‐MPI with the CATGTR model, with invariant sites deleted (as recommended in the manual of v.1.5 for better Markov chain Monte Carlo (MCMC) mixing) and default settings for other options. Analyses on the chloroplast alignment were run until the chain reached convergence (usually after 10 000–20 000 cycles), and the first 10% of the chain was discarded as burn‐in. To perform BI analyses on the complete one‐to‐one orthologue set in a computationally tractable manner, we ran 25 gene jackknifing replicates without replacement, dividing the total number of genes over five subsets with five replicates. These subsampled replicates were run in phylobayes‐MPI, with a starting tree derived from an analysis sampling the 100 genes with the longest gene tree length, using the CATGTR model with constant sites deleted, for 1000 cycles each. We found that all 25 chains had converged after a few hundred cycles, and discarded the first 500 cycles of each as burn‐in. A majority‐rule consensus tree was constructed using sumtrees.py (from the Dendropy library (Sukumaran & Holder, 2010)) from 12 500 total posterior trees, representing the MCMC cycles 501–1000 of each replicate. BI analyses were also not partitioned, as the CATGTR model describes heterogeneity across alignments more accurately than partitioning by gene and/or codon as the substitution process also varies across gene sequences and codon positions. The LG4X and CATGTR models have been shown to provide a better fit to empirical amino acid sequence data (Lartillot & Philippe, 2004; Le et al., 2012) and amino acid sequences are more suitable for resolving deep divergences because they are less saturated with substitutions (silent substitutions are absent), and thus less prone to long branch attraction. We have not analysed nucleotide sequences for the large nuclear gene dataset to avoid costly computation time to generate analyses that would be inferior to those presented here.
For the multispecies coalescent analysis with astral, we used ML topologies (not bootstrapped gene trees; see Mirarab et al. (2014b)) of the one‐to‐one orthologue gene trees estimated with RAxML, using local posterior probability and quartet support to evaluate the inferred topology (Sayyari & Mirarab, 2016). We also used the polytomy test in astral (Sayyari & Mirarab, 2018) to evaluate whether a hard polytomy can be rejected for the relationships among subfamilies, where P < 0.05 is considered to reject the null hypothesis of a polytomy.
We used splitstree4 to draw a filtered supernetwork (Whitfield et al., 2008) of the 1103 one‐to‐one orthologues, using the 80% majority‐rule consensus trees to only include well‐supported bipartitions to infer the network. Gene trees were pruned to include only the N‐fixing clade of angiosperms. Furthermore, for the relatively densely sampled Papilionoideae and Caesalpinioideae, we discarded several taxa that were less well represented across gene trees. The mintrees parameter was set to 552 (at least 50% of the number of orthologues) and the maximum distortion parameter to 0.
Counting supporting bipartitions for key nodes across gene trees
Using a custom Python script (Notes S1), numbers of matching and alternative bipartitions across the RT gene trees were counted for the nodes labelled A–H in Figs 3 and 4(b) (see later), to assess monophyly of each of the subfamilies and combinations (clades) of subfamilies, against the outgroup, across all gene trees. For each gene tree, we first assessed whether all six groups (five subfamilies plus the outgroup) are present and gene trees with missing groups were not taken into account. Next, we evaluated whether the gene tree includes a matching bipartition for the family, each subfamily and all possible combinations of subfamilies. A matching bipartition means that all taxa of a subfamily or combination of subfamilies are separated from all other taxa in the gene tree, including the outgroup, thus constituting support for that clade to be monophyletic. For combinations of subfamilies, the subfamilies themselves do not necessarily need to be monophyletic, but all taxa within the combined subfamilies should be separated from all other taxa to constitute a matching bipartition, and thus constitute a supported clade in the gene tree. For a clade to be well supported, we expect matching bipartitions for a majority of gene trees. For clades to be poorly supported, we expect gene trees either to be uninformative as a result of low phylogenetic signal or to contain significant conflicting bipartitions, and hence relatively low numbers of matching bipartitions. All counts were done for ML gene trees of RT homologues, and with 50 and 80% bootstrap cut‐offs.
Figure 3.
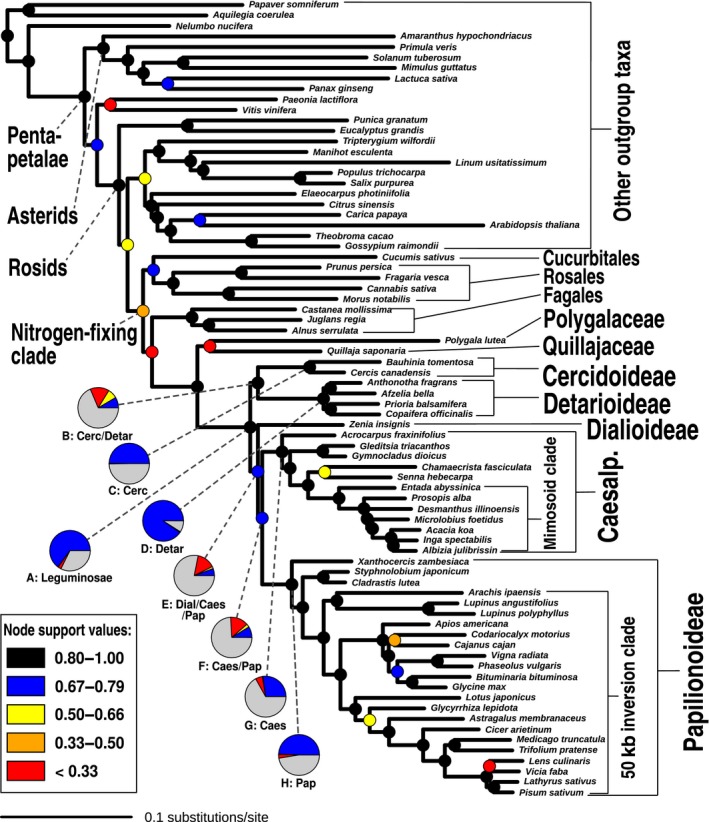
Maximum likelihood phylogeny of legumes estimated with raxml under the LG4X model from a concatenated alignment of 1103 nuclear orthologues. Internode certainty all (ICA) values are indicated with coloured symbols on nodes for simplicity of presentation (see Supporting Information Fig. S5 for actual support values for all nodes). For the first four divergences in the legume family, as well as the subfamily nodes for the four subfamilies represented by more than one accession (nodes labelled A–H), pie charts indicate the proportions of gene trees supporting the relationship shown (blue), supporting the most prevalent conflicting bipartition (yellow), supporting other conflicting bipartitions (red) and uninformative genes (i.e. no bootstrap support (BS) and/or missing relevant taxa; grey). Numbers of bipartitions for the pie charts are derived from phyparts analyses with a 50% BS filter. Labelled nodes A–H are analysed in more detail in Fig. 6. Abbreviations for subfamilies: Cerc, Cercidoideae; Detar, Detarioideae; Dial, Dialioideae; Caes, Caesalpinioideae; Pap, Papilionoideae.
Figure 4.
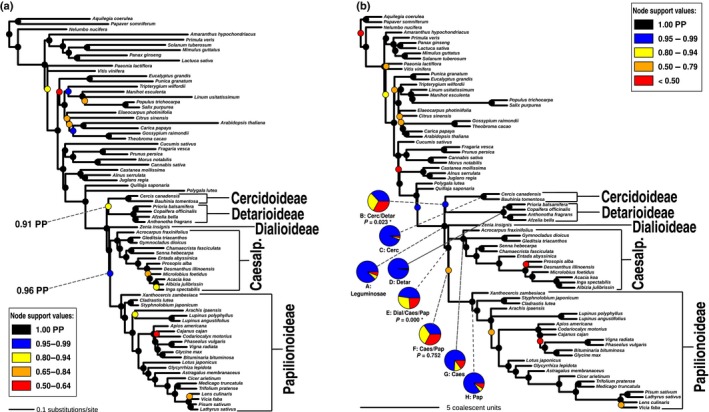
Bayesian and multispecies coalescent analyses yield congruent relationships, identical to those in Fig. 3 obtained with maximum likelihood (ML) analysis of nuclear data. (a) Bayesian gene jackknifing majority‐rule consensus tree of concatenated alignments of c. 220 genes per replicate. Support indicated by coloured circles on nodes represents posterior probability (PP) averaged over 25 replicates for 500 posterior trees each (in total, 12 500 posterior trees). (b) Phylogeny estimated under the multispecies coalescent with astral from ML gene trees. Support indicated by coloured symbols on nodes represents local posterior probability. Pie charts show relative quartet support for the first (blue) and the two (yellow and red) alternative quartets. P‐values for the polytomy test are given for nodes B, E and F below the respective pie charts for those nodes. Significance (P ≤ 0.05) is indicated with an asterisk (see Supporting Information Figs S6, S7 for phylogenetic trees with all PP and quartet support values indicated). Abbreviations for subfamlies: Cerc, Cercidoideae; Detar, Detarioideae; Dial, Dialioideae; Caes, Caesalpinioideae; Pap, Papilionoideae.
Results
The chloroplast alignment includes 72 protein‐coding genes, for 157 taxa (including 111 legume species; Table S1), with a total aligned length of 75 282 bp or 25 094 amino acid residues. From transcriptomes and fully sequenced genomes, we gathered 9282 homologous nuclear‐encoded gene clusters for 76 taxa, including 42 legume species (Table S2). From these clusters, we extracted protein alignments of 1103 one‐to‐one orthologues for species tree inference with a total aligned length of 325 134 amino acids when concatenated, and 7621 RT homologues for additional gene tree inference. The alignments, gene trees and species trees are available in TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S25315) and Datasets [Link], [Link], [Link], [Link], [Link], [Link].
We used different accessions for the chloroplast and nuclear datasets for some species that are present in both (nine altogether; see Table S1), meaning they are probably derived from different individuals of the same species. This is unlikely to cause conflicting results as we focus on deep‐branching relationships, and moreover their phylogenetic positions are fully congruent in the chloroplast and nuclear species trees.
Inferring the species tree
Our analyses reveal that both the chloroplast and nuclear datasets resolve all subfamilies as monophyletic with (nearly) full support (Figs S1–S7). The relationships among the subfamilies are less robustly resolved (Figs 2, 3, 4, S1–S7), with, in particular, the position of the root of the family and the Caesalpinioideae‐Papilionoideae sister relationship receiving low support in some analyses, and indeed a polytomy could not be rejected for the latter. The clade consisting of Papilionoideae, Caesalpinioideae and Dialioideae is recovered in all analyses, with Duparquetia as the sister group to this clade as inferred from chloroplast data. Duparquetia is not sampled for nuclear data. Transcriptome or genome sequencing is necessary for this taxon to confirm the relationship found by chloroplast data. The position of the root of the legume family (i.e. the relationships between Cercidoideae, Detarioideae and the clade comprising the remaining subfamilies) is more difficult to resolve, presumably because of the long stem branch, and here the chloroplast and nuclear datasets estimate conflicting topologies. The chloroplast alignment weakly supports Cercidoideae as sister to the rest of the family (Figs 2c, S1–S4), except in the BI analysis of nucleotide sequences. This suggests that the sister‐group relationship of Cercidoideae with the rest of the family is the most likely rooting as inferred from chloroplast data, but given the low BS values and lack of resolution in the BI analysis of nucleotide sequences, phylogenetic signal in the chloroplast data with regard to the root node appears to be limited.
Figure 2.
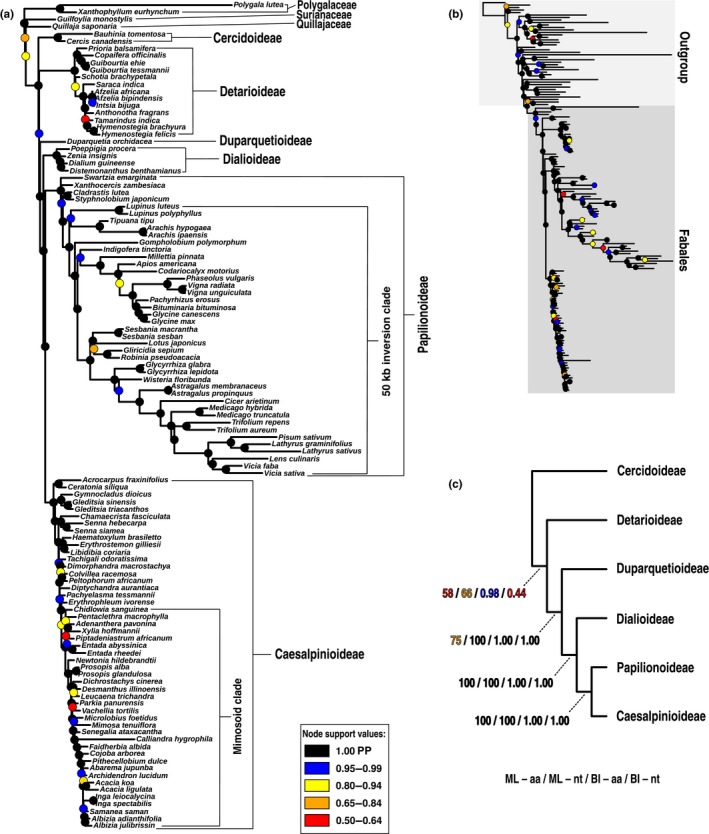
Phylogeny of legumes based on Bayesian analyses of 72 protein‐coding chloroplast genes under the CATGTR model in phylobayes. (a) Majority‐rule consensus tree of the amino acid alignment, showing only the Fabales portion of the tree, outgroup taxa pruned; (b) complete tree including outgroup taxa; (c) simplified tree showing support for subfamily relationships with different inference methods (ML, maximum likelihood; BI, Bayesian inference) and sequence types (aa, amino acids; nt, nucleotides). Majority‐rule consensus trees for both the amino acid and nucleotide alignments with tip labels for all taxa and support values indicated are included in Supporting Information Figs S1 and S2. In (a) and (b), coloured circles indicate node support in posterior probabilities (PP).
In contrast to the chloroplast phylogeny, in all analyses of the 1103 nuclear one‐to‐one orthologues, we recover a sister‐group relationship between Cercidoideae and Detarioideae, with this clade sister to the clade comprising Dialioideae, Caesalpinioideae and Papilionoideae (note that Duparquetioideae was not sampled) (Figs 3, 4, S5–S7). We inferred a ML tree of the concatenated alignment with the LG4X model, and calculated ICA values from bootstrapped gene trees on this topology (Figs 3c, S5), for which only gene tree bipartitions that received at least 80% BS were considered. The internode certainty metric was introduced to assess phylogenetic conflict among loci and identify internodes with high certainty, particularly in phylogenomic studies where bootstrap values are often inflated (Salichos & Rokas, 2013). The sister‐group relationship between Cercidoideae and Detarioideae is well supported, receiving an ICA value of 0.85. A Bayesian jackknifing analysis with the CATGTR model infers a nearly identical topology to the ML topology (Figs 4a, S6), with posterior probability of 0.91 in support of this same relationship. The multispecies coalescent species tree inferred with astral (Mirarab et al., 2014a), which accounts for ILS, is also consistent with that relationship (Figs 4b,c, S7), with the Cercidoideae/Detarioideae clade supported by a local posterior probability of 0.95 (Sayyari & Mirarab, 2016), and a polytomy is rejected for this node (Fig. 4b). In summary, all analyses of nuclear protein alignments lend strong support for a sister‐group relationship between Cercidoideae and Detarioideae.
The other contentious relationship, the sister‐group relationship between Caesalpinioideae and Papilionoideae, is fully supported in the chloroplast analyses (Fig. 2c), well supported in the Bayesian jackknife analysis of nuclear orthologues (Fig. 4a) and also more prevalent than the second most prevalent bipartition among gene trees (Fig. 6d; see later). However, the ICA value for this node is low (0.70) relative to the other nodes along the backbone (Figs 3, S5), and astral did not reject a polytomy for the relationships among these two subfamilies and Dialioideae.
Figure 6.
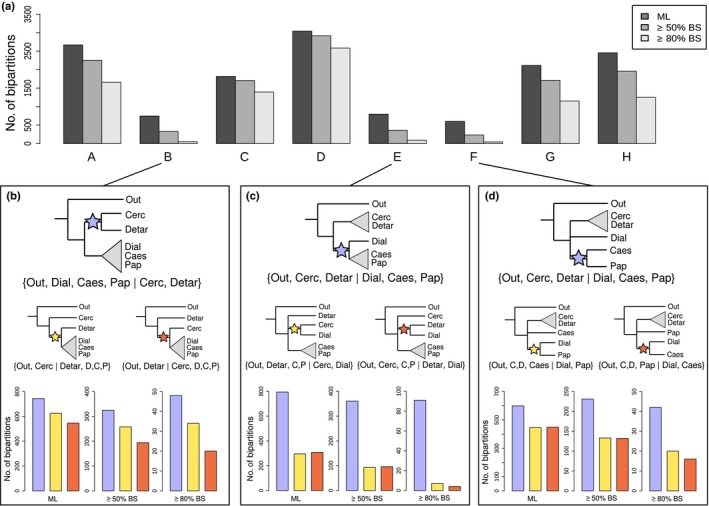
Leguminosae and its subfamilies are each supported by a large fraction of gene trees, in contrast to relationships among the subfamilies. (a) Prevalence of bipartitions that are equivalent to nodes A–H (see Figs 3, 4b) among the 3473 gene trees inferred from the rooted ingroup homologue clusters (including one‐to‐one orthologues) in which all five subfamilies and the outgroup were included. Numbers of bipartitions are shown as counted from the best‐scoring maximum likelihood (ML) gene trees as well as taking only bipartitions with ≥ 50% and ≥ 80% bootstrap support (BS) into account, as indicated in the legend. (b–d) Prevalence of bipartitions for nodes B, E and F plotted next to the most common alternative bipartitions. The locations of the stars in the illustrations indicate the internodes of the phylogeny that are equivalent to the bipartitions for which counts are plotted below, as counted from the ML estimates and for bipartitions with ≥ 50% or ≥ 80% BS. Colours of the stars correspond to the colours of the bars in the bar plots. Abbreviations in tree plots in (b–d) are as follows: Out, outgroup; Cerc, Cercidoideae; Detar, Detarioideae; Dial, Dialioideae; Caes, Caesalpinioideae; Pap, Papilionoideae; C; D, Cercidoideae + Detarioideae; D; C; P, Dialioideae + Caesalpinioideae + Papilionoideae; C; P, Caesalpinioideae + Papilionoideae.
The splitstree network (Fig. 5) shows relationships that are largely in line with the nuclear species tree, but is not entirely tree‐like, including along the backbone of the family where edge lengths are shorter than elsewhere in the network.
Figure 5.
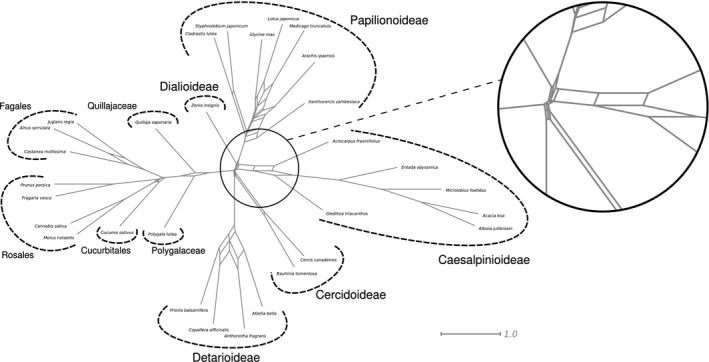
Filtered supernetwork inferred from the 1103 one‐to‐one orthologues, with extremely short internal edges around the origin of the legumes, highlighting their near‐simultaneous divergence.
Evaluation of gene tree support and conflict
While the chloroplast and nuclear phylogenies show different topologies with regard to the first two dichotomies within legumes, all analyses of the nuclear data (ML, BI and multispecies coalescent) yield the same topology at the base of the family (Figs 3, 4), showing a sister‐group relationship of Cercidoideae and Detarioideae and a clade comprising the remaining three sampled subfamilies as their sister clade. Because the nuclear dataset comprises 1103 loci sampled from across the nuclear genome that are therefore probably largely unlinked, while the recombination‐free chloroplast genome constitutes just a single locus, the nuclear topology should be considered a more realistic estimate of the species tree topology. However, when evaluating gene tree conflict, it is clear that many conflicting bipartitions exist, with the most prevalent being nearly as frequent across gene trees as compatible bipartitions (Fig. 3). The quartet support calculated by astral is also low (37%, with alternative quartet supports 33% and 30%; Fig. 4b). The relationships among the remaining three sampled subfamilies are also supported by significantly fewer bipartitions and lower quartet support than, for example, the legume crown node (pie charts in Figs 3, 4b).
We also sought to evaluate in a more intuitive way how much support and conflict there are among gene trees for the deepest divergences in the legume family. For nodes A–H in Fig. 3, we counted how often a bipartition equivalent to that node in the species tree is encountered across gene trees, and how often those bipartitions received at least 50% or 80% BS. We did this on all RT homologues (n = 7621) in which all subfamilies and the outgroup were represented by at least one taxon, leading to 3473 gene trees being considered. This shows that the legume family as a whole (node A), and the four subfamilies for which more than one taxon was sampled (nodes C, D, G and H), are all found to be monophyletic across the majority of gene trees (Fig. 6a; Table S3), and those bipartitions mostly receive at least 50% or 80% BS (Fig. 6a). Nodes B, E and F (i.e. the relationships among the subfamilies) are recovered in many fewer gene trees, especially when considering only bipartitions with at least 50 or 80% BS. Expressing these differences in percentages of the total number of gene trees (n = 3473), this difference becomes especially stark, with nodes A, C, D, G and H receiving at least 80% BS in 33.14–74.43% of gene trees, while the same BS value is found in only 1.38%, 2.62% and 1.21% of gene trees for nodes B, E and F, respectively. For these latter three nodes, we checked how often the most important conflicting bipartitions were present (Fig. 6b–d; Table S3). These conflicting bipartitions are each less prevalent than those found by the concatenated ML and BI analyses as well as by astral. This confirms that the recovered topology represents the relationships among legume subfamilies that is supported by the largest fraction of the genomic data used here, despite lack of phylogenetic signal across these nodes (Fig. 3) and significant and well‐supported gene tree conflict (Fig. 6b–d).
Discussion
Resolving the deep‐branching relationships in the Leguminosae
Previous phylogenetic studies aimed at resolving deep relationships in legumes have relied on only a few chloroplast markers (Doyle et al., 1997; Wojciechowski et al., 2004; Lavin et al., 2005; Bruneau et al., 2008; LPWG, 2017), but here we show that even 72 protein‐coding genes from the chloroplast genome fail to consistently resolve the root node with high support (Fig. 2c). Furthermore, substitution rate variation as evident from branch length disparity among legume subfamilies (Fig. 2a) (as previously shown for matK and rbcL by Lavin et al., 2005), implies that while the chloroplast genome may be a useful marker to resolve relationships within Papilionoideae (particularly within the 50 kb‐inversion clade), it is of limited use in other subfamilies, particularly Caesalpinioideae (Fig. 2a). Clearly, moving beyond the chloroplast genome and analysing nuclear gene data is necessary to improve phylogenetic hypotheses for the legume family, as found for other parts of the plant tree of life where chloroplast data have proven insufficiently informative (e.g. to resolve Mesangiosperms; Moore et al., 2010; Li et al., 2019). Nuclear data are also essential to detect conflicting signals across genomes. Using nuclear gene data, we recovered a best‐supported topology for the subfamily relationships (Figs 3, 4) that is different from the weakly supported chloroplast topology (Fig. 2), and also quantified the strength of phylogenetic signal for alternative topologies (Fig. 6).
We show that the difficulty of obtaining resolution for deep divergences in the legume family is in part caused by lack of phylogenetic signal in the chloroplast genome and a large fraction of the sampled nuclear genes (Fig. 3), with too few substitutions having accumulated along the deepest short internodes, leading to only a small fraction of the gene trees showing strong support (≥ 80% BS) for relationships among these (Fig. 6; Table S3). However, for a proportion of those genes that have sufficient phylogenetic signal, we find strongly supported conflicting evolutionary histories. Alongside methodological issues such as poor orthology inference for a number of genes, this conflict is probably caused by ILS (Pamilo & Nei, 1988; Maddison, 1997). Indeed, strong gene tree conflict caused by ILS is thought to be common when internodes are short owing to rapid diversification and this provides an explanation as to why many relationships are contentious at all taxonomic levels (e.g. Pollard et al., 2006; Suh et al., 2015; Moore et al., 2017; but see Scornavacca & Galtier, 2017 and Richards et al., 2018). Ancient hybridization could also lead to gene tree conflict; however, for such deep divergences as the earliest dichotomies in the legume family, it will be difficult to distinguish among deep coalescence and postspeciation gene flow. However, given the similar quartet frequencies for alternative topologies in the astral analysis for each of these dichotomies (Fig. 4b), ILS is probably predominant.
Taken together, this could suggest that a fully bifurcating tree is an inadequate representation of the initial radiation of the legumes. As we show, genes have many different evolutionary histories across the backbone (Table S3), while the species tree merely represents the dominant evolutionary history. In the case of complete lack of phylogenetic signal, or equally prevalent conflicting evolutionary histories without a single dominant one, this would constitute a hard polytomy, implying (nearly) instantaneous divergence of three or more lineages, as demonstrated for Neoaves (Suh, 2016). In the legumes, there does appear to be one dominant evolutionary history in the relationships among subfamilies supported by a larger fraction of gene trees (Fig. 6), suggesting that the deep‐branching relationships can be represented by a fully bifurcating topology. A hard polytomy at the root node of the legumes is also rejected by astral, but the same test did not reject a polytomy among Dialioideae, Caesalpinioideae and Papilionoideae. This is unexpected because the relationships among these have been recovered in previous studies (Bruneau et al., 2008; LPWG, 2017), and are recovered in all our analyses (Figs 2, 3, 4, S1–S7) with generally high support (Figs 2c, 4a, S1–S4, S6). The bipartition counts (Fig. 6d) also suggest that a hard polytomy can probably be rejected for the relationships among these three subfamilies. However, the ICA value for a sister‐group relationship of Caesalpinioideae and Papilionoideae is lower than for Cercidoideae and Detarioideae (0.70 vs 0.85) and support is even weaker in the astral analysis (0.58 pp). As the levels of conflict are similar to that for the position of the root of the legumes (Fig. 5b,d), the lower support and failure to reject a polytomy may be caused by deeper gene coalescences than for the Cercidoideae/Detarioideae clade and/or introgression shortly after divergence. The reticulate pattern observed at the base of Caesalpinioideae in the supernetwork (Fig. 5) might also indicate a hybrid origin of that subfamily, which merits further research. With denser taxon sampling, in particular for Dialioideae, for which we sampled just one species, it may also be possible to reject a hard polytomy across this clade.
A further complication potentially affecting phylogeny reconstruction is the occurrence of whole‐genome duplications (WGDs) in the early evolution of the legumes (Cannon et al., 2015; Stai et al., 2019), which could lead to issues with orthologue detection. Although the homologue trees inferred here during the orthologue selection procedure are suitable to test the placements of WGDs on the phylogeny, this is beyond the scope of this study and is addressed elsewhere (E. J. M. Koenen et al., unpublished).
In conclusion, we show that the most likely legume species tree is ((Cercidoideae,Detarioideae),(Duparquetioideae,(Dialioideae,(Caesalpinioideae,Papilionoideae)))). That legumes diversified rapidly following their origin was previously shown by Lavin et al. (2005), but here we demonstrate in greater detail that it is the relationships among subfamilies that are represented by particularly short internodes, generating conflicting relationships across gene trees indicative of ILS, with long stem lineages subtending each subfamily and the family as a whole (Figs 2, 3, 4, 6, S1–S7). This latter finding contrasts with those of Lavin et al. (2005) who inferred a rather short stem lineage for the family, probably an artefact of fixing the age of the stem node in their dating analyses. Nevertheless, the branching order among the subfamilies is rather insignificant, as further highlighted by the supernetwork (Fig. 5), which shows short edges and reticulation along the backbone of the family, indicative of a near‐simultaneous divergence of the subfamilies. Over the past decade, there was considerable debate about how many and which subfamilies of legumes should be recognized (LPWG, 2013b, 2017). That Leguminosae comprise six (nearly) simultaneously originating lineages, as demonstrated here, strongly supports the outcome of that debate, that is, the recognition of six legume subfamilies (LPWG, 2017).
Implications for our understanding of the evolution of legume diversity and traits
The near‐simultaneous divergence of the six main legume lineages is highly relevant for understanding the evolution of legume diversity and the appearance of key traits. Over the last few decades, the prevailing characterization of legume evolution has been that of mimosoids and papilionoids as derived clades that evolved from a paraphyletic grade of caesalpinioid legumes (e.g. LPWG, 2013a). This led to the misplaced characterization of several caesalpinioid lineages as in some way ‘basal’ or ‘early‐diverging’ (see LPWG, 2013a and references therein). Such characterizations are commonly made, but are in reality phylogenetic misinterpretations, given that basal nodes are ancestral nodes and at each bifurcating node two sister groups diverge from each other concurrently, neither of them earlier (Crisp & Cook, 2005). Species‐poor successive sister‐groups of species‐rich clades are often mistakenly referred to as basal or early‐diverging, and this appears also to have been the case in legumes, where the mimosoids and papilionoids have (vastly) more species than other lineages such as Cercidoideae, Detarioideae, Duparquetioideae and Dialioideae. This can lead to the erroneous assumption that lineages such as Cercidoideae, Detarioideae and Dialioideae have retained more ancestral traits than the species‐rich mimosoid and papilionoid clades (Crisp & Cook, 2005).
Near‐simultaneous divergence of the six subfamilies, and a rather insignificant branching order among them, provide additional arguments to abandon the idea of ‘early‐diverging’ lineages in legumes. Most trait evolution likely occurred along the long stem lineages of the family and subfamilies, rather than as derived legume traits having evolved in a stepwise fashion across the first divergences in the family. In comparative analyses, the branching order among subfamilies is unlikely to be meaningful and it should be effectively considered a polytomy with respect to trait evolution. We therefore suggest that typical legume traits evolved along the stem lineage of the family and were shared by the earliest stem relatives of each subfamily (i.e. the earliest stem relatives of each subfamily probably had similar traits).
The near‐simultaneous divergence of subfamilies suggests that many traits shared across legume subfamilies (Table 1 in LPWG, 2017) could be plesiomorphic, having been independently lost or modified in some subfamilies and retained in others. An alternative hypothesis is that these traits are not ancestral to all legumes and have evolved independently in different lineages, leading to homoplasy. Somewhat intermediate is the hypothesis of a shared cryptic precursor trait that can lead to deep homology, where similar traits evolved independently from a shared genetic basis (Shubin et al., 2009; Scotland, 2010). For instance, this could potentially explain the homoplasious distribution of extrafloral nectaries across legumes (Marazzi et al., 2012), which are present in several subfamilies but are different in structure and location, casting doubt on a single origin and prompting the possibility of a shared genetic precursor (Marazzi et al., 2012, 2019).
However, the precursor trait hypothesis may be motivated more by the notion that massive parallel loss of a trait is less parsimonious than assuming a few more independent gains. For instance, the evolution of N fixation in root nodules, a trait that is especially prominent in legumes, has been suggested to be driven by a cryptic precursor in the N‐fixing clade of angiosperms (Werner et al., 2014), with five independent gains in legumes, within subfamilies Caesalpinioideae and Papilionoideae, being most parsimonious (Doyle, 2016). Recent genomic evidence, however, supports a single origin of nodulation shared by the whole N‐fixing clade of angiosperms, with massive parallel losses in each of the four orders (Cucurbitales, Fabales, Fagales and Rosales) that make up the clade (van Velzen et al., 2018a; Griesmann et al., 2018). This suggests that the legume ancestor was also a nodulator, and given the rapid initial divergence of legumes documented here, stem relatives of all subfamilies probably also had the ability to nodulate, but nodulation was presumably lost in parallel along the long stem lineages or early in the crown group divergences of Cercidoideae, Detarioideae, Duparquetioideae and Dialioideae, in which no nodulating species are known. Determining when and why nodulation has been lost in all but two legume subfamilies will be important for understanding the causes of massive parallel loss of nodulation in the N‐fixing clade of angiosperms (van Velzen et al., 2018b).
Examples of other traits that are either plesiomorphic or homoplasious among and/or within subfamilies include wood with vestured pits (also present in some Polygalaceae (Jansen et al., 2001) and absent in Cercidoideae, Duparquetia and most Dialioideae (LPWG, 2017)); ectomycorrhizal symbiosis (known to occur in Detarioideae, Caesalpinioideae and Papilionoideae (Smith et al., 2011)); and floral symmetry that is variable across all nonmonotypic subfamilies (Cardoso et al., 2013; Bruneau et al., 2014; LPWG, 2017; Ojeda et al., 2019). These and other traits are candidates for comparative (genomic) analyses based on the new phylogenetic framework presented here, to test the hypothesis that several key legume traits are ancestral with multiple independent losses rather than independent gains.
Finally, our findings are also relevant for inferring the placements of WGDs and reconstructing the ancestral legume genome. For example, the recent suggestion by Stai et al. (2019) that Cercis could represent the genome duplication status of the ancestral legume is in part based on placement of Cercidoideae as sister to the rest of the legumes, which we show is only poorly supported in the chloroplast alignment and not the most likely species tree topology based on nuclear genes.
Concluding remarks
In this study, we present some of the first phylogenetic analyses using genome‐scale data for the Leguminosae, sampling representatives of all six subfamilies. Although our results show overwhelming support for monophyly of the family and each of the five nonmonotypic subfamilies, there is both a paucity of phylogenetic signal across the majority of genes and strongly conflicting relationships found across a small proportion of gene trees regarding relationships among them. This suggests that the six main lineages of legumes originated in quick succession, or nearly simultaneously, with significant implications for understanding the evolution of legume diversity and traits.
We also show that it is essential in phylogenomic studies to explicitly evaluate conflicting phylogenetic signals across the genome. By taking into account alternative topologies with high BS across gene trees (Fig. 6), the phylogenomic complexity of the initial radiation of the legumes is revealed. More generally, this study adds to an increasing understanding of the limits to phylogenetic resolution, suggesting that genome‐scale data may yield only relatively minor enhancements in topological robustness, and highlighting the role of rapid successive deep divergences in causing lack of phylogenetic signal and gene tree conflict across the Tree of Life.
Author contributions
EJMK and CEH designed the research; EJMK carried out the research and wrote the manuscript; DIO, RS, JM, FTB, JJW, CK, OJH, RTP, AB and CEH contributed to data collection, analysis and interpretation, as well as to writing of the final version of the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Dataset S1 The concatenated nucleotide alignment of 72 protein‐coding genes from the chloroplast genome in NEXUS format.
Dataset S2 The concatenated amino acid alignment of 72 protein‐coding genes from the chloroplast genome in NEXUS format.
Dataset S3 ZIP file containing amino acid alignments of 1103 one‐to‐one orthologues in NEXUS format.
Dataset S4 ZIP file containing amino acid alignments of 7621 RT homologues in NEXUS format.
Dataset S5 ZIP file containing 1103 gene trees estimated from one‐to‐one orthologues in newick format, with bootstrap values and branch lengths.
Dataset S6 ZIP file containing 7621 gene trees estimated from RT homologues in newick format, with bootstrap values and branch lengths.
Fig. S1 ML topology as inferred by raxml from amino acid alignment of chloroplast genes under the LG4X model.
Fig. S2 Bayesian majority‐rule consensus tree inferred with phylobayes from amino acid alignment of chloroplast genes under the CATGTR model.
Fig. S3 ML topology as inferred by raxml from nucleotide alignment of chloroplast genes under the GTR + G model.
Fig. S4 Bayesian majority‐rule consensus tree inferred with phylobayes from nucleotide alignment of chloroplast genes under the CATGTR model.
Fig. S5 ML topology as inferred by raxml from a concatenated alignment of 1103 nuclear genes, under the LG4X model.
Fig. S6 Bayesian gene jackknifing majority‐rule consensus tree inferred with phylobayes from a concatenated alignment of 1103 nuclear genes.
Fig. S7 Phylogeny estimated under the multispecies coalescent with astral.
Notes S1 Python script for counting bipartitions.
Table S1 Accession information for taxa included in the chloroplast alignment.
Table S2 Accession information for taxa included in the nuclear genomic and transcriptomic data set.
Table S3 Counts of bipartitions representing nodes A–H (see Fig. 3) and conflicting bipartitions representing other subfamily relationships among 3473 gene trees.
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant 31003A_135522 to CEH), the Department of Systematic and Evolutionary Botany, University of Zurich, the Natural Sciences and Engineering Research Council of Canada (grant to AB), the UK National Environment Research Council (grant NE/1027797/1 to RTP) and the Fonds de la Recherche Scientifique of Belgium (grant J.0292.17 to OH). We thank the S3IT of the University of Zurich for use of the ScienceCloud computational infrastructure and the Functional Genomics Center Zurich (FGCZ) for library preparation and sequencing. We thank Robin van Velzen, Pascal‐Antoine Christin, Donovan Bailey and two anonymous reviewers for insightful comments that improved the manuscript.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau A, Mercure M, Lewis GP, Herendeen PS. 2008. Phylogenetic patterns and diversification in the caesalpinioid legumes. Botany 86: 697–718. [Google Scholar]
- Bruneau A, Klitgaard BB, Prenner G, Fougere‐Danezan M, Tucker SC. 2014. Floral evolution in the Detarieae (Leguminosae): phylogenetic evidence for labile floral development in an early‐diverging legume lineage. International Journal of Plant Sciences 175: 392–417. [Google Scholar]
- Cannon SB, McKain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN Jr, Rolf M, Kutchan T. 2015. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Molecular Biology and Evolution 32: 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso D, de Queiroz LP, Pennington RT, de Lima HC, Fonty E, Wojciechowski MF, Lavin M. 2012. Revisiting the phylogeny of papilionoid legumes: New insights from comprehensively sampled early‐branching lineages. American Journal of Botany 99: 1991–2013. [DOI] [PubMed] [Google Scholar]
- Cardoso D, Pennington RT, de Queiroz LP, Boatwright JS, Van Wyk B‐E, Wojciechowski MF, Lavin M. 2013. Reconstructing the deep‐branching relationships of the papilionoid legumes. South African Journal of Botany 89: 58–75. [Google Scholar]
- Criscuolo A, Gribaldo S. 2010. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evolutionary Biology 10: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp MD, Cook LG. 2005. Do early branching lineages signify ancestral traits? Trends in Ecology and Evolution 20: 122–128. [DOI] [PubMed] [Google Scholar]
- Doyle JJ.1995. DNA data and legume phylogeny: a progress report In: Crisp MD, Doyle JJ, eds. Advances in legume systematics part 7: phylogeny. Richmond, UK: Royal Botanic Gardens, Kew, 11–30. [Google Scholar]
- Doyle JJ. 2016. Chasing unicorns: nodulation origins and the paradox of novelty. American Journal of Botany 103: 1865–1868. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL, Ballenger JA, Dickson EE, Kajita T, Ohashi H. 1997. A phylogeny of the chloroplast gene rbcL in the Leguminosae: taxonomic correlations and insights into the evolution of nodulation. American Journal of Botany 84: 541–554. [PubMed] [Google Scholar]
- Dugas DV, Hernandez D, Koenen EJ, Schwarz E, Straub S, Hughes CE, Jansen RK, Nageswara‐Rao M, Staats M, Trujillo JT, Hajrah NH. 2015. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP . Scientific Reports 5: 16958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad‐Toh K, Friedman N, Regev A. 2011. Trinity: reconstructing a full‐length transcriptome without a genome from RNA‐Seq data. Nature Biotechnology 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook M, Billault‐Penneteau B, Lauressergues D, Keller J, Imanishi L, Roswanjaya YP. 2018. Phylogenomics reveals multiple losses of nitrogen‐fixing root nodule symbiosis. Science 361: p.eaat1743. [DOI] [PubMed] [Google Scholar]
- Jansen S, Baas P, Smets E. 2001. Vestured pits: their occurrence and systematic importance in eudicots. Taxon 50: 135–167. [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution 34: 772–773. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. 2004. A Bayesian mixture model for across‐site heterogeneities in the amino‐acid replacement process. Molecular Biology and Evolution 21: 1095–1109. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Systematic Biology 62: 611–615. [DOI] [PubMed] [Google Scholar]
- Lassmann T, Hayashizaki Y, Daub CO. 2009. TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics 25: 2839–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M, Herendeen PS, Wojciechowski MF. 2005. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Systematic Biology 54: 575–594. [DOI] [PubMed] [Google Scholar]
- Le Q, Dang C, Gascuel O. 2012. Modeling protein evolution with several amino acid replacement matrices depending on site rates. Molecular Biology and Evolution 29: 2921–2936. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Schrire B, Mackinder B, Lock M. 2005. Legumes of the world. Richmond, UK: Royal Botanic Gardens Kew. [Google Scholar]
- Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, Gitzendanner MA, Fritsch PW, Cai J, Luo Y et al 2019. Origin of angiosperms and the puzzle of the Jurassic gap. Nature Plants 5: 461–470. [DOI] [PubMed] [Google Scholar]
- LPWG (Legume Phylogeny Working Group) . 2013a. Legume phylogeny and classification in the 21st century: progress, prospects and lessons for other species‐rich clades. Taxon 62: 217–248. [Google Scholar]
- LPWG (Legume Phylogeny Working Group) . 2013b. Towards a new classification system for legumes: progress report from the 6th International Legume Conference. South African Journal of Botany 89: 3–9. [Google Scholar]
- LPWG (Legume Phylogeny Working Group) . 2017. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 66: 44–77. [Google Scholar]
- Maddison WP. 1997. Gene trees in species trees. Systematic Biology 46: 523–536. [Google Scholar]
- Magee AM, Aspinall S, Rice DW, Cusack BP, Sémon M, Perry AS, Stefanović S, Milbourne D, Barth S, Palmer JD, Gray JC. 2010. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Research 20: 1700–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazzi B, Ané C, Simon MF, Delgado‐Salinas A, Luckow M, Sanderson MJ. 2012. Locating evolutionary precursors on a phylogenetic tree. Evolution 66: 3918–3930. [DOI] [PubMed] [Google Scholar]
- Marazzi B, Gonzalez AM, Delgado‐Salinas A, Luckow MA, Ringelberg J, Hughes CE. 2019. Extrafloral nectaries in Leguminosae: phylogenetic distribution, morphological diversity and evolution. Australian Systematic Botany 32: 409–458. [Google Scholar]
- McKey D. 1994. Legumes and nitrogen: the evolutionary ecology of a nitrogen‐demanding lifestyle In: Sprent JI, McKey D, eds. Advances in legume systematics 5: the nitrogen factor. Richmond, UK: Royal Botanic Gardens, Kew, 211–228. [Google Scholar]
- Mirarab S, Reaz R, Bayzid MdS, Zimmermann T, Swenson MS, Warnow T. 2014a. ASTRAL: genome‐scale coalescent‐based species tree estimation. Bioinformatics 30: i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Bayzid MS, Warnow T. 2014b. Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Systematic Biology 65: 366–380. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. 2010. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Acadamy of Sciences, USA 107: 4623–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AJ, Vos JMD, Hancock LP, Goolsby E, Edwards EJ. 2017. Targeted enrichment of large gene families for phylogenetic inference: phylogeny and molecular evolution of photosynthesis genes in the portullugo clade (Caryophyllales). Systematic Biology 67: 367–383. [DOI] [PubMed] [Google Scholar]
- Morgan CC, Foster PG, Webb AE, Pisani D, McInerney JO, O'Connell MJ. 2013. Heterogeneous models place the root of the placental mammal phylogeny. Molecular Biology and Evolution 30: 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda DI, Koenen E, Cervantes S, de la Estrella M, Banguera‐Hinestroza E, Janssens SB, Migliore J, Demenou B, Bruneau A, Forest F, Hardy OJ. 2019. Phylogenomic analyses reveal an exceptionally high number of evolutionary shifts in a florally diverse clade of African legumes. Molecular Phylogenetics and Evolution 137: 156–167. [DOI] [PubMed] [Google Scholar]
- Pamilo P, Nei M. 1988. Relationships between gene trees and species trees. Molecular Biology and Evolution 5: 568–583. [DOI] [PubMed] [Google Scholar]
- Pollard DA, Iyer VN, Moses AM, Eisen MB. 2006. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genetics 2: e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJP. 2011. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE 6: e22594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards EJ, Brown JM, Barley AJ, Chong RA, Thomson RC. 2018. Variation across mitochondrial gene trees provides evidence for systematic error: How much gene tree variation is biological? Systematic Biology 67: 847–860. [DOI] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. 2003. Genome‐scale approaches to resolving incongruence in molecular phylogenies. Nature 425: 798. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Delsuc F, Galtier N, Douzery EJ. 2013. Less is more in mammalian phylogenomics: AT‐rich genes minimize tree conflicts and unravel the root of placental mammals. Molecular Biology and Evolution 30: 2134–2144. [DOI] [PubMed] [Google Scholar]
- Salichos L, Rokas A. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497: 327–331. [DOI] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S. 2016. Fast coalescent‐based computation of local branch support from quartet frequencies. Molecular Biology and Evolution 33: 1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S. 2018. Testing for polytomies in phylogenetic species trees using quartet frequencies. Genes 9: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scornavacca C, Galtier N. 2017. Incomplete lineage sorting in mammalian phylogenomics. Systematic Biology 66: 112–120. [DOI] [PubMed] [Google Scholar]
- Scotland RW. 2010. Deep homology: a view from systematics. Bioessays 32: 438–449. [DOI] [PubMed] [Google Scholar]
- Shen XX, Hittinger CT, Rokas A. 2017. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nature Ecology & Evolution 1: 0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubin N, Tabin C, Carroll S. 2009. Deep homology and the origins of evolutionary novelty. Nature 457: 818. [DOI] [PubMed] [Google Scholar]
- Simion P, Philippe H, Baurain D, Jager M, Richter DJ, Di Franco A, Roure B, Satoh N, Queinnec E, Ereskovsky A, Lapebie P. 2017. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Current Biology 27: 958–967. [DOI] [PubMed] [Google Scholar]
- Simon MF, Grether R, de Queiroz LP, Skema C, Pennington RT, Hughes CE. 2009. Recent assembly of the Cerrado, a Neotropical plant diversity hotspot, by in situ evolution of adaptations to fire. Proceedings of the National Academy of Sciences, USA 106: 20359–20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ME, Henkel TW, Aime MC, Fremier AK, Vilgalys R. 2011. Ectomycorrhizal fungal diversity and community structure on three co‐occurring leguminous canopy tree species in a Neotropical rainforest. New Phytologist 192: 699–712. [DOI] [PubMed] [Google Scholar]
- Smith SA, Moore MJ, Brown JW, Yang Y. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evolutionary Biology 15: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stai JS, Yadav A, Sinou C, Bruneau A, Doyle JJ, Fernández‐Baca D, Cannon SB. 2019. Cercis: a non‐polyploid genomic relic within the generally polyploid legume family. Frontiers in Plant Science 10: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML Version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh A, Smeds L, Ellegren H. 2015. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biology 13: e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh A. 2016. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zoologica Scripta 45: 50–62. [Google Scholar]
- Sukumaran J, Holder MT. 2010. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26: 1569–1571. [DOI] [PubMed] [Google Scholar]
- van Velzen R, Holmer R, Bu F, Rutten L, van Zeijl A, Liu W, Santuari L, Cao Q, Sharma T, Shen D, Roswanjaya Y. 2018a. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen‐fixing rhizobium symbioses. Proceedings of the National Academy of Sciences, USA 115: E4700–E4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velzen R, Doyle JJ, Geurts R. 2018b. A resurrected scenario: single gain and massive loss of nitrogen‐fixing nodulation. Trends in Plant Science 24: 49–57. [DOI] [PubMed] [Google Scholar]
- Werner GD, Cornwell WK, Sprent JI, Kattge J, Kiers ET. 2014. A single evolutionary innovation drives the deep evolution of symbiotic N2‐fixation in angiosperms. Nature Communications 5: 4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Cameron SA, Huson D, Steel M. 2008. Filtered Z‐closure supernetworks for extracting and visualizing recurrent signal from incongruent gene trees. Systematic Biology 57: 939–947. [DOI] [PubMed] [Google Scholar]
- Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, Ruhfel BR. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences, USA 111: E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AV, Boykin LM, Howell KA, Nevill PG, Small I. 2015. The complete sequence of the Acacia ligulata chloroplast genome reveals a highly divergent clpP1 gene. PLoS ONE 10: e0125768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski MF, Lavin M, Sanderson MJ. 2004. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well‐supported subclades within the family. American Journal of Botany 91: 1846–1862. [DOI] [PubMed] [Google Scholar]
- Yang Y, Smith SA. 2014. Orthology inference in nonmodel organisms using transcriptomes and low‐coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31: 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics 30: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Dataset S1 The concatenated nucleotide alignment of 72 protein‐coding genes from the chloroplast genome in NEXUS format.
Dataset S2 The concatenated amino acid alignment of 72 protein‐coding genes from the chloroplast genome in NEXUS format.
Dataset S3 ZIP file containing amino acid alignments of 1103 one‐to‐one orthologues in NEXUS format.
Dataset S4 ZIP file containing amino acid alignments of 7621 RT homologues in NEXUS format.
Dataset S5 ZIP file containing 1103 gene trees estimated from one‐to‐one orthologues in newick format, with bootstrap values and branch lengths.
Dataset S6 ZIP file containing 7621 gene trees estimated from RT homologues in newick format, with bootstrap values and branch lengths.
Fig. S1 ML topology as inferred by raxml from amino acid alignment of chloroplast genes under the LG4X model.
Fig. S2 Bayesian majority‐rule consensus tree inferred with phylobayes from amino acid alignment of chloroplast genes under the CATGTR model.
Fig. S3 ML topology as inferred by raxml from nucleotide alignment of chloroplast genes under the GTR + G model.
Fig. S4 Bayesian majority‐rule consensus tree inferred with phylobayes from nucleotide alignment of chloroplast genes under the CATGTR model.
Fig. S5 ML topology as inferred by raxml from a concatenated alignment of 1103 nuclear genes, under the LG4X model.
Fig. S6 Bayesian gene jackknifing majority‐rule consensus tree inferred with phylobayes from a concatenated alignment of 1103 nuclear genes.
Fig. S7 Phylogeny estimated under the multispecies coalescent with astral.
Notes S1 Python script for counting bipartitions.
Table S1 Accession information for taxa included in the chloroplast alignment.
Table S2 Accession information for taxa included in the nuclear genomic and transcriptomic data set.
Table S3 Counts of bipartitions representing nodes A–H (see Fig. 3) and conflicting bipartitions representing other subfamily relationships among 3473 gene trees.