

Abstract
Historical events, habitat preferences, and geographic barriers might result in distinct genetic patterns in insular versus mainland populations. Comparison between these two biogeographic systems provides an opportunity to investigate the relative role of isolation in phylogeographic patterns and to elucidate the importance of evolution and demographic history in population structure. Herein, we use a genotype‐by‐sequencing approach (GBS) to explore population structure within three species of mastiff bats (Molossus molossus, M. coibensis, and M. milleri), which represent different ecological histories and geographical distributions in the genus. We tested the hypotheses that oceanic straits serve as barriers to dispersal in Caribbean bats and that isolated island populations are more likely to experience genetic drift and bottlenecks in comparison with highly connected ones, thus leading to different phylogeographic patterns. We show that population structures vary according to general habitat preferences, levels of population isolation, and historical fluctuations in climate. In our dataset, mainland geographic barriers played only a small role in isolation of lineages. However, oceanic straits posed a partial barrier to the dispersal for some populations within some species (M. milleri), but do not seem to disrupt gene flow in others (M. molossus). Lineages on distant islands undergo genetic bottlenecks more frequently than island lineages closer to the mainland, which have a greater exchange of haplotypes.
In this manuscript, we use the genotype‐by‐sequencing approach on three species of Mastiff bats with different ecological histories and geographical distributions to explore population genetics parameters and better understand the role of geographic barriers in the dispersal and gene flow in bats. Our analyses show that the population structure of each species varies according to environmental factors, levels of population isolation, and demographic histories. We show that oceanic straights seem to pose a partial barrier for some species and that more isolated lineages on islands tend to undergo genetic bottlenecks more than connected lineages closer to the mainland.

1. INTRODUCTION
Evolutionary geographic patterns and demographic histories of species are largely affected by historical events, habitat preferences, and geographic barriers (Bernatchez & Wilson, 1999; Querejeta & Castresana, 2018; Soltis, Morris, McLachlan, Manos, & Soltis, 2006). Moreover, several mechanisms might result in genetic isolation, such as geography and ecology, which often interact to shape the current pattern of diversity, but also making it difficult to distinguish the primary diversification factors (Wang & Bradburd, 2014). Populations in early stages of speciation provide good model systems to study these diversification mechanisms (Schield et al., 2015). Such groups have not yet developed secondary reproductive isolating mechanisms, and the primary genetic precursors of speciation are more easily discernable (Good, Dean, & Nachman, 2008; Orr, 1995).
Evolutionary processes might also act differently on insular and mainland populations, according to their degree of ecological and geographic isolation (Garcia‐Verdugo, Caujapé‐Castells, Mairal, & Monroy, 2018; Ortiz‐Ramírez, Sánchez‐González, Castellanos‐Morales, Ornelas, & Navarro‐Sigüenza, 2018). Populations on islands are more isolated, have a smaller meta‐population size, and are more vulnerable to habitat disturbance than mainland populations, which might alter insular population structure (Leisler & Winkler, 2015; Losos & Ricklefs, 2009; Spilani et al., 2019). Although alternative drivers of phylogeographic and population structure have been proposed for continental (Gray et al., 2019; Kalkvik, Stout, Hoffman, & Parkinson, 2018) and island populations (Čandek, Agnarsson, Binford, & Kuntner, 2018), few studies have explicitly compared the patterns between these two biogeographic systems (e.g., Pons et al., 2019). This comparison provides an opportunity to investigate the relative role of isolation, habitat selection, and demographic history in shaping phylogeographic patterns (Kalkvik et al., 2018; Sexton, Hangartner, & Hoffmann, 2014), especially using closely related species with similar life histories.
The Caribbean archipelago comprises numerous islands that differ in size, age, habitat, and level of isolation from other islands and from the mainland. In addition, this archipelago is located in proximity to two continents (North and South America), providing a varied landscape for testing phylogeographic hypotheses (Ricklefs and Bermingham, 2008). The Caribbean is divided into two main regions. The Lesser Antilles are located on the eastern margin of the Caribbean tectonic plate, and most of these islands were formed more than 20 Ma ago by volcanic activity and have never been connected (Bender et al., 1979; Donnelly, 1988; Pindell, 1994). In contrast, the Greater Antilles are much older and were formed during the separation of North and South America 170 Ma ago, with many islands remaining united until the Eocene (Iturralde‐Vinent & MacPhee, 1999; Kerr, Iturralde‐vinent, Saunders, Babbs, & Tarney, 1999; Pindell and Barrett, 1990). Pitman, Cande, Labrecque, and Pindell (1993) hypothesized that during dry periods in the Eocene, a land bridge formed connecting the Greater Antilles with Middle America, potentially resulting in faunal exchange between these landmasses. Iturralde‐Vinent and MacPhee (1999) also proposed that a short‐lived land bridge connected the Greater Antilles to northwestern South America during the Oligocene, promoting more recent faunal exchange.
Individual species and genera of bats are some of the few extant, native mammals in the Neotropics that occur on both the continental mainland and Caribbean islands. Bats are the second most speciose order of mammals and occupy almost all parts of the globe (Patterson, Willig, & Stevens, 2003), and their broad distribution renders them useful in comparative phylogeographic analyses. They are the only mammals capable of true flight and the dispersal ability of some groups allows these bats to colonize large geographic areas, including oceanic islands (Dávalos, 2007; Russell et al., 2016; Speer et al., 2017). Dispersal is essential to promote gene flow within a species, and the study of barriers that could isolate populations may provide important insights regarding how current and historical evolutionary processes effect speciation (Miller‐Butterworth, Jacobs, & Harley, 2003; Tam et al., 2005). Bats with high dispersal abilities usually exhibit little genetic structure among populations due to high rates of gene flow (Carstens, Sullivan, Davalos, Larsen, & Pedersen, 2004; McCracken, McCracken, & Vawter, 1994; Pumo, Goldin, Elliot, Phillips, & Genoways, 1988). These bats are potentially less affected by habitat disturbance and genetic fragmentation than more sedentary groups (Ibáñez, García‐Mudarra, Ruedi, Stadelmann, & Juste, 2006; Meyer, Kalko, & Kerth, 2009), although some exceptions have been reported in species of Molossidae inhabiting insular systems (Speer et al., 2017).
In the Caribbean, there are more than 60 species of bats (Dávalos, 2004, 2006; Loureiro, Gregorin, & Perini, 2018; Velazco & Patterson, 2013), but not much is known about the capacity of the different species to disperse among islands. Koopman (1977) suggested that oceanic straits present functional barriers for dispersal in bats in the Caribbean. Similarly, Genoways (1998) proposed that migration between islands was unlikely and predicted that gene flow among island populations was infrequent. Populations of bats are vulnerable to Caribbean hurricanes and volcanic eruptions, which may reduce population sizes and possibly result in accidental dispersal (Pedersen, 1998; Pedersen, Genoways, & Freeman, 1996). Therefore, even in the absence of regular inter‐island migration, the genetic diversification among some island populations could be muted by episodic gene flow. Likewise, populations from small and distant islands might be expected to be subjected to genetic drift more frequently than populations from large and less isolated islands, which can potentially decrease the genetic variability on small islands (Nei & Tajima, 1981). Previous studies have shown that although bats have significant capacity for dispersal, ocean straits may act as a barrier for some groups (Carstens et al., 2004; Fleming & Racey, 2013; García‐Mudarra, Ibáñez, & Juste., 2009; Larsen et al., 2012; Speer et al., 2017), but may not impose a strong barrier for others (Carstens et al., 2004; García‐Mudarra et al., 2009; Larsen et al., 2007; Pumo et al., 1988). This pattern might also have been affected by lower sea levels during the Pleistocene that shortened overwater distance, decreasing the oceanic barrier among some islands (Velazco & Patterson, 2013).
Mastiff bats of the genus Molossus represent an ideal model system for the study of population structuring on a broad geographic scale. Molossus are common aerial insectivores that inhabit a large range of habitats, from dry and humid semideciduous forests and tropical rainforests to pastures and savannas (Eger, 2008; Reid, 2009). Many species in this genus are well‐adapted to anthropogenic modifications, and they can be numerous in urban areas and degraded habitats (Taylor et al., 2019). Molossus is nonmigratory, but many species are widely distributed and occur on both sides of prominent geographic barriers (e.g., Andes Mountains, Caribbean Sea) (Dolan, 1989; López‐González & Presley, 2001). Several species of Molossus are environmental generalists and are broadly distributed in the Neotropics, including on islands in the Caribbean. In contrast, other species in the genus are restricted to either mainland or Caribbean islands and prefer specific types of habitats, such as dry grasslands (Taylor et al., 2019). The extensive distribution of Molossus throughout the Neotropics, including the Caribbean, suggests a strong colonizing ability and capacity to fly or to be carried by wind currents and storm systems over water. Although no studies have measured vagility in Molossus, other molossids have been reported to fly up to 160 km in a single night (McCracken et al., 2016), to reach speeds over 50 km/hr, to fly for up to 10 hr without resting (Marques, Rainho, Carapuço, Oliveira, & Palmeirim, 2004), and to migrate long distances (Cockrum, 1969; Glass 1958; Russell, Medellín, & Mccracken, 2005). Additionally, bats in the family Molossidae have relatively long, narrow wings with a reduced area, resulting in high wing loadings and high aspect ratios (Norberg & Rayner, 1987). This suite of adaptations is commonly associated with fast, long‐distance flight and enhanced dispersal abilities (Peterson, Eger, & Mitchell, 1995; Taylor et al., 2012; Burns & Broders, 2014).
Although phylogenetic relationships among some clades of Molossus are uncertain, 14 species are currently recognized (Lindsey & Ammerman, 2016; Lim, Loureiro, Upham, & Brocca, 2017; Loureiro, Gregorin, et al., 2018; Loureiro, Engstrom, & Lim, 2019; Loureiro et al., in review), but relationships among populations within species have not been examined. We investigated population structure in three species of Molossus, which represent the different ecological requirements and geographic distributions found in the genus: M. coibensis, M. molossus, and M. milleri. The most widespread species of Molossus is M. molossus, being present in both the mainland and Caribbean islands; M. coibensis is broadly distributed on the mainland, but is absent from the Caribbean, and M. milleri is the most broadly distributed species of the genus that is restricted to the Caribbean.
Molossus coibensis is one of the smallest species of Molossus and the second most widespread species of the genus, occurring from southern Mexico to southeastern Brazil (Dolan, 1989; Eger, 2008; Loureiro, Gregorin, et al., 2018; Reid, 2009). This species occurs in a variety of habitats, such as urban areas, evergreen forest, and dry and humid semideciduous forest, and it is only found in the mainland (Taylor et al., 2019).
Molossus molossus is larger than M. coibensis (Dolan, 1989) and is the most common and broadly distributed species of the genus, occurring from Argentina to the southern United States and the Lesser Antilles (Barquez, Mares, & Braun, 1999; Dolan, 1989; Eger, 2008; Fabian & Gregorin, 2007). This species also is present on both the west and east sides of the Andes. Several geographic populations were originally described as subspecies of M. molossus based on morphological characters but have been relegated as synonyms based on molecular and further morphological analyses (Dolan, 1989; Loureiro et al., 2019; Loureiro, Gregorin, et al., 2018). For instance, Loureiro et al. (in review), based on a molecular phylogeny, demonstrated that the subspecies M. m. debilis from Nevis and M. m. pygmaeus from Curacao group with other populations from South America within the M. molossus clade do not appear to be distinct lineages.
Several other supposed subspecies of M. molossus have been described that are confined to one or a few Caribbean islands (Dolan, 1989; Loureiro, Gregorin, et al., 2018). However, based on morphological characters, all Caribbean species and subspecies of Molossus were previously synonymized under the name M. molossus (Dolan, 1989; Eger, 2008; Simmons, 2005). Recent studies based on mitochondrial and nuclear genes, however, demonstrated that M. verrilli from the Dominican Republic, and M. milleri from Cuba, the Cayman Islands, and Jamaica are distinct species, restricting the distribution of M. molossus in the Caribbean to the Lesser Antilles and Puerto Rico (Lim et al., 2017; Loureiro et al., 2019; Loureiro, Gregorin, et al., 2018). M. milleri is morphologically similar to M. verrilli and M. molossus and occupies both forests and urban areas in the Greater Antilles (Taylor et al., in press).
Species within Molossus have low genetic variability, and mitochondrial and nuclear markers are often insufficient to resolve the relationship among some morphologically distinct species (Loureiro et al., 2019). However, the use of a next generation sequencing approach (NGS) has shown promise for resolving relationships and population structure in recently diversified groups in which the rate of genetic change is low (Cronin et al., 2015; Enk et al., 2016; Kusza et al., 2014; Lozier, 2014). One of these approaches is the genotyping‐by‐sequencing (GBS) method, which sequences many small tags in the genome flanking restriction sites. By this method, thousands of single nucleotide polymorphisms (SNPs) are recovered, vastly increasing the size of the overall dataset compared to typical Sanger methods. With this large dataset, consistent variance can be detected among genetically similar groups that are not revealed by standard gene sequencing approaches. In addition, genomic‐scale SNP data provide powerful options for testing patterns of genetic structure and demographic trends and to estimate population parameters for identifying changes in population size over time (Excoffier, Dupanloup, Huerta‐Sánchez, Sousa, & Foll, 2013; Gutenkunst, Hernandez, Williamson, & Bustamante, 2009; Lozier, 2014). These estimates may be important in a conservation context because they can indicate if a population has the potential to undergo inbreeding depression or has the genetic bandwidth to adapt to future environmental changes (Sovic, Carstens, & Gibbs, 2016).
Herein, we use the GBS approach on three species of Mastiff bats (M. molossus, M. coibensis, and M. milleri) with different ecological histories and geographical distributions to explore population genetic parameters and better understand the role of geographic barriers in dispersal and gene flow in bats. We tested the hypothesis proposed by Koopman (1977) and supported by Genoways (1998) that oceanic straits serve as barriers to dispersal by Caribbean bats. If this hypothesis is correct, we would expect significant genetic structuring and low gene flow among island populations and between islands and mainland populations when compared to levels of variation within island populations or within the mainland.
We also tested the hypothesis proposed by Nei and Tajima (1981) that more isolated populations are likely to experience genetic drift and bottlenecks relative to less isolated ones. To support this hypothesis, we would expect to find a decrease in the effective population size in island populations and a constant or increasing effective population size on the mainland. We would also expect to find that island populations with a mainland source are less affected by bottlenecks than insular populations isolated from other landmasses. As a result of different isolation patterns, we expect divergent phylogeographic structuring in mainland and insular species based on geographic barriers, habitat selection, and historical fluctuation in climate.
2. MATERIAL AND METHODS
2.1. Sample collection and library preparation
We obtained tissue samples from 62 M. molossus from South America, Middle America, and the Lesser Antilles, 20 M. coibensis from South and Middle America, and 19 M. milleri from the Greater Antilles (Figure 1; Appendix 1). Tissues samples included skeletal muscle, liver, heart, and kidney and were preserved in 95% ethanol or were frozen in liquid nitrogen upon collection of the specimen in the field. DNA extraction was conducted using Qiagen DNeasy extraction kit (Qiagen, Inc.) following standard protocols. Genomic DNA quality was checked by visual inspection on an agarose gel, and the DNA concentration was measured using a Nanodrop spectrophotometer (Nanodrop Technologies). We used 30 μl of DNA samples with concentrations higher than 100 ng/μl for library preparation and for the genotyping‐by‐sequencing approach following the protocol described by Elshire et al. (2011). All libraries were sequenced on an Illumina HiSeq 2000 in the Cornell Institute of Genomic Diversity (IGD).
Figure 1.
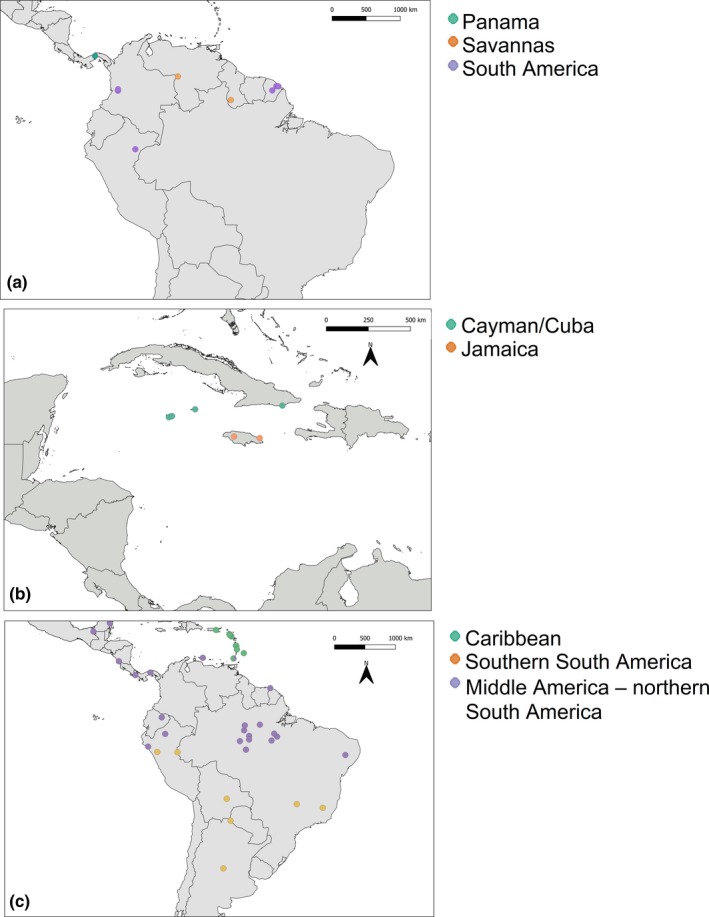
Maps of study areas and sampling location. (a) M. coibensis, (b) M. milleri, (c) M. molossus. Colors represent populations found in the structure and DAPC analyses
2.2. Genotyping
De novo genotyping was performed using the Universal Network‐Enable Analysis Kit (UNEAK) pipeline, available on TASSEL 3.0 software (Bradbury et al., 2007). The sequences were trimmed to a 64 bp length, and shorter reads were discarded. In this pipeline, identical reads are clustered into tags and all unique tags are merged. No reference genome or GBS reference sequences of any species of Molossus are available. The phylogenetically closest genome available is from the family Vespertilionidae, which diverged from molossids in the Eocene (Lack, Roehrs, Stanley, Ruedi, & Ven Den Bussche, 2010; Soria‐Carrasco & Castresana, 2012; Teeling, Jones, & Rossiter, 2016). All three species were pooled and aligned for the reference genotyping before the dataset for each species was filtered and analyzed separately. Quality control and filtering of the reference genotypes of each species sample were also conducted on TASSEL 3.0 (Bradbury et al., 2007). Tags with depth lower than seven were treated as missing. The minimum allele frequency (MAF) value of 0.02 removed almost half of the SNPs from the original dataset of both the de novo and reference genotyping analyses, while any value above 0.02 had a very small impact on number of SNPs (Figure S1), which could suggest that these removed SNPs might represent informative rare alleles, rather than sequencing errors. The increase of the MAF value may cause an underestimation of heterozygotes because it may remove rare alleles, instead of the removal of sequencing errors, with the loss of biological information (Ni and Stoneking, 2016). Kim et al. (2011) argued that for rare SNPs (e.g., MAF < 0.01) it is not easy to differentiate between sequencing errors and a true rare allele, and alleles with less the 1% of MAF should be discarded. Linck and Battey (2019) showed that highly accurate population inferences are reached when relatively rare alleles are included (minimum allele count 2% to 8%). Therefore, in this study we set the MAF value at 2%.
We discarded individuals with more than 20% missing data. We also removed invariant SNPs and those with more than 10% missing data. The minimum heterozygosity proportion was set to 0.01. To remove linked sites in the alignment, SNPs <128 bp apart were removed. We tested for deviations of Hardy–Weinberg equilibrium in each dataset using TASSEL 3.0 (Bradbury et al., 2007). We estimated kinship between individuals within each species by exploring the relationship between identity by state (IBS), when two or more individuals share similar nucleotide sequences, using TASSEL 3.0 to ensure results were not conflicted by kinship (Rodríguez‐Ramilo & Wang, 2012).
2.3. Population structure analysis
We assigned individuals to populations under an admixture model with correlated allele frequencies using Structure v.2.3.4 (Pritchard, Stephens, & Donnelly, 2000). This model assumes that the common ancestor of all populations passed part of its genotype to all its descendants (Falush, Stephens, & Pritchard, 2003). The possible number of populations (K) in each species was estimated. To rule out population substructuring within samples of individuals for each species, we allowed K to range from 1 to a number in excess of the geographic locations. For example, we obtained samples of M. milleri from four different Caribbean islands, but we allowed K to vary between 1 and 10 (Figure S2). Five runs for each K were analyzed under a model of admixture and correlated allele frequency for 10 million generations each. The log likelihoods for each K value were averaged among runs and verified using log (Alpha) plots by interaction and ln L (K) by interaction. We used Structure Harvest v0.694 (Earl & vonHoldt, 2011) to assess the most likely number of populations, using the results of the Structure analyses. Patterns of individual assignment to clusters were also used to make an optimal inference regarding the K value for each species.
We assessed population differentiation of each species by conducting a principal component analysis (PCA) of pairwise individual genetic distances among populations and discriminant analysis of principal components (DAPC). We conducted these analyses converting the observed SNP data into principal components that summarize the variation between samples using the R package poppr (Kamvar, Tabima, & Grünwald, 2014). The relationships among clades within each species were investigated through a coalescence approach, which accounts for differences in genealogical histories of individual loci using the program SVDquartets (Chifman & Kubatkoin 2015) implemented in PAUP 4.0 (Swofford, 2003). Four independent runs were conducted to assess topological convergence, each including 500 bootstrap replicates and exhaustive quartet sampling. Trees were visualized using FigTree v. 1.4.3. We also generated a genetic distance tree to represent the genetic relatedness of the samples based on the UPGMA algorithm, with 500 bootstrap replicates. After the populations were defined, we visualized the posterior assignment of each sample using a composite stacked bar plot.
To quantify differentiation of allele frequency, we calculated the effective number of migrants (Nm) (Barton & Slatkinf, 1986) using Genepop 4.7.0 (Rousset, 2018). We also calculated the observed and expected heterozygosity, and the pairwise fixation index (Fst) between populations using Arlequin 3.5 (Excoffier & Lischer, 2010). The Fst was performed by calculating genetic distance based on all markers (after quality control and filtering) using a weighted analysis of variance (Cockerham, 1973; Weir & Cockerham, 1984). An analysis of molecular variance (AMOVA) for all pairs of populations was used to compute the divergence among populations using Arlequin 3.5.
2.4. Demographic inference
We generated joined population folded site frequency spectra (SFS) for each population within a species using Arlequin 3.5 (Excoffier & Lischer, 2010) with 100 bootstraps. To infer historical demography patterns in all populations, each SFS was imported into FastSimCoal26 (Excoffier et al., 2013) and the likelihood of three demographic models was compared using the likelihood ratio test (LRT) and Akaike's information criterion (AIC). The maximum log likelihood given the observed SFS was calculated to determine how well each model fit the data. The first model represents a constant population size, the second model represents a population expansion, and the third model imposes a bottleneck in each population. The last two models assume changes in population size and also calculate the present population size, the ancestral population size, and the time in number of generations since the growth or decline began.
All parameters used in the demographic models were selected from a uniform distribution, and we considered the mutation rate as 2.5 × 10−8 per nucleotide per generation. This value was calculated from human genomic data (Keightley, 2012; Nachman & Crowell, 2000) and is thought to provide a conservative estimate of population genetic parameters in mammals, including bats (Sovic et al., 2016). We calculated the confidence interval for each variable by performing a parametric bootstrap using point estimates from each parameter. For each model, we performed 100 runs (500,000 simulations per run) on FastSimCoal26 (Excoffier et al., 2013) and used the highest likelihood value in the LRT and AIC comparison. To ensure simulations were not stuck in a local maxima, individual runs of FastSimCoal26 under each model were tested for consistency and similar likelihood.
3. RESULTS
3.1. GBS data
A total of 436,086 SNPs were sequenced, and tags among individuals were aligned de novo. The species were then analyzed separately, and after initial filtering, the final GBS dataset with unlinked SNPs and only polymorphic sites consisted of 20 samples with 8,930 SNPs for M. coibensis, 54 samples with 7,311 SNPs for M. molossus, and 19 samples with 3,505 SNPs for M. milleri. Tests for Hardy–Weinberg equilibrium revealed significant linkage (p < .01) for seven pairs of loci in M. molossus, one pair of loci in M. coibensis, and no loci in M. milleri. One locus of each linked pair was removed to avoid bias in the results due to linked SNPs. Linkage disequilibrium is not common within subpopulations, except between very close or adjacent sites. In addition, isolated populations are less likely to exhibit strong correlations among loci (Pritchard et al., 2000). The initial removal of SNPs <128 bp apart likely removed almost all linked loci from our dataset, since only a few linked loci were found after the application of this filter. In addition, the low admixture among populations within all three study species might have also reduced linkage disequilibrium in our data. The IBS analysis did not reveal any closely related individuals in any of the three species, and no individuals were discarded (Figure S2).
3.2. Population structure analyses
The Structure analyses indicated a best fit of K = 3 for M. coibensis, K = 3 for M. molossus, and K = 2 for M. milleri (Figures 1 and 2a; Figure S3). Structure plots based on higher K values did not show any substructuring within populations (Figure S4). The composite stacked bar plots reveal that M. coibensis has three distinct populations: Panama, the savannas of Venezuela and Guyana, and rainforests of other South American countries (French Guiana, Ecuador, and Peru) (Figure 2a). M. molossus was also divided into three populations. The first population represents northern South America and Middle America with samples from northeast Brazil, Ecuador, French Guiana, Guyana, Mexico, Nicaragua, Panama, Bonaire, Grenada, and western Peru; the second population represents southern South America with individuals from Argentina, southeastern Brazil, Bolivia, Paraguay, and eastern Peru; and the third population comprises Caribbean samples from the Lesser Antilles and Puerto Rico. However, one of the four samples from Grenada (TTU18551) has almost 100% of its genotypes shared with the continental population and not with the Lesser Antilles (Figure 2a). The other three individuals from Grenada, as well as the samples from Martinique, had a higher probability of belonging to the Caribbean population, but they also share part of their genotypes with the Middle America–northern South America population. The samples from M. milleri were divided into two populations: one from Jamaica and one from the Cayman Islands and Cuba (Figure 2a).
Figure 2.
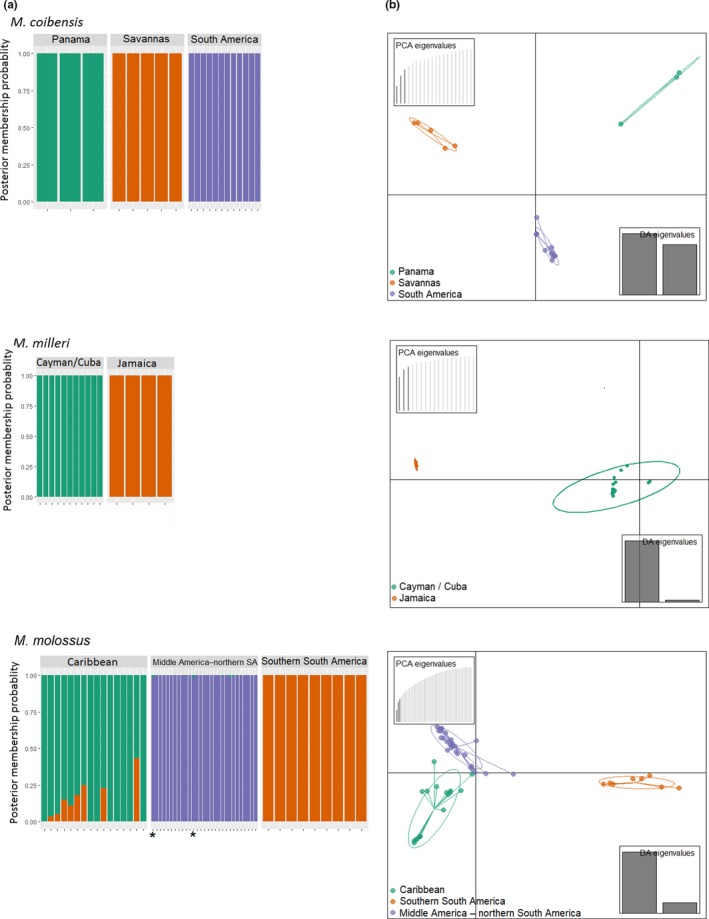
(a) Composite stacked bar plots under assumptions of K = 2 for M. milleri and K = 3 for M. coibensis and M. molossus. Each vertical bar along the x‐axis represents the genotype of an individual. Colors represent the haplotypes of each specimen. The y‐axis indicates the posterior probability of a genotype belonging to one or more clusters. * shows the individuals ROM125468 from Bonaire and TTU18551 from Grenada with migrant haplotypes. (b) Discriminant analysis of principal component (DAPC) with 95% confidence ellipses among populations within three species of Molossus: (a) Molossus coibensis, (b) M. milleri, and (c) M. molossus
The PCA and DAPC show similar patterns of pairwise genetic distance within each species (Figure 2b; Figures S5 and S6). The individuals of M. coibensis were separated into three main groups. The population from the savannas of Venezuela and Guyana appears more similar to Panama than to the other South American population in the PCA (Figure S5), but all three populations are equally distance in the DAPC space (Figure 2b). In M. molossus, samples were divided into three groups. The group from southern South America appears distinct, and the populations from Middle America–northern South America and from the Caribbean slightly overlap in the PCA but not in the DAPC (Figure 2b and Figure S5). In M. milleri, samples were divided into two distinct populations, one from Jamaica and the other from the Cayman Islands and Cuba (Figure 2b; Figure S5).
3.3. Phylogenetic relationships
The phylogenetic and distance trees generated for each species also support the genetic structure found in the other analyses, although there were minor differences in relationships within each population between the analyses (Figure 3; Figure S7). In M. coibensis, individuals were primarily clustered by geographic location, and the three main groups had 100% bootstrap support (Figure 3a; Figure S7a). The individuals from savannas in Guyana and Venezuela cluster together and then group with those from Panama. In the third group, samples from rainforest in French Guiana cluster with those from Peru and Ecuador.
Figure 3.
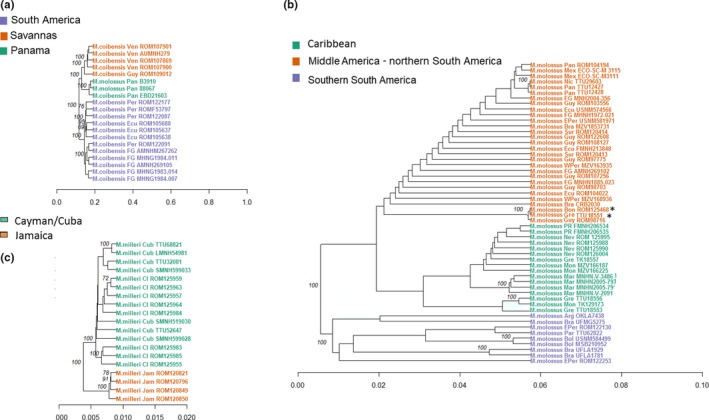
Distance trees of genetic similarity for three species of Molossus: (a) M. coibensis, (b) M. molossus, and (c) M. milleri. Nodes with greater than 90% bootstrap support are indicated. Colors represent population clusters, and * indicates samples where geographic location is different from population cluster
The phylogenetic and distance tree for M. molossus has three large clusters (Figure 3b; Figure S7b). The first group is composed of individuals from Middle America, northern South America, and from off‐shore islands (ROM125468 from Bonaire and TTU18551 from Grenada). The Caribbean group joined the Middle America–northern South America cluster, and grouped individuals from Puerto Rico and Lesser Antilles, including the other three samples from Grenada. The third group, comprising individuals from Argentina and southeastern Brazil, Paraguay, Bolivia, and western Peru, was an outlier to the two other groups. In the M. milleri trees, the Greater Antilles samples clustered into a Jamaica group and a Cayman + Cuba group (Figure 3c; Figure S7c).
3.4. Population diversity
The AMOVA results indicated significant genetic differentiation between each pair of populations within each analyzed species (p < .01; Table S1). Observed heterozygosity was lower than expected heterozygosity for all populations, except for M. molossus in the Middle America–northern South America population (Table 1). Designating a threshold of Fst > 0.2 as high population genetic differentiation (Frankham, Bradshaw, & Brook, 2014), pairwise comparisons of Fst between the southern South America and the Middle America–northern South America populations, and between the Caribbean and southern South America populations of M. molossus suggest some isolation (Table 1). Pairwise Fst was low between populations of M. coibensis, between the two populations of M. milleri, and between the Middle America–northern South America and the Caribbean populations of M. molossus, suggesting low levels of differentiation (Table 1). The estimated number of migrants was above one for all population pairs within species, except between M. molossus from southern South America and the Caribbean (0.699, Table 1).
Table 1.
Quantitat parameters of population structure for three species of Molossus: M. coibensis, M. molossus, and M. milleri
| Species | Fst | Nm | Population | Ho | He |
|---|---|---|---|---|---|
| M. coibensis | Panama × Savannas = 0.127 | Panama × Savannas = 3.437 | Panama | 0.34 (0.24) | 0.44 (0.11) |
|
Panama × SA = 0.180 Savannas × SA = 0.176 |
Panama × SA = 2.145 Savannas × SA = 2.341 |
Savannas | 0.28 (0.20) | 0.35 (0.14) | |
| SA | 0.11 (0.22) | 0.22 (0.11) | |||
| M. molossus |
Middle America–northern SA × Caribbean = 0.122 Middle America–northern SA × Southern SA = 0.300 Caribbean × Southern SA = 0.417 |
Middle America–northern South America × Caribbean = 3.598 Middle America–northern SA × Southern SA = 1.167 Caribbean × Southern SA = 0.699 |
Middle America–northern SA | 0.96 (0.05) | 0.51 (0.02) |
| Caribbean | 0.29 (0.31) | 0.36 (0.15) | |||
| Southern SA | 0.13 (0.15) | 0.38 (0.13) | |||
| M. milleri | 0.148 | 2.878 | Jamaica | 0.38 (0.34) | 0.52 (0.15) |
| Cuba/ Cayman Islands | 0.50 (0.16) | 0.85 (0.03) |
Numbers in parentheses represent the standard deviation of each value.
Abbreviations: Fst, pairwise fixation index; He, mean expected heterozygosity; Ho, mean observed heterozygosity; Nm, number of migrants between populations; SA, South America.
3.5. Demographic inferences
Likelihoods of each run in FastSimCoal under different demographic models were similar, indicating a convergence of the runs within a population and adequate searching of sample space. However, analyses suggest different demographic histories and qualitatively divergent parameter estimates among populations (Tables 2 and 3). The constant population size model was the best fit for M. coibensis from savannas and Panama and for M. molossus from southern South America. The South America population of M. coibensis and the Caribbean population of M. molossus had a higher likelihood value for the expansion model, but these values did not significantly differ from the constant model (Table 2). In contrast, the population expansion model was the best fit and significantly different from the constant model for M. molossus from the Middle America–northern South America population. The two groups of M. milleri had a different demographic history. For both populations of this species, the best fit model was the bottleneck, which was significantly different than the constant size model (Table 2).
Table 2.
LRT and AIC for demographic models describing populations of three species of Molossus: M. coibensis, M. molossus, and M. milleri
| Species | Population | Max ln L | Model | np | ln L | Null | LRT | df | p | AIC |
|---|---|---|---|---|---|---|---|---|---|---|
| M. coibensis | Panama | Constant | 1 | −1,418.02 | 10.3037 | |||||
| −1,176.198 | Expansion | 3 | −1,422.82 | Constant | 9.61 | 2 | .000 | 10.3063 | ||
| Bottleneck | 3 | −1,542.16 | Constant | 248.27 | 2 | .000 | 10.3997 | |||
| M. coibensis | Savannas | Constant | 1 | −502.121 | 9.4016 | |||||
| −354.110 | Expansion | 3 | −503.690 | Constant | 3.14 | 2 | .043 | 9.4065 | ||
| Bottleneck | 3 | −553.180 | Constant | 102.12 | 2 | .000 | 9.4857 | |||
| M. coibensis | South America | Constant | 1 | −156.000 | 8.3862 | |||||
| −148.583 | Expansion | 3 | −155.696 | Constant | 0.61 | 2 | .738 | 8.3845 | ||
| Bottleneck | 3 | −167.388 | Constant | 22.78 | 2 | .000 | 8.4474 | |||
| M. molossus | Middle America–northern South America | Constant | 1 | −504.119 | 9.4051 | |||||
| −498.010 | Expansion | 3 | −500.981 | Constant | 6.28 | 2 | .043 | 9.3996 | ||
| Bottleneck | 3 | −505.959 | Constant | 3.68 | 2 | .159 | 9.4082 | |||
| M. molossus | Caribbean | Constant | 1 | −501.756 | 9.4010 | |||||
| −344.215 | Expansion | 3 | −502.261 | Constant | 1.01 | 2 | .604 | 9.4019 | ||
| Bottleneck | 3 | −548.154 | Constant | 92.80 | 2 | .000 | 9.4778 | |||
| M. molossus | Southern South America | Constant | 1 | −1,312.743 | 10.2364 | |||||
| −1,269.769 | Expansion | 3 | −1,326.488 | Constant | 6.24 | 2 | .000 | 10.2454 | ||
| Bottleneck | 3 | −1,452.235 | Constant | 276.98 | 2 | .000 | 10.3241 | |||
| M. milleri | Cayman/Cuba | Constant | 1 | −28.577 | ||||||
| −16.184 | Expansion | 3 | −28.471 | Constant | 0.21 | 2 | .899 | 6.9081 | ||
| Bottleneck | 3 | −25.088 | Constant | 6.98 | 2 | .031 | 6.7989 | |||
| M. milleri | Jamaica | Constant | 1 | −380.054 | 9.1569 | |||||
| −349.914 | Expansion | 3 | −380.848 | Constant | 1.59 | 2 | .452 | 9.1594 | ||
| Bottleneck | 3 | −360.748 | Constant | 38.61 | 2 | .000 | 9.1157 |
Bold numbers indicate statistical significance (p < .05).
Abbreviations: df, degrees of freedom; ln L, ln likelihood; Max ln L, maximum log likelihood given the observed data; Np, number of parameters; Null, null model (constant model); p, probability that the bottleneck and expansion models are significantly different from the null model.
Table 3.
Mean values and confidence intervals (CI) of present population size, ancestral population size, and the number of generations for observed population change for three species of Molossus: M. coibensis, M. molossus, and M. milleri
| Species | Population | Population size | Ancestral population size | Number of generations | |
|---|---|---|---|---|---|
| M. coibensis | Panama | Lower CI | 65,360 | 65,360 | 0 |
| Mean | 66,845 | 66,845 | 0 | ||
| Higher CI | 67,617 | 67,617 | 0 | ||
| M. coibensis | Savannas | Lower CI | 59,037 | 59,037 | 0 |
| Mean | 62,774 | 62,774 | 0 | ||
| Higher CI | 65,327 | 65,327 | 0 | ||
| M. coibensis | South America | Lower CI | 65,739 | 65,739 | 0 |
| Mean | 67,421 | 67,421 | 0 | ||
| Higher CI | 69,216 | 69,216 | 0 | ||
| M. molossus | Middle America–northern South America | Lower CI | 67,883 | 49,033 | 67,981 |
| Mean | 67,992 | 49,037 | 100,540 | ||
| Higher CI | 68,158 | 49,495 | 123,170 | ||
| M. molossus | Caribbean | Lower CI | 44,249 | 44,249 | 0 |
| Mean | 57,260 | 57,260 | 0 | ||
| Higher CI | 69,301 | 69,301 | 0 | ||
| M. molossus | Southern South America | Lower CI | 63,466 | 63,466 | 0 |
| Mean | 66,243 | 66,243 | 0 | ||
| Higher CI | 70,550 | 70,550 | 0 | ||
| M. milleri | Cayman/Cuba | Lower CI | 14,916 | 65,978 | 163,714 |
| Mean | 27,411 | 67,921 | 298,996 | ||
| Higher CI | 42,250 | 69,169 | 442,312 | ||
| M. milleri | Jamaica | Lower CI | 3,155 | 65,241 | 148,549 |
| Mean | 3,791 | 66,633 | 184,471 | ||
| Higher CI | 4,578 | 68,013 | 206,784 |
Populations of M. coibensis appeared to have a constant effective population size through time (Table 3). The populations of M. molossus from southern South America and the Caribbean also did not appear to vary with time (Table 3). The best fit model assumes that the current population sizes of these groups did not change significantly over time and are the same as the ancestral population sizes. However, the M. molossus group from Middle America–northern South America increased about 14% in the last ~100 generations. The populations of M. milleri had the lowest current population size of any Molossus, especially for Jamaica. A bottleneck occurred earlier in the population from Cayman and Cuba in comparison with the group from Jamaica and had reduced population sizes of 40% and 56%, respectively (Table 3).
4. DISCUSSION
We examined three species of the mastiff bat (Molossus) with distinct geographic and ecological distribution patterns and found that their population structures have a low admixture of haplotypes and vary with habitat preferences, level of population isolation, and historical fluctuations in climate, resulting in mainland and insular species having different phylogeographic patterns. Mainland geographic barriers such as the Andes did not correspond to lineage breaks, except for the Amazon River, which acts as a filter barrier for M. molossus and also correlates with differences between rainforest and savanna habitats. Oceanic straits pose a partial barrier for some bats but not others, isolating populations of M. milleri between islands in the Greater Antilles, but with gene flow occurring among island populations of M. molossus within the Lesser Antilles. M. molossus populations from the mainland are distinct from those in the Lesser Antilles, indicating a degree of isolation. However, migrants from the continent found in the archipelago (such as Grenada) demonstrate ongoing gene flow between these two regions. For this group of bats, oceanic straits appear to act as a partial, filter barrier to dispersal. Our dataset also is consistent with the expectation that more isolated lineages on islands undergo genetic bottlenecks more frequently than lineages closer to the mainland.
4.1. Mainland populations
Geographic barriers in the Neotropics can disrupt dispersal in some birds (Weir, Faccio, Pulido‐Santacruz, Barrera‐Guzmán, & Aleixo, 2015; Weir & Price, 2011) and bats (Cuadrado‐Ríos & Mantilla‐Meluk, 2016; Dias, Santos Júnior, Perini, & Santos, 2017). The two species of bats examined in this study that occur in the mainland, M. coibensis and M. molossus, show little concordance between genetic structure and geological barriers. For example, both species fail to show phylogeographic structuring associated with the Andes or the Panamanian land bridge, and haplotypes are widespread across these potential geographic barriers. An exception is found within M. molossus, wherein two distinct groups are for the most part separated by the Amazon River, other than samples from Pernambuco, south of the Amazon River in northeastern Brazil, which group within the Middle America–northern South America population. The riverine barrier hypothesis proposes that rivers act as a barrier to gene flow, promoting divergence between populations on opposite banks (Wallace, 1852). The Amazon River in particular, which originated during the Miocene and attained its present course during the Pliocene, is thought to have contributed to allopatric speciation and population differentiation in many taxa (Baker et al., 2014; Nazareno, Dick, & Lohmann, 2017). Indeed, Amazonian rivers seem to be acting as dispersal barriers to several taxa of volant (Hayes & Sewlal, 2004) and nonvolant animals (Ayres & Clutton‐Brock, 1992; Bonvicino, Lindbergh, Faria, & Bezerra, 2012; Valdez & D'Elía, 2013). Conversely, rivers might not always act as barriers to gene flow in bats or small mammals. Many groups of bats have been reported to use smaller rivers as landmarks for orientation and migration pathways (Furmankiewicz & Kucharska, 2009; Serra‐Cobo, Lopez‐Roig, Marqués‐Bonet, & Lahuerta, 2000), and as preferred foraging habitats (Smith & Racey, 2008), suggesting that rivers might also act as dispersal corridors. In addition, the genetic patterns of some terrestrial small mammals do not corroborate the riverine hypothesis, with population substructure more pronounced along the length of the margins of the rivers, rather than between opposite sides (Patton & Da Silva, 1998; Patton, Silva, & Malcolm, 1994). In M. molossus, the Amazon River appears to act as a partial barrier to gene flow, despite the vagility of this bat, in accord with the riverine barrier hypothesis.
An additional explanation is that ecological factors also likely play an important role in population patterning. The division between these two populations of M. molossus is consistent with the transition between two large biomes in South America. The Middle America–northern South America population comprises individuals primarily distributed in tropical and subtropical rainforests, whereas the southern South America population inhabits mostly savannas, seasonal tropical forest, and agricultural landscapes. Exceptions are samples from eastern Peru, which occupy tropical forest. This distinction is supported by genetic differentiation based on the fixation index, whereby the southern South American population has higher Fst values in pairwise comparisons to the two other populations of M. molossus. Some groups of rodents and marsupials have a similar distribution and are restricted to each of these different habitats (Almeida, Bonvicino, & Cordeiro‐Estrela, 2007; Voss, 1991). Within each M. molossus lineage from the mainland, there is low intra‐population genetic divergence, a lack of obvious phylogeographic structure, and high levels of gene flow. This pattern is perhaps not surprising in a species with high dispersal abilities (Burns & Broders, 2014; Norberg & Rayner, 1987; Speer et al., 2017; Taylor et al., 2012).
Molossus coibensis was structured as three populations from: (a) tropical forests in Panama; (b) tropical forests of northern South America; and (c) savannas of Venezuela and Guyana. The Amazonian region of South America is composed primarily of rainforest, but patches of savanna are distributed from Venezuela to Suriname and southern Brazil (Haffer, 2002; Vanzolini & Williams, 1970). Repeated cycles of expansion and fragmentation of savannas in the Neotropics occurred most recently in the last 2 My, due to changes in temperature and sea level (Bennett, 1990), promoting opportunities for periodic connection and isolation between populations. Previous studies have reported the importance of the Amazonian savannas in the divergence of lineages of rodents (Bonvicino, Gonalves, Oliveira, Oliveira, & Mattevi, 2009), bats (Lim, 2010), lizards (Gainsbury & Colli, 2003), and birds (Naka, Cohn‐Haft, Mallet‐Rodrigues, Santos, & Torres, 2006). The specimens of the savanna population of M. coibensis were collected in the Llanos of Venezuela and Rupununi of Guyana, which are separated by a large region of rainforest (800 km) and by the Guiana Highland plateau (Lim & Lee, 2018). Although these geographic barriers could potentially decrease gene flow between these two savanna regions, other populations of different species of bats from the families Phyllostomidae, Vespertilionidae, Emballonuridae, and Molossidae are also united in phylogeographic analyses (Lim & Lee, 2018). These savanna regions were likely connected in the recent past (Sarmiento, 1983), and the distributional patterns observed today are the result of sundering of a common paleoenvironment rather than long‐distance dispersal across large expanses of Amazonian forest (Da Silva & Bates, 2002; Haffer, 1997; Sarmiento, 1983).
Previous studies based on mitochondrial and nuclear genes have reported that the population of M coibensis from the savannas formed a distinct clade relative to M. coibensis from forests of South and Middle America, suggesting that this population might belong to a putative new species (Lim & Engstrom, 2001; Lim & Lee, 2018; Loureiro et al., 2019). Although this population is genetically distinctive, it has a low degree of isolation with a high number of immigrants per generation (Table 1). Therefore, our data suggest that there is gene flow among these structured populations of M. coibensis, and the savanna population likely does not represent a distinct species.
4.2. Caribbean populations
Koopman (1977) and Genoways (1998) hypothesized that oceanic straits act as migration barriers in the Caribbean, which may result in low rates of gene flow among islands, and serve to isolate populations. In the Caribbean, M. molossus and M. milleri had distinct patterns of population differentiation, with evidence of oceanic straits acting as barriers to gene flow within M. milleri from the Greater Antilles, but not within M. molossus from the Lesser Antilles. The two populations of M. milleri are distinctive, but our analysis shows that there is some gene flow between Jamaica and Cayman/Cuba, and isolation is incomplete. Similarly, Muscarella, Murray, Ortt, Russell, and Fleming (2011), Carstens et al. (2004), and Larsen et al. (2012) found varying population structuring in different species of phyllostomid bats in the Caribbean, ranging from some species having monophyletic populations confined to individual islands, and other species lacking any genetic structuring among islands. Similar patterns have also been observed in other groups of volant animals, such as butterflies (Davies & Bermiham., 2001) and birds (Khimoun et al., 2016).
Two individuals from off‐shore islands (Bonaire and Grenada) had higher genetic similarity with the Middle America–northern South America group than to the Caribbean population. Bonaire is located approximately 80 km off the coast of Venezuela, and geologically considered a part of the continent, and Grenada is located about 160 km north of Trinidad, but geologically considered part of the Lesser Antilles. Due to the proximity of both islands to South America, they share similar fauna and flora with the mainland (Baker & Genoways, 1978), and these results are not unexpected, especially for Bonaire. However, some specimens from Grenada shared more haplotype similarity with other Lesser Antilles populations and clustered within the Caribbean group. These results indicate that there are two different haplotypes in Grenada, but because only one of our specimens from this island grouped within the Middle America–northern South America population, it suggests at least infrequent dispersal from the mainland (Pedersen et al., 1996). Speer et al. (2017) also reported mixed mainland/islands populations of the bat Tadarida brasiliensis in the Bahamas. The authors suggested that this pattern may have derived from independent colonization of the archipelago by divergent mainland lineages and that the genetic structure found is not due to isolation by oceanic barriers. Carstens et al. (2004) also reported distinct higher levels of genetic diversity among some populations of bats within individual Lesser Antilles islands, such as Montserrat, Nevis, and St. Kitts, and also suggested that this pattern could be derived from multiple colonization.
Some of the Caribbean islands are very small (e.g., Cayman Brac—19 km long), and in some islands, Molossus was only caught in one or a few nearby geographic locations. Therefore, it was not possible to test for differences in gene flow among populations within individual islands compared with gene flow between different islands separated by similar geographic distance. However, in the islands where we could examine potential population differentiation, no population structuring was found within the same island, except for Grenada, which suggests lower genetic variation and higher gene flow within as compared to among different islands or between island and mainland populations.
4.3. Demographic histories
All mainland populations showed a constant or expanding effective population size through time, whereas the isolated Caribbean populations showed a higher probability of experiencing bottlenecks. These results support the hypothesis that island populations are more susceptible to bottlenecks than their continental relatives (Luikart, Sherwin, Steele, & Allendorf, 1998). Bottlenecks might cause heterozygosity deficiency in natural populations (Nei and Graur, 1984), decrease the genetic diversity of a population through random genetic drift (Groombridge, Jones, Bruford, & Nichols, 2000), reduce reproductive function (Madsen, Shine, Olsson, & Wittzell, 1999), and be involved in the speciation process (Mayr, 1963). Population persistence is highly connected to its evolutionary potential, which is enhanced by genetic variation (Frankel & Soulém, 1981; Newman & Pilson, 1997). Thus, level of genetic variance has direct implications for conservation management (Bouzat, 2010), especially in determining the minimum viable sizes of wild populations (Lande, 1988). These estimates may be important in a conservation context and can indicate if a population has the potential to undergo inbreeding depression or has the genetic breadth to adapt to future environmental changes (Sovic et al., 2016).
The recent divergence times associated with population expansion and bottlenecks among different lineages suggest that historical changes in climate during the Pleistocene affected the present phylogeographic patterns in these bats. M. coibensis showed no change in effective population size over time in its three populations. However, the other two species had a more complex demographic history. In M. molossus from the mainland, the lineage from southern South America showed no evidence of change in population size through time and the Middle America–northern South America population was a best fit expansion demographic model. Our analysis suggests that effective population size of the Middle America–northern South America lineage has increased about 14% over the last ~100k generations. Pacifici et al. (2013) estimated the generation length for Molossus as approximately 3.9 years. Using this estimation, M. molossus from Middle America–northern South America started to expand about 392 ka years ago. This period coincides with the middle Pleistocene, characterized by interglacial conditions possibly interrupted by climatic events associated with small‐scale glaciations (Rabassa and Clapperton, 1990). It has been hypothesized that a rise in global temperature 450 and 600 ka ago produced the longest and warmest interglacial episode during the Pleistocene (Verzi, Deschamps, & Tonni, 2004), which resulted in a distributional expansion of several species (Kozma, Melsted, Magnusson, & Hoglund, 2016; Lambeck, Yokoyama, & Purcell, 2002; Martizez‐Freiria, Velo‐Anton, & Brito, 2015; Vrba, 1985).
Species in the Caribbean showed contrasting patterns of demographic history. Both populations of M. milleri (from Jamaica, and Cuba and Cayman Islands) closely fit the bottleneck model, and the population of M. molossus from the Lesser Antilles showed a higher likelihood for the constant population size model. Considering the same generation length of 3.9 years estimated for M. molossus and for other species of the genus (Pacifici et al., 2013), the effective population size decline in M. milleri started around 1.1 mya in the group from Cuba and the Cayman Islands, and about 719 ka in the group from Jamaica. These dates correspond to the early and beginning of the middle Pleistocene, respectively, which was characterized by several cycles of glacial and interglacial climates (Raymo, Ganley, Carter, Oppo, & McManus, 1998). These cycles likely caused fluctuations in temperature and sea level, resulting in the extinction of some Caribbean bat species during the late Pleistocene (Dávalos & Russell, 2012; Morgan, 2001). Therefore, we hypothesize that climate change in the early Pleistocene likely generated ecological changes and repetitive fluctuations in size of island landmasses, affecting population sizes of these bats. However, the confidence intervals of ancestral and extant effective population sizes overlapped within the two populations of M. milleri and caution is needed in the interpretation of these data. Two alternative scenarios, the anthropogenic colonization during the Quaternary (Valente, Etienne, & Dávalos, 2017) and the Holocene last glacial–interglacial transition (Soto‐Centeno & Steadman, 2015), have also been proposed to explain the extinction of bats in the Caribbean and show that fluctuations in population sizes might be more complex than previously thought.
The differences in demographic history among Caribbean lineages of Molossus might be explained by historical biogeography and current degree of isolation. M. milleri is restricted to the Greater Antilles, with no apparent connection to the mainland. In contrast, M. molossus from the Lesser Antilles receives migrants from northern South America, and mainland and island populations exhibit low levels of population differentiation. For example, the two specimens from Bonaire and Grenada with mainland haplotypes are indicative of gene flow between the areas. This haplotype exchange might have resulted from accidental and isolated events or indicates that the continent is acting as a source population of migrants to the Caribbean population, in particular for Bonaire because of its proximity to Venezuela. Populations that are isolated on islands might decrease drastically in size due to environmental changes and catastrophic events (Pedersen, 1998; Pedersen et al., 1996), and genetic drift in these small populations may result in the loss or decrease of some allele frequencies (Keller et al., 2001). Increased levels of immigration can lead to very different genetic outcomes from those expected in isolated populations. Island lineages with immigration sources can recover much faster from a bottleneck than isolated populations, despite increases in average inbreeding on islands (Keller et al., 2001).
Buerlke and Gompert (2012) found that population allele frequencies were more variable for smaller samples of individuals (2–10), which could potentially affect results in both populations structuring and demographic analyses. However, Pluzhnikov and Donnelly (1996) and Beerli (2004) argue that the effect of small sample size on the estimates of population structuring and gene flow is minimum, except that the confidence intervals are somewhat larger with fewer individuals. Nadeau et al. (2011) proposed that a deeper genetic coverage could ameliorate the effect of a small number of individuals. In addition, other studies with next generation sequencing have conducted similar population genetic and demographic analyses using different population sizes (from 5 to 27 individuals) and found consistent results (Sovic, Fries, Martin, & Gibbs, 2018). Robison, Coffman, Hickerson, and Gutenkunst (2014) also found accurate parameter and demographic estimates for populations with more ancient demographic events (in the order of 0.5Ne generations ago) in small numbers of sampled individuals. In our data, when the models that included changes in population size had a higher likelihood, all the events (bottlenecks or expansions) occurred more than 0.5Ne generations ago, corroborating with Robison et al. (2014).
Our study of phylogeographic patterns in mainland and island populations in a group of highly mobile bats (Molossus) found that the Amazon River and ecological habitats (rainforest and savanna), but not the Andes Mountains, have an effect on the genetic structuring of M. molossus in South America. By contrast, oceanic barriers in the Greater Antilles play a role in isolation of some species (M. milleri), and that these isolated populations are more subject to bottlenecks and therefore vulnerable to environmental change. We expect these patterns to be even more pronounced in populations of bats with lower dispersal abilities, such as fruit and nectar feeding phyllostomids. Demographic research on the bat fauna as a whole would provide important information relevant to biological conservation in the Caribbean as climate change and environment vulnerability accelerate.
CONFLICT OF INTEREST
None declared.
AUTHORS CONTRIBUTION
L. Loureiro, M. Engstrom, and B. Lim collected the data through field trips in several Neotropics countries. L. Loureiro conducted sequencing and analyses. M. Engstrom and B. Lim verified the analytical methods and supervised the findings of this work. All authors contributed to the interpretation of the results. L. Loureiro wrote the manuscript with support from M. E. and B. Lim.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes). Neotropical fieldwork has been primarily funded by the Royal Ontario Museum with additional financial support in Ecuador by Ecuambiente Consulting Group and in Guyana by Conservation International and funding through the Academy of Natural Sciences, Philadelphia. We thank the following curators and collection support staff that provided access or loaned specimens: R. Gregorin (UFLA), F. A. Perini (UFMG), B. D. Patterson (FNMH), C. J. Conroy (MVZ), M. Campbell (MSB), B. S. Coyner (Sam Noble Museum), N. B. Simmons (AMNH), H. J. Garner (TTU), C. Lopez‐Gonzalez (Instituto Politécnico Nacional, Mexico City), J. Juste (CSIC), A. L. Gardner (NMNH/USMN), M. de Vivo and J. G. Barros (MZUSP), C. G. Costa (MCN‐PUC Minas), G. Graciolli and M. Bordignon (UFMS), E. Morielle‐Versute (UNESP), L. Peracchi (UFRRJ), and J. A. Oliveira (MNRJ). We also thank Oliver Haddrath for providing constructive feedback on this manuscript.
APPENDIX 1.
SPECIMEN VOUCHERS, SPECIES IDENTIFICATION, COUNTRY OF ORIGIN, LOCALITY, AND COORDINATES FOR MOLOSSUS USED IN THE GENETIC ANALYSES
| Sample name | Genus | Species | Country | Locality | Latitude | Longitude |
|---|---|---|---|---|---|---|
| ROM 105,638 | Molossus | coibensis | Ecuador | Napo | 39.000 | −76.270 |
| ROM 105,637 | Molossus | coibensis | Ecuador | Napo | 39.000 | −76.270 |
| ROM 105,688 | Molossus | coibensis | Ecuador | Napo | 3.900 | −76.270 |
| MHNG 19,884.011 | Molossus | coibensis | French Guiana | Cacao | 4.568 | −52.468 |
| MNHG 19,883.014 | Molossus | coibensis | French Guiana | Cacao | 4.568 | −52.468 |
| MHNG 1984.007 | Molossus | coibensis | French Guiana | Kaw | 4.550 | −52.167 |
| AMNH 267,262 | Molossus | coibensis | French Guiana | Cayenne | 3.9332 | −53.088 |
| AMNH 269,105 | Molossus | coibensis | French Guiana | Cayenne | 3.9332 | −53.088 |
| ROM 119,012 | Molossus | coibensis | Guyana | Dadanawa Ranch | 2.4927 | −59.313 |
| ROM 53,797 | Molossus | coibensis | Peru | Loreto | 4.899 | −73.650 |
| PAN 88,067 | Molossus | coibensis | Panama | Gamboa | 9.116 | −79.699 |
| PAN B3910 | Molossus | coibensis | Panama | Gamboa | 9.116 | −79.699 |
| EBD 21,603 | Molossus | coibensis | Panama | Gamboa | 9.116 | −79.699 |
| ROM 122,087 | Molossus | coibensis | Peru | Loreto | 4.899 | −73.650 |
| ROM 122,091 | Molossus | coibensis | Peru | Loreto | 4.899 | −73.650 |
| ROM 122,177 | Molossus | coibensis | Peru | Loreto | 4.899 | −73.650 |
| ROM 107,901 | Molossus | coibensis | Venezuela | Amazonas | 6.030 | −67.250 |
| ROM 107,900 | Molossus | coibensis | Venezuela | Amazonas | 6.030 | −67.250 |
| ROM 107,869 | Molossus | coibensis | Venezuela | Amazonas | 6.030 | −67.250 |
| AUMNH 279 | Molossus | coibensis | Venezuela | Amazonas | 6.030 | −67.250 |
| ROM 125,985 | Molossus | milleri | Cayman Islands | Cayman Brac | 19.699 | −79.856 |
| ROM 125,983 | Molossus | milleri | Cayman Islands | Cayman Brac | 19.699 | −79.856 |
| ROM 125,955 | Molossus | milleri | Cayman Islands | Grand Cayman | 19.314326 | −81.169 |
| ROM 125,963 | Molossus | milleri | Cayman Islands | Grand Cayman | 19.332 | −81.104 |
| 125,957 ROM | Molossus | milleri | Cayman Islands | Grand Cayman | 19.314 | −81.169 |
| ROM 125,959 | Molossus | milleri | Cayman Islands | Grand Cayman | 19.271 | −81.281 |
| ROM 125,984 | Molossus | milleri | Cayman Islands | Cayman Brac | 19.699 | −79.856 |
| ROM 125,964 | Molossus | milleri | Cayman Islands | Grand Cayman | 19.332 | −81.104 |
| SMNH 59,028 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| SMNH 519,030 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| SMNH 599,033 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| LMNH 54,981 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| TTU 52,647 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| TTU 32,081 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| TTU 68,821 | Molossus | milleri | Cuba | Guantanamo Bay | 19.900 | −75.150 |
| TTU 22,195 | Molossus | milleri | Jamaica | St. Ann | 18.436 | −77.201 |
| ROM 120,849 | Molossus | milleri | Jamaica | Portland | 18.14347 | −76.373 |
| ROM 120,796 | Molossus | milleri | Jamaica | Saint Elizabeth | 18.227 | 77.75446 |
| ROM 120,821 | Molossus | milleri | Jamaica | Portland | 18.14347 | −76.373 |
| ROM 120850 | Molossus | milleri | Jamaica | Portland | 18.14347 | −76.373 |
| ROM 120798 | Molossus | milleri | Jamaica | Saint Elizabeth | 18.227 | 77.75446 |
| OKLA 7,438 | Molossus | molossus | Argentina | Jujuy | −32.600 | −63.883 |
| TTU 151411 | Molossus | molossus | Barbados | St Thomas Parish | 13.194 | −59.543 |
| USNM 584,499 | Molossus | molossus | Bolivia | Santa Cruz | −17.786 | −63.181 |
| MSB 210,952 | Molossus | molossus | Bolivia | Santa Cruz | −17.786 | −63.181 |
| ROM 125,468 | Molossus | molossus | Bonaire | Playa Frans | 12.202 | −68.262 |
| MZV 1,853,731 | Molossus | molossus | Brazil | Pernambuco | −8.472 | −37.947 |
| UFLA 1781 | Molossus | molossus | Brazil | Minas Gerais | −19.717 | −42.750 |
| CRB 2030 | Molossus | molossus | Brazil | Not provided | Not provided | Not provided |
| UFLA 1929 | Molossus | molossus | Brazil | Minas Gerais | −19.717 | −42.750 |
| UFMG 5,275 | Molossus | molossus | Brazil | Minas Gerais | −18.919 | −48.277 |
| ROM 104,022 | Molossus | molossus | Ecuador | Napo | −3.990 | −76.270 |
| FMNH 213,848 | Molossus | molossus | Ecuador | Tiguino | −0.462 | −76.993 |
| USNM 574,566 | Molossus | molossus | Ecuador | Orellana | −0.466 | −76.987 |
| AMNH 269,102 | Molossus | molossus | French Guiana | Cayenne | −3.933 | −53.088 |
| MHNG 1885.023 | Molossus | molossus | French Guiana | Awala‐Yalimapo | −5.7411 | −53.928 |
| MHNH 2004‐356 | Molossus | molossus | French Guiana | Angoulème | −5.400 | −53.650 |
| MHNG 1972‐021 | Molossus | molossus | French Guiana | Cacao | −4.568 | −52.468 |
| TTU 18,551 | Molossus | molossus | Grenada | St George | 12.056 | −61.748 |
| TTU 18,553 | Molossus | molossus | Grenada | St George | 12.056 | −61.748 |
| TTU 18,556 | Molossus | molossus | Grenada | St George | 12.056 | −61.748 |
| TTU 18,557 | Molossus | molossus | Grenada | St George | 12.056 | −61.748 |
| ROM 97,775 | Molossus | molossus | Guyana | Upper Takutu‐Upper Essequibo | −3.230 | −59.480 |
| ROM 98,716 | Molossus | molossus | Guyana | Barima‐Waini | −7.340 | −59.090 |
| ROM 122,608 | Molossus | molossus | Guyana | Upper Takutu‐Upper Essequibo | −2.182 | −59.337 |
| ROM 107,256 | Molossus | molossus | Guyana | Potaro‐Siparuni | −4.400 | −58.410 |
| ROM 103,556 | Molossus | molossus | Guyana | Upper Demerara‐Berbice | −5.180 | −58.420 |
| ROM 98,703 | Molossus | molossus | Guyana | Barima‐Waini | −7.340 | −59.090 |
| ROM 108,127 | Molossus | molossus | Guyana | Cuyuni‐Mazaruni | −5.520 | −60.370 |
| MHNH 2005‐791 | Molossus | molossus | Martinique | Le Precheur | 14.800 | −61.217 |
| MHNH 2055‐793 | Molossus | molossus | Martinique | Le Precheur | 14.800 | −61.217 |
| MHNH V‐2091 | Molossus | molossus | Martinique | Morne Rouge | 14.767 | −61.133 |
| MNHN/V3486 | Molossus | molossus | Martinique | Morne Rouge | 14.767 | −61.133 |
| ECO‐SC‐M 3,111 | Molossus | molossus | Mexico | Tabasco | 17.800 | −91.533 |
| ECO‐SC‐M 3,115 | Molossus | molossus | Mexico | Tabasco | 17.800 | −91.533 |
| ECO‐SC‐M 5675 | Molossus | molossus | Mexico | Quintana Roo | 19.578 | −88.045 |
| MZV 166,187 | Molossus | molossus | Montserrat | Belham River | 16.749 | −62.193 |
| MZV 166,225 | Molossus | molossus | Montserrat | Belham River | 16.749 | −62.193 |
| TTU 129,173 | Molossus | molossus | Montserrat | Belham River | 16.749 | −62.193 |
| TTU 151275 | Molossus | molossus | St. Lucia | Castries | 13.996 | −61.006 |
| ROM 125,988 | Molossus | molossus | Nevis | Newcastle | 17.191 | −62.586 |
| ROM 125,990 | Molossus | molossus | Nevis | Newcastle | 17.196 | −62.596 |
| ROM 125,995 | Molossus | molossus | Nevis | Newcastle | 17.196 | −62.596 |
| ROM 126,004 | Molossus | molossus | Nevis | Pond Hill | 17.124 | −62.593 |
| TTU 29,603 | Molossus | molossus | Nicaragua | Rivas | 11.470 | −86.125 |
| ROM 104,194 | Molossus | molossus | Panama | Gamboa | 9.060 | −79.420 |
| TTU 12,427 | Molossus | molossus | Panama | Chiriqui | 8.517 | −82.617 |
| TTU 12,428 | Molossus | molossus | Panama | Chiriqui | 8.517 | −82.617 |
| TTU 62,822 | Molossus | molossus | Paraguay | Boqueron | −22.452 | −62.348 |
| TTU 129172 | Molossus | molossus | Paraguay | Boqueron | −22.452 | −62.348 |
| ROM 122,253 | Molossus | molossus | Peru | Loreto | −9.899 | −73.650 |
| MZV 163,935 | Molossus | molossus | Peru | Loreto | −9.899 | −73.650 |
| ROM 122256 | Molossus | molossus | Peru | Loreto | −9.899 | −73.650 |
| MZV 168,936 | Molossus | molossus | Peru | Lambayerque | −6.700 | −79.900 |
| LMNH 7545 | Molossus | molossus | Peru | Amazonas | −9.825 | −77.948 |
| USNM 581,971 | Molossus | molossus | Peru | Cordillera Del Condor | −4.095 | −78.393 |
| ROM 122,131 | Molossus | molossus | Peru | Loreto | −9.899 | −73.650 |
| FMNH 206,534 | Molossus | molossus | Puerto Rico | Vieques Island | 18.125 | −65.442 |
| FMNH 206,535 | Molossus | molossus | Puerto Rico | Vieques Island | 18.125 | −65.442 |
| TTU 151296 | Molossus | molossus | St. Lucia | Castries | 13.996 | −61.006 |
| ROM 120,414 | Molossus | molossus | Suriname | Sipaliwini | −2.026 | −56.124 |
| ROM 120,413 | Molossus | molossus | Suriname | Sipaliwini | −2.026 | −56.124 |
Samples that were excluded due to high missing data.
Loureiro LO, Engstrom MD, Lim BK. Comparative phylogeography of mainland and insular species of Neotropical molossid bats (Molossus). Ecol Evol. 2020;10:389–409. 10.1002/ece3.5903
DATA AVAILABILITY STATEMENT
VCF files with SNP data are available at https://doi.org/10.5061/dryad.d7wm37pxc
REFERENCES
- Adam, R. A. & Pedersen, S. C. (2013). Bat Evolution, Ecology, and Conservation (pp. 1–547). Switzerland, AG: Springer Nature. [Google Scholar]
- Almeida, F. C. , Bonvicino, C. R. , & Cordeiro‐Estrela, P. (2007). Phylogeny and temporal diversification of Calomys (Rodentia, Sigmodontinae): Implications for the biogeography of an endemic genus of the open/dry biomes of South America. Molecular Phylogenetics and Evolution, 42, 449–466. 10.1016/j.ympev.2006.07.005 [DOI] [PubMed] [Google Scholar]
- Ayres, J. M. , & Clutton‐Brock, T. H. (1992). River boundaries and species range size in Amazonian primates. The American Naturalist, 140, 531–537. 10.1086/285427 [DOI] [PubMed] [Google Scholar]
- Baker, P. A. , Fritz, S. C. , Dick, C. W. , Eckert, A. J. , Horton, B. K. , Manzoni, S. , … Battisti, D. S. (2014). The emerging field of geogenomics: Constraining geological problems with genetic data. Earth Science Reviews, 135, 38–47. 10.1016/j.earscirev.2014.04.001 [DOI] [Google Scholar]
- Baker, R. J. , & Genoways, H. H. (1978). Zoogeography of antillean bats. Academy of Natural Sciences of Philadelphia, 13, 53–97. [Google Scholar]
- Barquez, R. M. , Mares, M. A. , & Braun, J. K. (1999). The bats of Argentina. Special Publications, the Museum, Texas Tech University, 42, 1–275. [Google Scholar]
- Barton, N. H. , & Slatkinf, M. (1986). A Quasi‐equilibrium theory of the distribution of rare alleles in a. Genetics, 56, 409–415. [DOI] [PubMed] [Google Scholar]
- Beerli, P. (2004). Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Molecular Ecology, 13(4), 827–836. [DOI] [PubMed] [Google Scholar]
- Bender, M. L. , Fairbanks, R. G. , Taylor, F. W. , Matthews, R. K. , Goddard, J. G. , & Broecker, W. S. (1979). Uranium series dating of the pleistocene reef tracts of Barbados, West Indies. Geological Society of America Bulletin, 90(6), 577–594. 10.1130/0016-7606(1979)90<577:UDOTPR>2.0.CO;2 [DOI] [Google Scholar]
- Bennett, K. D. (1990). Paleontological society milankovitch cycles and their effects on species in ecological and evolutionary time. Paleobiology, 16, 11–21. [Google Scholar]
- Bernatchez, L. , & Wilson, C. C. (1999). Diadromy and genetic diversity in Neartic and Palearctic fishes: A reply. Molecular Ecology, 8, 527–528. [Google Scholar]
- Bonvicino, C. R. , Gonalves, P. R. , De Oliveira, J. A. , De Oliveira, L. F. B. , & Mattevi, M. S. (2009). Divergence in zygodontomys (Rodentia: Sigmodontinae) and distribution of Amazonian Savannas. Journal of Heredity, 100, 322–328. 10.1093/jhered/esn105 [DOI] [PubMed] [Google Scholar]
- Bonvicino, C. R. , Lindbergh, S. M. , Faria, M. B. , & Bezerra, A. M. R. (2012). The Eastern boundary of the Brazilian cerrado: A hotspot region. Zoological Studies, 51, 1207–1218. [Google Scholar]
- Bouzat, J. L. (2010). Conservation genetics of population bottlenecks: The role of chance, selection, and history. Conservation Genetics, 11(2), 463–478. 10.1007/s10592-010-0049-0 [DOI] [Google Scholar]
- Bradbury, P. J. , Zhang, Z. , Kroon, D. E. , Casstevens, T. M. , Ramdoss, Y. , & Buckler, E. S. (2007). TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. 10.1093/bioinformatics/btm308 [DOI] [PubMed] [Google Scholar]
- Buerlke, C. A. , & Gompert, Z. (2012). Population genomics based on low coverage sequencing: How low should we go? Molecular Ecology, 22(11), 3028–3035. [DOI] [PubMed] [Google Scholar]
- Burns, L. E. , & Broders, H. G. (2014). Correlates of dispersal extent predict the degree of population genetic structuring in bats. Conservation Genetics, 15, 1371–1379. 10.1007/s10592-014-0623-y [DOI] [Google Scholar]
- Čandek, K. , Agnarsson, I. , Binford, G. J. , & Kuntner, M. (2018). Caribbean golden orbweaving spiders maintain gene flow with North America. Preprint. BioRxiv. 10.1101/454181 [DOI] [Google Scholar]
- Carstens, B. C. , Sullivan, J. , Davalos, L. M. , Larsen, P. A. , & Pedersen, S. C. (2004). Exploring population genetic structure in three species of Lesser Antillean bats. Molecular Ecology, 13, 2557–2566. 10.1111/j.1365-294X.2004.02250.x [DOI] [PubMed] [Google Scholar]
- Chifman, J. , & Kubatko, L. (2015). Identifiability of the unrooted species tree topology under the coalescent model with time‐reversible substitution processes, site‐specific rate variation, and invariable sites. Journal of Theoretical Biology, 374, 35–47. 10.1016/j.jtbi.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Clapperton, C. M. (1990). Quaternary glaciations in the southern hemisphere: An overview. Quaternary Science Reviews, 9, 299–304. 10.1016/0277-3791(90)90024-5 [DOI] [Google Scholar]
- Cockerham, C. C. (1973). Analyses of gene frequencies of mates. Genetics, 74, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockrum, E. L. (1969). Migration in the guano bat, Tadarida brasiliensis . Miscellaneous Publication (University of Kansas Museum of Natural History), 51, 303–336. [Google Scholar]
- Cronin, M. A. , Cánovas, A. , Bannasch, D. L. , Oberbauer, A. M. , MeDrano, J. F. , & Ostrander, E. (2015). Single nucleotide polymorphism (SNP) variation of wolves (Canis lupus) in Southeast Alaska and comparison with wolves, dogs, and Coyotes in North America. Journal of Heredity, 106, 26–36. 10.1093/jhered/esu075 [DOI] [PubMed] [Google Scholar]
- Cuadrado‐Ríos, S. , & Mantilla‐Meluk, H. (2016). Timing the evolutionary history of tent‐making bats, genus Uroderma (Phyllostomidae): A biogeographic context. Mammalian Biology, 81, 579–586. 10.1016/j.mambio.2016.07.045 [DOI] [Google Scholar]
- Da Silva, J. M. , & Bates, J. M. (2002). Biogeographic patterns and conservation in the South American Cerrado: A tropical savanna hotspot. BioScience, 52(3), 225–233. [Google Scholar]
- Dávalos, L. M. (2004). Phylogeny and biogeography of Caribbean mammals. Biological Journal of the Linnean Society, 373–394. 10.1111/j.1095-8312.2003.00302.x [DOI] [Google Scholar]
- DÁValos L. M. (2006). The geography of diversification in the mormoopids (Chiroptera: Mormoopidae). Biological Journal of the Linnean Society, 88, 101–118. [Google Scholar]
- Dávalos, L. M. (2007). Short‐faced bats (Phyllostomidae: Stenodermatina): A Caribbean radiation of strict frugivores. Journal of Biogeography, 34, 364–375. 10.1111/j.1365-2699.2006.01610.x [DOI] [Google Scholar]
- Dávalos, L. M. , & Russell, A. L. (2012). Deglaciation explains bat extinction in the Caribbean. Ecology and Evolution, 2, 3045–3051. 10.1002/ece3.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, N. , & Bermingham, E. (2001). The historical biogeography of two Caribbean butterflies lepidoptera: Heliconiidae) as inferred from genetic variation at multiple loci. Evolution, 56(3), 573–589. 10.1111/j.0014-3820.2002.tb01368.x [DOI] [PubMed] [Google Scholar]
- Dias, C. A. R. , Santos Júnior, J. E. , Perini, F. A. , & Santos, F. R. (2017). Biogeographic scenarios for the diversification of a widespread Neotropical species, Glossophaga soricina (Chiroptera: Phyllostomidae). Systematics and Biodiversity, 15, 440–450. 10.1080/14772000.2016.1271060 [DOI] [Google Scholar]
- Dolan, P. G. (1989). Systematics of Middle American mastiff bats of the genus Molossus . Special Publications, the Museum, Texas Tech University, 29, 1–71. 10.5962/bhl.title.142636 [DOI] [Google Scholar]
- Donnelly, T. W. (1988). Geologic constraints on Caribbean biogeography In Liebherr J. K. (Ed.), Zoogeography of Caribbean insects (pp. 15–37). Ithaca, NY: Cornell University Press. [Google Scholar]
- Earl, D. A. , & vonHoldt, B. M. (2011). Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Eger, J. L. (2008). Family Molossidae P. Gervais, 1856 In Gardner A. L. (Ed.), Mammals of South America (pp. 399–440). Chicago, IL: University of Chicago Press. [Google Scholar]
- Elshire, R. J. , Glaubitz, J. C. , Sun, Q. , Poland, J. A. , Kawamoto, K. , Buckler, E. S. , & Mitchell, S. E. (2011). A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE, 6, 1–10. 10.1371/journal.pone.0019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enk, J. , Devault, A. , Widga, C. , Saunders, J. , Szpak, P. , Southon, J. , … Poinar, H. (2016). Mammuthus population dynamics in late pleistocene North America: Divergence, phylogeography, and introgression. Frontiers in Ecology and Evolution, 4, 1–13. 10.3389/fevo.2016.00042 [DOI] [Google Scholar]
- Excoffier, L. , Dupanloup, I. , Huerta‐Sánchez, E. , Sousa, V. C. , & Foll, M. (2013). Robust demographic inference from genomic and SNP data. PLoS Genetics, 9, e1003905 10.1371/journal.pgen.1003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Fabian, M. E. , & Gregorin, R. (2007). Família Molossidae In Reis N. R., Peracchi A. L., Pedro W. A., & Lima I. P. (Eds.), Morcegos do Brasil (pp. 149–166). Londrina, Brazil: Universidade Federal de Londrina. [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics., 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, T. H. , & Racey, P. A. (2013). Island Bats. Isl: Bats; 10.7208/chicago/9780226253312.001.0001 [DOI] [Google Scholar]
- Frankel, O. H. , & Soulém, M. E. (1981). Conservation and evolution. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Frankham, R. , Bradshaw, C. G. A. , & Brook, B. W. (2014). Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biological Conservation, 70, 56–63. 10.1016/j.biocon.2013.12.036 [DOI] [Google Scholar]
- Furmankiewicz, J. , & Kucharska, M. (2009). Migration of bats along a large river valley in Southwestern Poland. Journal of Mammalogy, 90(6), 1310–1317. 10.1644/09-MAMM-S-099R1.1 [DOI] [Google Scholar]
- Gainsbury, A. M. , & Colli, G. R. (2003). Lizard Assemblages from Natural Cerrado Enclaves in Southwestern Amazonia: The Role of Stochastic Extinctions and Isolation. Biotropica, 35(4), 503–519. 10.1111/j.1744-7429.2003.tb00607.x [DOI] [Google Scholar]
- García‐Mudarra, J. L. , Ibáñez, C. , & Juste, J. (2009). The Straits of Gibraltar: Barrier or bridge to Ibero‐Moroccan bat diversity? Biological Journal of the Linnean Society, 96(2), 434–450. 10.1111/j.1095-8312.2008.01128.x [DOI] [Google Scholar]
- García‐Verdugo, C. , Caujapé‐Castells, J. , Mairal, M. , & Monroy, P. (2018). How repeatable is microevolution on islands? Patterns of dispersal and colonization‐related plant traits in a phylogeographical context. Annals of Botany, 123(3), 557–568. 10.1093/aob/mcy191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genoways, H. H. (1998). Two new subspecies of bats of the genus Sturnira from the Lesser Antilles, West Indies (p. 176). Occasional Papers ‐ The Museum, Texas Tech University. [Google Scholar]
- Glass, B. P. (1958). Returns of Mexican freetail bats banded in Oklahoma. Journal of Mammalogy., 39, 435–437. [Google Scholar]
- Good, J. M. , Dean, M. D. , & Nachman, M. W. (2008). A complex genetic basis to X‐linked hybrid male sterility between two species of house mice. Genetics, 179, 2213–2228. 10.1534/genetics.107.085340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, L. N. , Barley, A. J. , Poe, S. , Thomson, R. C. , Nieto‐Montes de Oca, A. , & Wang, I. J. (2019). Phylogeography of a widespread lizard complex reflects patterns of both geographic and ecological isolation. Molecular Ecology, 28, 644–657. 10.1111/mec.14970 [DOI] [PubMed] [Google Scholar]
- Groombridge, J. J. , Jones, C. G. , Bruford, M. W. , & Nichols, R. A. (2000). ‘Ghost’ alleles of the Mauritius kestrel. Nature, 403, 616 10.1038/35001148 [DOI] [PubMed] [Google Scholar]
- Gutenkunst, R. N. , Hernandez, R. D. , Williamson, S. H. , & Bustamante, C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5, e1000695 10.1371/journal.pgen.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffer, J. (1997). Alternative models of vertebrate speciation in Amazonia: an overview. Biodiver. and Conserv., 6, 451–476. [Google Scholar]
- Haffer, J. (2002). A rare hybrid manakin (Aves, Pipridae) and the origin of vertebrate species in Amazonia. Rudolstadter Naturhistorische Schriften, 4, 47–73. [Google Scholar]
- Hayes, F. E. , & Sewlal, J. A. N. (2004). The Amazon river as a dispersal barrier to passerine birds: Effects of river width, habitat and taxonomy. Journal of Biogeography, 31, 1809–1818. 10.1111/j.1365-2699.2004.01139.x [DOI] [Google Scholar]
- Ibáñez, C. , García‐Mudarra, J. L. , Ruedi, M. , Stadelmann, B. , & Juste, J. (2006). The Iberian contribution to cryptic diversity in European bats. Acta Chiropterologica, 8, 277–297. 10.3161/1733-5329(2006)8[277:tictcd]2.0.co;2 [DOI] [Google Scholar]
- Iturralde‐Vinent, M. A. , & MacPhee, R. (1999). Paleogeography of the Caribbean Region: Implications for Cenozoic Biogeography. Bulletin of the American Museum of Natural History, 238, 1–95. [Google Scholar]
- Kalkvik, H. M. , Stout, I. J. , Hoffman, E. A. , & Parkinson, C. L. (2018). Colonization and divergence: Phylogeography and population genetics of the Atlantic coast beach mice. Systematics and Biodiversity, 16, 757–773. 10.1080/14772000.2018.1486339 [DOI] [Google Scholar]
- Kamvar, Z. N. , Tabima, J. F. , & Grünwald, N. J. (2014). Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ, 2, e281 10.7717/peerj.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D. (2012). Rates and fitness consequences of new mutations in humans. Genetics, 190, 295–304. 10.1534/genetics.111.134668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, L. F. , Jeffery, K. J. , Arcese, P. , Beaumont, M. A. , Hochachka, W. M. , Smith, J. N. M. , & Bruford, M. W. (2001). Immigration and the ephemerality of a natural population bottleneck: Evidence from molecular markers. Proceedings of the Royal Society B‐Biological Sciences, 268, 1387–1394. 10.1098/rspb.2001.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, A. C. , Iturralde‐vinent, M. A. , Saunders, A. D. , Babbs, T. L. , & Tarney, J. (1999). A new plate tectonic model of the Caribbean. Geological Society of America Bulletin, 7606, 1581–1599. 10.1130/0016-7606(1999)111<1581 [DOI] [Google Scholar]
- Khimoun, A. , Arnoux, E. , Martel, G. , Pot, A. , Eraud, C. , Conde, B. , … Garnier, S. (2016). Contrast patterns of genetic differentiation across eight bird species in the Lesser Antilles. Genetica, 10.1007/s10709-016-9883-4 [DOI] [PubMed] [Google Scholar]
- Kim, S. Y. , Lohmueller, K. E. , Albrechtsen, A. , Li, Y. , Korneliussen, T. , Tian, G. , … Jorgensen, T. (2011). Estimation of allele frequency and association mapping using next-generation sequencing data. BMC bioinformatics, 12(1), 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman, K. F. (1977). Zoogeography In Baker, R. J. , Jones Jr., J. K. , & Carter, D. C. (Eds.), Biology of bats of the new world family phyllostomidae (pp. 39–47). Lubbock, TX: Special publications, the Museum, Texas Tech University. [Google Scholar]
- Kozma, R. , Melsted, P. , Magnusson, K. P. , & Hoglund, J. (2016). Looking into the past – the reaction of three grouse species to climate change over the last million years using whole genome sequences. Molecular Ecology, 25, 570–580. 10.1111/mec.13496 [DOI] [PubMed] [Google Scholar]
- Kusza, S. , Podgórski, T. , Scandura, M. , Borowik, T. , Jávor, A. , Sidorovich, V. E. , … Jędrzejewska, B. (2014). Contemporary genetic structure, phylogeography and past demographic processes of wild boar Sus scrofa population in Central and Eastern Europe. PLoS ONE, 9, e91401 10.1371/journal.pone.0091401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack, J. B. , Roehrs, P. , Stanley, C. E. J. , Ruedi, M. , & Ven Den Bussche, R. A. (2010). Molecular phylogenetics of Myotis indicate familial‐level divergence for the genus Cistugo (Chiroptera). Journal of Mammalogy, 91, 976–992. 10.1644/09-MAMM-A-192.1.Key [DOI] [Google Scholar]
- Lambeck, K. , Yokoyama, Y. , & Purcell, T. (2002). Into and out of the last glacial maximum: Sea‐level change during oxygen isotope stages 3 and 2. Quaternary Science Reviews, 21, 343–360. 10.1016/S0277-3791(01)00071-3 [DOI] [Google Scholar]
- Lande, R. (1988). Genetics and demography in biological conservation. Sciences, 241(4872), 1455–1460. [DOI] [PubMed] [Google Scholar]
- Larsen, P. A. , Hoofer, S. R. , Bozeman, M. C. , Pedersen, S. C. , Genoways, H. H. , Phillips, C. J. , … Baker, R. J. (2007). Phylogenetics and phylogeography of the Artibeus jamaicensis complex based on cytochrome-b DNA sequences. J. Mammal., 88, 712–727. [Google Scholar]
- Larsen, R. J. , Larsen, P. A. , Genoways, H. H. , Catzeflis, F. M. , Geluso, K. , Kwiecinski, G. G. , … Baker, R. J. (2012). Evolutionary history of Caribbean species of Myotis, with evidence of a third Lesser Antillean endemic. Mammalian Biology, 77, 124–134. 10.1016/j.mambio.2011.11.003 [DOI] [Google Scholar]
- Leisler, B. , & Winkler, H. (2015). Evolution of island warblers: Beyond bills and masses. Journal of Avian Biology, 46, 236–244. 10.1111/jav.00509 [DOI] [Google Scholar]
- Lim, B. K. (2010). Adaptive radiation of neotropical emballonurid bats: Molecular phylogenetics and evolutionary patterns in behaviour and morphology In Pontarotti P. (Ed.), Evolutionary biology ‐ concepts, molecular and morphological evolution (pp. 283–299). Heidelberg, Berlin: Springer‐Verlag. [Google Scholar]
- Lim, B. K. , & Engstrom, M. D. (2001). Species diversity of bats (Mammalia: Chiroptera) in Iwokrama Forest, Guyana, and the Guianan subregion: Implications for conservation. Biodiversity and Conservation, 10, 613–657. 10.1023/A:1016660123189 [DOI] [Google Scholar]
- Lim, B. K. , & Lee, T. E. (2018). Community ecology and phylogeography of bats in the Guianan savannas of northern South America. Diversity, 10, 1–15. 10.3390/d10040129 [DOI] [Google Scholar]
- Lim, B. K. , Loureiro, L. O. , Upham, N. S. , & Brocca, J. L. (2017). Phylogeography of Dominican Republic bats and implications for systematic relationships in the neotropics. Journal of Mammalogy, 98, 986–993. 10.1093/jmammal/gyw147 [DOI] [Google Scholar]
- Linck, E. , & Battey, C. J. (2019). Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol Ecol Resour., 19, 639–647. 10.1111/1755-0998.12995 [DOI] [PubMed] [Google Scholar]
- Lindsey, L. L. , & Ammerman, L. K. (2016). Patterns of genetic diversification in a widely distributed species of bat, Molossus molossus. Occ. Pap. Texas Tech Univ. Museum., 339, 1–16. [Google Scholar]
- López‐González, C. , & Presley, S. J. (2001). Taxonomic status of Molossus bondae J. A. Allen, 1904 (Chiroptera: Molossidae), with description of a new subspecies. Journal of Mammalogy, 82, 760–774. [Google Scholar]
- Losos, J. B. , & Ricklefs, R. E. (2009). Adaptation and diversification on islands. Nature, 457, 830–836. 10.1038/nature07893 [DOI] [PubMed] [Google Scholar]
- Loureiro, L. O. , Engstrom, M. D. , & Lim, B. K. (2019). Not all Molossus are created equal: Genetic variation in the mastiff bat reveals diversity masked by conservative morphology. Acta Chiropterologica, 21, 51–64. [Google Scholar]
- Loureiro, L. O. , Gregorin, R. , & Perini, F. A. (2018). Diversity, morphological phylogeny, and distribution of bats of the genus Molossus E. Geoffroy, 1805 (Chiroptera, Molossidae) in Brazil. Zoosystema, 40, 425–452. 10.5252/zoosystema2018v40a18 [DOI] [Google Scholar]
- Loureiro, L. O. , Engstrom, M. D. , & Lim, B. K. (in review). Single nucleotide polymorphisms (SNPs) provide unprecedented resolution of species boundaries, phylogenetic relationships, and genetic diversity in the mastiff bats (Molossus). Molecular Phylogenetics and Evolution. 143, 106690. [DOI] [PubMed] [Google Scholar]
- Taylor, P. , Lim, B. , Pennay, M. , Kinston, T. , Loureiro, L. , & Moras, L. (2019). Family Molossidae. Genus Molossus In Wilson D. E., & Mittermeier R. A. (Eds.), Handbook of Mammals of the World (pp. 598–673). Barcelona, Spain: Lynx Edicions. [Google Scholar]
- Loureiro, L. O. , Lim, B. K. , & Engstrom, M. D. (2018). A new species of mastiff bat (Chiroptera, Molossidae, Molossus) from Guyana and Ecuador. Mammalian Biology, 90, 10–21. 10.1016/j.mambio.2018.01.008 [DOI] [Google Scholar]
- Lozier, J. D. (2014). Revisiting comparisons of genetic diversity in stable and declining species: Assessing genome‐wide polymorphism in North American bumble bees using RAD sequencing. Molecular Ecology, 23, 788–801. 10.1111/mec.12636 [DOI] [PubMed] [Google Scholar]
- Luikart, G. , Sherwin, W. B. , Steele, B. M. , & Allendorf, F. W. (1998). Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Molecular Ecology, 7(8), 963–974. 10.1046/j.1365-294x.1998.00414.x [DOI] [PubMed] [Google Scholar]
- Madsen, T. , Shine, R. , Olsson, M. , & Wittzell, H. (1999). Restoration of an inbred adder population. Nature., 402, 34–35. [Google Scholar]
- Marques, J. T. , Rainho, A. , Carapuço, M. , Oliveira, P. , & Palmeirim, J. M. (2004). Foraging behaviour and habitat use by the European free‐tailed bat Tadarida teniotis . Acta Chiropterologica, 6(1), 99–110. 10.3161/1508110042176680 [DOI] [Google Scholar]
- Martizez‐Freiria, F. , Velo‐Anton, G. , & Brito, J. C. (2015). Trapped by climate: interglacial refuge and recent population expansion in the endemic Iberian adder Vipera seoanei . Diversity and Distributions, 21, 331–344. 10.1111/ddi.12265 [DOI] [Google Scholar]
- Mayr, E. (1963). Animal species and evolution. Cambridge, MA: Harvard University Press. [Google Scholar]
- Mccracken, G. F. , Mccracken, M. K. , & Vawter, A. T. (1994). Genetic structure in migratory populations of the bat Tadarida brasiliensis mexicana . Journal of Mammalogy, 75(2), 500–514. 10.2307/1382574 [DOI] [Google Scholar]
- McCracken, G. F. , Safi, K. , Kunz, T. , Dechmann, D. , Swartz, S. , & Wikelski, M. (2016). Airplane tracking documents the fastest flight speeds recorded for bats. Royal Society Open Science, 3, 160398 10.1098/rsos.160398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C. F. , Kalkol, E. K. , & Kerth, G. (2009). Small‐scale fragmentation effects on local genetic diversity in two phyllostomid bats with different dispersal abilities in panama. Biotropica, 41(1), 95–102. 10.1111/j.1744-7429.2008.00443.x [DOI] [Google Scholar]
- Miller‐Butterworth, C. M. , Jacobs, D. S. , & Harley, E. H. (2003). Strong population substructure is correlated with morphology and ecology in a migratory bat. Nature, 424, 187–191. 10.1038/nature01742 [DOI] [PubMed] [Google Scholar]
- Morgan, G. S. (2001). Patterns of extinction in West Indian bats In Woods C. A., & Sergile F. E. (Eds.), Biogeography of the West Indies: patterns and perspectives, (2nd ed., pp. 369–407). Boca Raton, FL: CRC Press. [Google Scholar]
- Muscarella, R. A. , Murray, K. L. , Ortt, D. , Russell, A. L. , & Fleming, T. H. (2011). Exploring demographic, physical, and historical explanations for the genetic structure of two lineages of greater Antillean bats. PLoS ONE, 6, 1–13. 10.1371/journal.pone.0017704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman, M. W. , & Crowell, S. L. (2000). Estimate of the mutation rate per nucleotide in humans. Genetics, 156, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau, N. J. , Whibley, A. , Jones, R. T. , Davey, J. W. , Dasmahapatra, K. K. , Baxter, S. W. , … Jiggins, C. D. (2011). Evidence for genomic islands of divergence among hybridizing Heliconius butterflies obtained by large‐scale targeted sequencing. Philosophical Transactions of the Royal Society B: Biological Sciences, 367, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka, L. N. , Cohn‐Haft, M. , Mallet‐Rodrigues, F. , Santos, M. P. D. , & Torres, M. D. F. (2006). The avifauna of the Brazilian state of Roraima: Bird distribution and biogeography in the Rio Branco basin. Revista Brasileira De Ornitologia, 14, 197–238. [Google Scholar]
- Nazareno, A. G. , Dick, C. W. , & Lohmann, L. G. (2017). Wide but not impermeable: Testing the riverine barrier hypothesis for an Amazonian plant species. Molecular Ecology, 26(14), 3636–3648. 10.1111/mec.14142 [DOI] [PubMed] [Google Scholar]
- Nei, M. , Graur, D. (1984). Extent of protein polymorphism and the neutral mutation theory. Evol. Biol, 17, 73–118. [Google Scholar]
- Nei, M. , & Tajima, F. (1981). Genetic drift and estimation of effective population size. Genetics, 98, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, D. , & Pilson, D. (1997). Increased probability of extinction due to decreased genetic effective population size: Experimental populations of Clarkia pulchella . Evolution, 51, 345–362. [DOI] [PubMed] [Google Scholar]
- Ni, S. , & Stoneking, M. (2016). Improvement in detection of minor alleles in next generation sequencing by base quality recalibration. BMC Genomics., 17, 1–12. 10.1186/s12864-016-2463-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg, U. M. , & Rayner, J. M. V. (1987). Ecological morphology and flight in bats (Mammalia; Chiroptera): Wing adaptations, flight performance, foraging strategy and echolocation. Philosophical Transactions of the Royal Society B: Biological Sciences, 316, 335–427. 10.1098/rstb.1987.0030 [DOI] [Google Scholar]
- Orr, H. A. (1995). The population genetics of speciation: The evolution of hybrid incompatibilities. Genetics, 139, 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz‐Ramírez, M. F. , Sánchez‐González, L. A. , Castellanos‐Morales, G. , Ornelas, J. F. , & Navarro‐Sigüenza, A. G. (2018). Concerted Pleistocene dispersal and genetic differentiation in passerine birds from the Tres Marías Archipelago, Mexico. The Auk, 135, 716–732. 10.1642/auk-17-190.1 [DOI] [Google Scholar]
- Pacifici, M. , Santini, L. , Di Marco, M. , Baisero, D. , Francucci, L. , Grottolo Marasini, G. , … Rondinini, C. (2013). Generation length for mammals. Nature Conservation, 5, 89–94. 10.3897/natureconservation.5.5734 [DOI] [Google Scholar]
- Patterson, B. D. , Willig, M. R. , & Stevens, R. D. (2003). Trophic strategies, niche partitioning, and patterns of ecological organization In Kunz T. H., & Fenton M. B. (Eds.), Bat ecology (pp. 536–579). Chicago, IL: University of Chicago Press. [Google Scholar]
- Patton, J. L. , & Da Silva, M. N. F. (1998). Rivers, refuges, and ridges: The geography of speciation in Amazonian mammals In Howard D. E., & Berlocher S. (Eds.), Endless forms: Species and speciation (pp. 202–213). New York, NY: Oxford University Press. [Google Scholar]
- Patton, J. L. , Silva, M. N. , & Malcolm, J. R. (1994). Gene genealogy and differentiation among arboreal spiny rats (Rodentia: Echimyidae) of the Amazon basin: A test of the Riverine barrier hypothesis. Evolution, 48, 1314–1323. 10.1111/j.1558-5646.1994.tb05315.x [DOI] [PubMed] [Google Scholar]
- Pedersen, S. C. (1998). Blown in, blown off, and blown up; the bats of Montserrat BWI. American Zoologist, 37, 17A. [Google Scholar]
- Peterson, R. L. , Eger, J. L. , & Mitchell, L. (1995). Chiropterès. Faune de Madagascar, 84, 1–204. [Google Scholar]
- Pedersen, S. C. , Genoways, H. H. , & Freeman, P. W. (1996). Notes on bats from Montserrat (Lesser Antilles) with comments concerning the effects of Hurricane Hugo. Mammalogy Papers: University of Nebraska State Museum Sci. York. [Google Scholar]
- Pindell, J. L. (1994). Evolution of the Gulf of Mexico and the Caribbean In Donovan S.K., & Jackson T.A. (Eds.), aribbean Geology: An Introduction (pp. 19-39) . Kingston, JM: University of the West Indies Publisher's Association. [Google Scholar]
- Pindell, J. L. , Barrett, S. (1990). Geological evolution of the Caribbean region: a plate tectonic perspective In Dengo G. & Case J. E. (Eds.), The geology of North America, The Caribbean region (pp. 405–432). Boulder, CO: Geological Society of America. [Google Scholar]
- Pitman III, W. C. , Cande, S. , Labrecque, J. , & Pindell, J. (1993). Fragmentation of Gondwana: The separation of Africa from South America InGoldblatt P. O. (Ed.), Biological relationships between Africa and South America (pp. 15–34). New Haven, CT: Yale University Press. [Google Scholar]
- Pluzhnikov, A. , & Donnelly, P. (1996). Optimal sequencing strategies for surveying molecular genetic diversity. Genetics, 144, 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons, J. M. , Cibois, A. , Fournier, J. , Fuchs, J. , Olioso, G. , & Thibault, J. C. (2019). Gene flow and genetic divergence among mainland and insular populations across the south‐western range of the Eurasian treecreeper (Certhia familiaris, Aves). Biological Journal of the Linnean Society, 126, 447–461. 10.1093/biolinnean/bly200 [DOI] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of Population Structure using Multilocus Genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumo, D. E. , Goldin, E. Z. , Elliot, B. , Phillips, C. J. , & Genoways, H. H. (1988). Mitochondrial DNA polymorphism in three Antillean island populations of the fruit bat, Artibeus jamaicensis . Molecular Biology and Evolution, 5(1), 79–89. 10.1093/oxfordjournals.molbev.a040477 [DOI] [PubMed] [Google Scholar]
- Querejeta, M. , & Castresana, J. (2018). Evolutionary history of the endemic water shrew Neomys anomalus: Recurrent phylogeographic patterns in semi‐aquatic mammals of the Iberian Peninsula. Ecology and Evolution, 8, 10138–10146. 10.1002/ece3.4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymo, M. E. , Ganley, K. , Carter, S. , Oppo, D. W. , & McManus, J. (1998). Millennial-scale climate instability during the early Pleistocene epoch. Nature, 392, 699–702. [Google Scholar]
- Rousset, F. (2018). GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. Journal of Heredity, 68(3), 248–249. 10.1093/oxfordjournals.jhered.a111573 [DOI] [Google Scholar]
- Reid, F. A. (2009). A field guide to the mammals of Central America and Southeast Mexico. New York, NY: Oxford University Press. [Google Scholar]
- Ricklefs, R. , & Bermingham, E. (2008). The West Indies as a laboratory of biogeography and evolution. Phil. Trans. R. Soc. B., 363, 2393–2413. 10.1098/rstb.2007.2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison, J. D. , Coffman, A. J. , Hickerson, M. , & Gutenkunst, R. N. (2014). Sampling strategies for frequency spectrum‐based population genomic inference. BMC Evolutionary Biology, 14(1), 1–16. 10.1186/s12862-014-0254-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Ramilo, S. T. , & Wang, J. (2012). The effect of close relatives on unsupervised Bayesian clustering algorithms in population genetic structure analysis. Molecular Ecology Resources, 12, 873–884. 10.1111/j.1755-0998.2012.03156.x [DOI] [PubMed] [Google Scholar]
- Russell, A. L. , Brown, V. A. , Utzurrum, R. C. B. , Brooke, A. P. , Wolf, L. A. , & Mccracken, G. F. (2016). Comparative Phylogeography of Pteropus samoensis and P. tonganus (Pteropodidae: Chiroptera) in the South Pacific. Acta Chiropterologica, 18, 325–335. 10.3161/15081109acc2016.18.2.002 [DOI] [Google Scholar]
- Russell, A. L. , Medellín, R. A. , & Mccracken, G. F. (2005). Genetic variation and migration in the Mexican free‐tailed bat (Tadarida brasiliensis mexicana). Molecular Ecology, 14, 2207–2222. 10.1111/j.1365-294X.2005.02552.x [DOI] [PubMed] [Google Scholar]
- Sarmiento, G. (1983).The savannas of tropical America InBourlière F. (Ed.), Tropical savannas (pp. 245–288). Amsterdam, the Netherlands: Elsevier. [Google Scholar]
- Schield, D. R. , Card, D. C. , Adams, R. H. , Jezkova, T. , Reyes‐Velasco, J. , Proctor, F. N. , … Castoe, T. A. (2015). Incipient speciation with biased gene flow between two lineages of the Western Diamondback Rattlesnake (Crotalus atrox). Molecular Phylogenetics and Evolution, 83, 213–223. 10.1016/j.ympev.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Serra‐Cobo, J. , Lopez‐Roig, M. , Marqués‐Bonet, T. , & Lahuerta, E. (2000). Rivers as possible landmarks in the orientation flight of Miniopterus schreibersii . Acta Theriologica, 4, 347–352. [Google Scholar]
- Sexton, J. P. , Hangartner, S. B. , & Hoffmann, A. A. (2014). Genetic isolation by enviroment or distance: which pattern of gene flow in most common? Evolution., 68, 1–15. 10.1111/evo.12258 [DOI] [PubMed] [Google Scholar]
- Simmons, N. B. (2005). Ordem Chiroptera In Wilson D. E., & Reeder D. M. (Eds.), Mammal species of the world: A taxonomic and geographic reference (3rd ed., pp. 312–529). Baltimore, MD: Johns Hopkins University Press. [Google Scholar]
- Smith, P. G. , & Racey, P. A. (2008). Natterer's bats prefer foraging in broad‐leaved woodlands and river corridors. Journal of Zoology, 275(3), 314–322. 10.1111/j.1469-7998.2008.00445.x [DOI] [Google Scholar]
- Soltis, D. E. , Morris, A. B. , McLachlan, J. S. , Manos, P. S. , & Soltis, P. S. (2006). Comparative phylogeography of unglaciated eastern North America. Molecular Ecology, 15, 4261–4293. 10.1111/j.1365-294X.2006.03061.x [DOI] [PubMed] [Google Scholar]
- Soria‐Carrasco, V. , & Castresana, J. (2012). Diversification rates and the latitudinal gradient of diversity in mammals. Proceedings of the Royal Society B‐Biological Sciences, 279, 4148–4155. 10.1098/rspb.2012.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto‐Centeno, J. A. , & Steadman, D. W. (2015). Fossils reject climate change as the cause of extinction of Caribbean bats. Scientific Reports, 7971, 1–7. 10.1038/srep07971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sovic, M. G. , Carstens, B. C. , & Gibbs, H. L. (2016). Genetic diversity in migratory bats: Results from RADseq data for three tree bat species at an Ohio windfarm. PeerJ, 4, e1647 10.7717/peerj.1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sovic, M. G. , Fries, A. , Martin, S. A. , & Gibbs, H. L. (2018). Genetic signatures of small effective population sizes and demographic declines in an endangered rattlesnake, Sistrurus catenatus . Evolut. Applications, 12, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer, K. A. , Petronio, B. J. , Simmons, N. B. , Richey, R. , Magrini, K. , Soto‐Centeno, J. A. , & Reed, D. L. (2017). Population structure of a widespread bat (Tadarida brasiliensis) in an island system. Ecology and Evolution, 7, 7585–7598. 10.1002/ece3.3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilani, L. , Bougiouri, K. , Antoniou, A. , Psonis, N. , Poursanidis, D. , Lymberakis, P. , & Poulakakis, N. (2019). Multigene phylogeny, phylogeography and population structure of Podarcis cretensis species group in south Balkans. Molecular Phylogenetics and Evolution, 138, 193–204. 10.1016/j.ympev.2019.05.026 [DOI] [PubMed] [Google Scholar]
- Swofford, D. L. (2003). PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Tam, S. M. , Mhiri, C. , Vogelaar, A. , Kerkveld, M. , Pearce, S. R. , & Grandbastien, M. A. (2005). Comparative analyses of genetic diversities within tomato and pepper collections detected by retrotransposon‐based SSAP, AFLP and SSR. Theoretical and Applied Genetics, 110, 819–831. 10.1007/s00122-004-1837-z [DOI] [PubMed] [Google Scholar]
- Taylor, P. J. , Stoffberg, S. , Monadjem, A. , Schoeman, M. C. , Bayliss, J. , & Cotterill, F. P. D. (2012). Four New Bat Species (Rhinolophus hildebrandtii Complex) Reflect Plio‐Pleistocene Divergence of Dwarfs and Giants across an Afromontane Archipelago. PLoS ONE, 7, e41744 10.1371/journal.pone.0041744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, E. C. , Jones, G. , Rossiter, S. J. (2016). Phylogeny, genes, and Hearing: Implications for the evolution of echolocation in bats In Fenton M. B., Grinnell A. D., Popper A. N., & Fay R. R. (Eds.), Bat bioacoustics (pp. 25–64). New York, NY: Springer Science + Business Media. [Google Scholar]
- Valdez, L. , & D'Elía, G. (2013). Differentiation in the Atlantic Forest: Phylogeography of Akodon montensis (Rodentia, Sigmodontinae) and the Carnaval‐Moritz model of Pleistocene refugia. Journal of Mammalogy, 94, 911–922. [Google Scholar]
- Valente, L. , Etienne, R. S. , & Dávalos, L. M. (2017). Recent extinctions disturb path to equilibrium diversity in Caribbean bats. Nature, 1, 1–7. 10.1038/s41559-016-0026 [DOI] [PubMed] [Google Scholar]
- Vanzolini, P. E. , & Williams, E. E. (1970). South American anoles: The geographic differentiation and evolution of the Anolis chrysolepis species group (Sauria, Iguanidae). Arquivos De Zoologia, 19, 1–298. [Google Scholar]
- Velazco, P. M. , & Patterson, B. D. (2013). Molecular phylogenetics and evolution diversification of the yellow‐shouldered bats, genus Sturnira (Chiroptera, Phyllostomidae ), in the new world tropics. Molecular Phylogenetics and Evolution, 68, 683–698. 10.1016/j.ympev.2013.04.016 [DOI] [PubMed] [Google Scholar]
- Verzi, D. H. , Deschamps, C. M. , & Tonni, E. P. (2004). Biostratigraphic and palaeoclimatic meaning of the Middle Pleistocene South American rodent Ctenomys kraglievichi (Caviomorpha, Octodontidae). Palaeogeography, Palaeoclimatology, Palaeoecology, 212, 315–329. 10.1016/S0031-0182(04)00328-1 [DOI] [Google Scholar]
- Voss, R. S. (1991). An introduction to the Neotropical muroid rodent genus Zygodontomys . Bulletin of the American Museum of Natural History, 210, 1–113. [Google Scholar]
- Vrba, E. S. (1985). Environment and evolution: Alternative causes of the temporal distribution of evolutionary events. South African Journal of Science, 81, 221–236. [Google Scholar]
- Wallace, A. R. (1852). On the monkeys of the Amazon. Proceedings Zoological Society of London, 20, 107–110. [Google Scholar]
- Wang, I. J. , & Bradburd, G. S. (2014). Isolation by environment. Molecular Ecology, 23, 5649–5662. 10.1111/mec.12938 [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358 10.2307/2408641 [DOI] [PubMed] [Google Scholar]
- Weir, J. T. , Faccio, M. S. , Pulido‐Santacruz, P. , Barrera‐Guzmán, A. O. , & Aleixo, A. (2015). Hybridization in headwater regions, and the role of rivers as drivers of speciation in Amazonian birds. Evolution, 69, 1823–1834. 10.1111/evo.12696 [DOI] [PubMed] [Google Scholar]
- Weir, J. T. , & Price, T. D. (2011). Limits to speciation inferred from times to secondary sympatry and ages of hybridizing species along a latitudinal gradient. American Naturalist, 177, 462–469. 10.1086/658910 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
VCF files with SNP data are available at https://doi.org/10.5061/dryad.d7wm37pxc