

Summary
Background
We evaluated antidrug antibody (ADA) development in patients with chronic plaque psoriasis from three clinical trials of tildrakizumab, a humanized anti‐interleukin‐23p19 monoclonal antibody (P05495, reSURFACE 1 and reSURFACE 2).
Objectives
To determine the effects of immunogenicity on the pharmacokinetics, efficacy and safety of tildrakizumab.
Methods
In 1400 (weeks 12–16) and 780 (weeks 52–64) evaluable patients randomized to tildrakizumab 100 or 200 mg, treatment‐emergent ADA‐positive (TE‐POS) patients were identified and characterized for neutralizing antibodies (NAbs). Pharmacokinetics, safety and efficacy were evaluated by ADA status.
Results
In patients treated with tildrakizumab 100 or 200 mg continuously, < 7% were inconclusive at 52–64 weeks. In long‐term data through 52–64 weeks, the incidence of TE‐POS was 6·5% (100 mg) and 8·2% (200 mg) and the incidence of TE‐POS NAb‐POS was 2·5% (100 mg) and 3·2% (200 mg). TE‐POS NAb‐POS patients had modestly increased median tildrakizumab clearance (36·5%) compared with ADA‐NEG patients. Percentage Psoriasis Area and Severity Index improvements in TE‐POS NAb‐POS vs. ADA‐NEG patients on continuous treatment through week 52 were 76% (n = 10) vs. 91% (n = 342) for 100 mg and 77% (n = 12) vs. 87% (n = 299) for 200 mg. The incidence of potential immunogenicity‐related adverse events did not indicate a clear trend in any positive ADA patient category compared with ADA‐NEG patients through weeks 52–64. The effects of ADA on pharmacokinetics, efficacy and safety at 12–16 weeks were also summarized.
Conclusions
ADA development with tildrakizumab treatment for 52–64 weeks was low; around 3% of patients developed TE‐POS NAb‐POS ADAs and showed lower serum concentrations and corresponding reduced efficacy. No relationship between ADAs and safety was observed.
What's already known about this topic?
Unwanted immune responses – for example immunogenicity and antidrug antibodies (ADAs) – have been observed with therapeutic monoclonal antibodies and can affect efficacy and safety.
Tildrakizumab is a humanized monoclonal antibody targeting interleukin‐23 and is currently approved for patients with plaque psoriasis.
What does this study add?
ADA development in tildrakizumab‐treated patients with psoriasis over 52 weeks was low.
The small proportion of patients who had treatment‐emergent ADAs and had neutralizing antibodies experienced lower serum tildrakizumab concentrations and reduced efficacy.
No relationship between ADAs and safety events was observed.
Short abstract
Linked Comment: https://doi.org/10.1111/bjd.18321.
https://doi.org/10.1111/bjd.18662 available online
Antidrug antibodies (ADAs) have been found in up to 30% of patients treated with anti‐tumour necrosis factor agents who are not responding to treatment, and in another 50% who lose clinical responses over time.1 These unwanted immune responses (i.e. immunogenicity) can be due to molecular structure, dosing regimen, patient characteristics and other factors,2 and can affect not only clinical response, but also safety issues including infusion reactions, serum sickness or anaphylactic reactions.3
More recently, biologic therapies that target the interleukin (IL)‐23/IL‐17 immunological pathway have been developed and have demonstrated clinically important treatment effects in patients with chronic plaque psoriasis.4, 5, 6 Tildrakizumab is a high‐affinity, humanized IgG1κ monoclonal antibody targeting the IL‐23p19 subunit of IL‐23. Tildrakizumab, at doses of 100 mg and 200 mg administered at weeks 0 and 4 and every 12 weeks thereafter, has demonstrated efficacy and safety in the treatment of moderate‐to‐severe chronic plaque psoriasis in clinical trials. Recently, tildrakizumab has been approved for use in the treatment of chronic plaque psoriasis by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).7, 8, 9, 10, 11
We evaluated immunogenicity with tildrakizumab in three large, randomized controlled clinical studies (NCT01225731, NCT01722331 and NCT01729754) both at the primary end points (12–16 weeks) and at the end of the base periods (52–64 weeks).7, 8 Treatment interruptions were not studied.
Patients and methods
Clinical trial designs
This evaluation included three studies: P05495 (phase IIb; n = 355), reSURFACE 1 (phase III; n = 772) and reSURFACE 2 (phase III; n = 1090). P05495 was a three‐part, randomized, double‐blind trial in adults with chronic plaque psoriasis. Participants were randomized to receive subcutaneous tildrakizumab (5, 25, 100 or 200 mg) or placebo at weeks 0 and 4 (part 1; 16 weeks) and every 12 weeks thereafter until week 52 (part 2), with a 20‐week washout (part 3).8 The two phase III studies (reSURFACE 1 and reSURFACE 2) were performed as a three‐part, parallel‐group, double‐blinded randomized controlled trial to assess the efficacy, safety and tolerability of tildrakizumab compared with placebo and etanercept. The phase III trials tested tildrakizumab 100 or 200 mg or placebo through 12 weeks in part 1, tildrakizumab 100 or 200 mg through 28 weeks in part 2, and 100 or 200 mg in part 3 through the end of the base period of each trial: 64 weeks (reSURFACE 1) or 52 weeks (reSURFACE 2). reSURFACE 2 additionally tested etanercept in parts 1 and 2 followed by tildrakizumab 200 mg in part 3.7
Primary efficacy end points included ≥ 75% reduction in Psoriasis Area and Severity Index (PASI 75) and Physician's Global Assessment (PGA) score of ‘clear’ or ‘minimal’ with at least a 2‐point reduction from baseline at week 12. PASI 75 was the primary end point in P05495, while PASI 75 and PGA were coprimary end points for reSURFACE 1 and 2.
Details of the assays for tildrakizumab, antitildrakizumab antibodies and neutralizing antibodies (NAbs) are provided in Table S1 (see Supporting Information). As the two ADA assays used in the phase IIb and III studies did not differ substantially in execution and performance, and to be consistent in the evaluation of results across assays, one uniform drug tolerance level (6 μg mL−1) was utilized based on the assay used for phase III. At tildrakizumab concentrations above 6 μg mL−1 the ADA assay was not able to detect 500 ng mL−1 of ADA‐positive control, hence 6 μg mL−1 was considered the drug tolerance level of the assay.
Immunogenicity assessment strategy
Patients with at least one ADA sample result following dosing with tildrakizumab were evaluable for the immunogenicity analysis. Tildrakizumab can interfere with the ADA assay at concentrations above the drug tolerance level of 6 μg mL−1, leading to the potential for false‐negative results. Therefore, the immunogenicity status of an individual patient was categorized as negative (ADA‐NEG) if all pretreatment and postdose samples tested negative in the ADA assay, and the concentration of tildrakizumab in the last postdose sample was less than the drug tolerance level. Patients were characterized as inconclusive if they did not have any ADA‐positive samples and the tildrakizumab concentration in the last sample was at or above the drug tolerance level. The inconclusive patients were designated in order to be transparent about the number of patients who had the potential to have false‐negative results in the ADA assay. A description of how patients were categorized for the immunogenicity assessment is provided in Table 1.
Table 1.
Definitions of patients’ immunogenicity status
| Patient status | Definition |
|---|---|
| Antidrug antibody negative (ADA‐NEG) | All pretreatment and postdose samples were negative in the ADA assay and the drug concentration in the last postdose sample was < 6 μg mL−1 |
| Inconclusive | All pretreatment and postdose samples were negative in the ADA assay and the drug concentration in the last postdose sample was ≥ 6 μg mL−1. This category represents patients who may have had drug interference in the ADA assay |
| Treatment emergent positive (TE‐POS) | Pretreatment sample was negative and at least one postdose sample was positive in the ADA assay (treatment induced positive) |
| Pretreatment and postdose samples were both positive in the ADA assay and the titre increased postdose by ≥ 2‐fold (treatment boosted positive) | |
| TE‐POS NAb‐POS | Treatment emergent positive and at least one sample was positive for NAbs |
| TE‐POS NAb‐NEG | Treatment emergent positive and negative for NAbs |
| Nontreatment emergent positive (non‐TE‐POS) | Pretreatment sample was positive and postdose samples were negative in the ADA assay |
| Pretreatment and postdose samples were positive in the ADA assay with a < 2‐fold increase in titre postdose | |
| Non‐TE‐POS NAb‐POS | Nontreatment emergent positive and at least one sample was positive for NAbs |
| Non‐TE‐POS NAb‐NEG | Nontreatment emergent positive and negative for neutralizing antibodies |
NAb, neutralizing antibody.
Primary end points were at week 16 (P05495) or week 12 (reSURFACE 1, reSURFACE 2); therefore, patients’ ADA status was summarized through these respective end points. A second summary was performed at the end of the base periods to determine the patients’ ADA status based on data through the last time point available: washout after week 52 for P05495 and reSURFACE 2 or week 64 for reSURFACE 1. The summary at the end of the base period was focused on patients who were treated with tildrakizumab 100 or 200 mg continuously in phase IIb and phase III, because this was the proposed clinical treatment paradigm and it enabled evaluation of the long‐term effects of ADA on pharmacokinetics (PK), efficacy and safety. Treatment interruptions were not studied.
Statistical methods
The proportion of patients with ADAs was determined using the number of ADA‐evaluable patients as the denominator. Efficacy and safety tables were prepared for different categories of patient immunogenicity status using the same methods as described previously.7, 8
Results
Patients included in the analysis
In P05495, there were 89 (100 mg) and 86 (200 mg) ADA‐evaluable patients in part 1, and 31 (100 mg) and 51 (200 mg) on continuous therapy through week 52. In reSURFACE 1, there were 308 (100 mg) and 305 (200 mg) ADA‐evaluable patients in part 1, and 135 (100 mg) and 160 (200 mg) patients on continuous therapy through 64 weeks. In reSURFACE 2, the numbers of ADA‐evaluable patients were 303 (100 mg) and 309 (200 mg) in part 1, and 234 (100 mg) and 169 (200 mg) in patients on continuous therapy through 52 weeks.
Antidrug antibody incidence
In patients treated with tildrakizumab 100 mg and 200 mg, the proportions of treatment‐emergent ADA‐positive (TE‐POS) patients were 4·3% and 4·1% through 12–16 weeks (primary end points) and 6·5% and 8·2% through 52–64 weeks, respectively (Table 2). The proportions of patients who were TE‐POS NAb‐POS were 0·6% for both doses through 12–16 weeks and 2·5% and 3·2% for 100 mg and 200 mg, respectively, through 52–64 weeks.
Table 2.
Antidrug antibody (ADA) incidence in patients treated with tildrakizumab 100 mg or 200 mg through weeks 12–16 and weeks 52–64 in P05495, reSURFACE 1 and reSURFACE 2
| Total evaluablea , b | ADA‐NEGc | Inconclusivec | Non‐TE‐POSc | TE‐POSc | ||
|---|---|---|---|---|---|---|
| ADA positive | NAb positive | ADA positive | NAb positive | |||
| Immunogenicity summary through weeks 12–16 for patients treated with 100 mg in part 1d | ||||||
| 700 | 603 (86·1) | 29 (4·1) | 38 (5·4) | 5 (0·7) | 30 (4·3) | 4 (0·6) |
| Immunogenicity summary through weeks 12–16 for patients treated with 200 mg in part 1d | ||||||
| 700 | 374 (53·4) | 255 (36·4) | 42 (6·0) | 6 (0·9) | 29 (4·1) | 4 (0·6) |
| Immunogenicity summary through weeks 52–64 for patients treated with 100 mg continuouslye | ||||||
| 400 | 342 (85·5) | 8 (2·0) | 24 (6·0) | 3 (0·8) | 26 (6·5) | 10 (2·5) |
| Immunogenicity summary through weeks 52–64 for patients treated with 200 mg continuouslye | ||||||
| 380 | 299 (78·7) | 25 (6·6) | 25 (6·6) | 4 (1·1) | 31 (8·2) | 12 (3·2) |
The data are given as n (%). NEG, negative; TE‐POS, treatment emergent ADA positive; NAb, neutralizing antibody. aIncludes patients treated with the listed dose of tildrakizumab (100 mg or 200 mg) in parts 1, 2 and 3 of reSURFACE 1 and reSURFACE 2 or in parts 1 and 2 of P05495. bIncludes patients with at least one ADA sample available after treatment with tildrakizumab. cDenominator is the total number of evaluable patients. dIn P05495, part 1 ended at week 16; in reSURFACE 1 and reSURFACE 2, part 1 ended at week 12. eIn P05495, the trial ended at week 52; in reSURFACE 1 and reSURFACE 2, the base portions of the trials ended at weeks 64 and 52, respectively.
Proportions of inconclusive patients
Through 12–16 weeks, the proportions of inconclusive patients were 4·1% and 36·4% for patients on 100 mg and 200 mg, respectively (Table 2). The higher proportion of inconclusive patients in part 1 relative to the end of the base period was due to the loading dose of tildrakizumab, which affected the interpretability of the assay as described in the methods. In the phase III trials, doses were given at weeks 0 and 4 (part 1) and every 12 weeks thereafter, and the primary end point was at week 12, where the immunogenicity data were summarized. The predose tildrakizumab concentrations at week 12 were higher than the steady‐state trough concentrations because they were collected 8 weeks after dosing at week 4, as opposed to 12 weeks after dosing at steady state. Although there was a higher proportion of inconclusive patients in the 200‐mg dose group (36·4%) at 12–16 weeks, the proportions of patients who were TE‐POS were similar for both dose levels (4·3% for 100 mg and 4·1% for 200 mg), suggesting that the results from the 200‐mg group may not have been biased by inconclusive patients. At the end of the base portion of the trials (52–64 weeks), the proportions of inconclusive patients were 2·0% and 6·6% for 100 mg and 200 mg continuously, respectively.
Maximum postdose titre values of all patients on tildrakizumab through 12–16 weeks and 52–64 weeks
The maximum postdose titres for evaluable positive patients were summarized for each immunogenicity patient category. TE‐POS NAb‐POS patients showed the highest median postdose titres (64) through 12–16 weeks, whereas the median titre for other positive categories was ≤ 2, suggesting a relationship between developing TE‐POS NAb‐POS ADAs and higher postdose titre responses. In patients completing 52–64 weeks of treatment, a similar trend was observed: the median postdose titre value for TE‐POS NAb‐POS patients was 32, whereas the median value for the other positive categories was 1 (Fig. 1). Note that titres were summarized for all evaluable patients at the end of the base period, rather than only those on 100 mg or 200 mg continuously.
Figure 1.
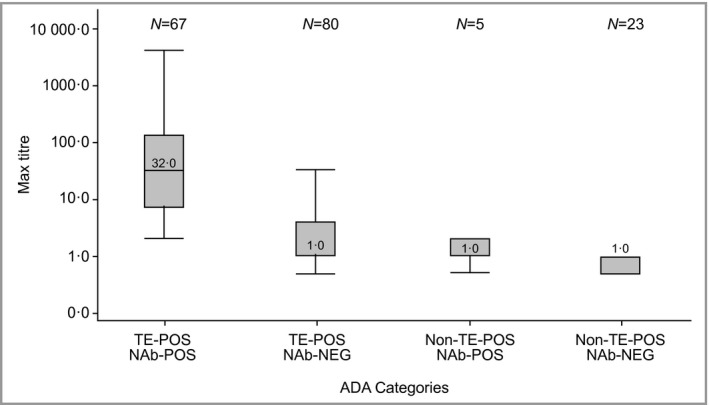
Maximum postdose titre values by antidrug antibody (ADA) patient category for all ADA‐evaluable patients through weeks 52–64. The boxes represent the interquartile range, the line in the box is the median and the whiskers are the 5th and 95th percentiles. TE‐POS, treatment emergent positive; NAb‐NEG, neutralizing antibody negative; NAb‐POS, neutralizing antibody positive.
Effect of antidrug antibodies on tildrakizumab pharmacokinetics through 52–64 weeks
The TE‐POS NAb‐POS subgroup experienced decreased mean (Fig. 2a, c) and individual (Fig. 2b, d) tildrakizumab concentrations and a modest increase (36·5%) in median drug clearance (Fig. 3), compared with ADA‐NEG patients who were on 100 mg or 200 mg continuously through weeks 52–64. Patients in other positive ADA categories and inconclusive patients were generally similar to ADA‐NEG patients with regard to mean tildrakizumab concentrations and clearance. The population PK analysis did not test ADA status as a covariate. An estimate of the typical population clearance (approximating the median population value) was 0·297 L per day across the phase I, phase IIb and phase III trials. The geometric mean (coefficient of variation) clearance in patients with psoriasis in the phase IIb and phase III studies was 0·32 L per day (38%).12
Figure 2.
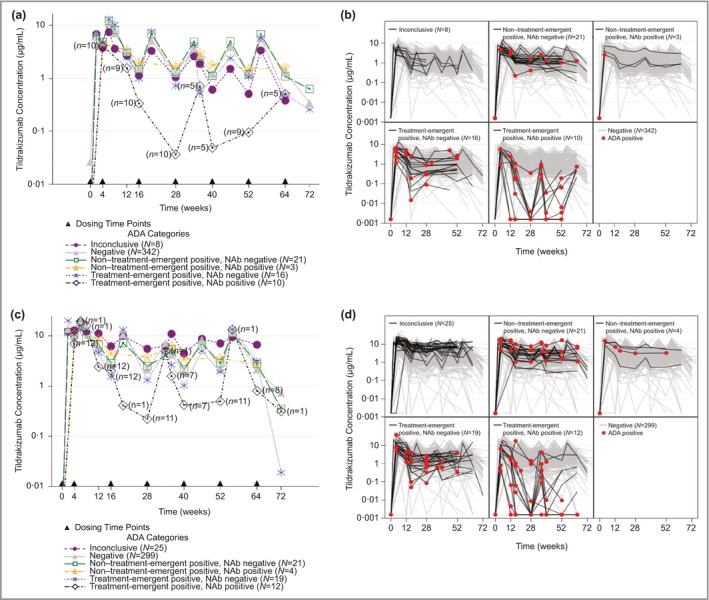
Effect of antidrug antibodies (ADAs) on tildrakizumab (MK‐3222) concentrations for patients treated with 100 mg or 200 mg continuously for 52–64 weeks. (a) Mean concentration–time profiles for patients treated with 100 mg. (b) Individual concentration–time profiles for patients treated with 100 mg. (c) Mean concentration–time profiles for patients treated with 200 mg. (d) Individual concentration–time profiles for patients treated with 200 mg. For (a) and (c), dosing is represented by a solid triangle along the x‐axis. The numbers on the treatment‐emergent‐positive, neutralizing antibody (NAb)‐positive profile represent the number of patients contributing to that time point. For (b) and (d), grey lines represent the concentration–time profile in ADA‐negative patients. Red filled circles represent individual samples that tested positive for ADAs. Continuous treatment with tildrakizumab means that patients treated with placebo or etanercept and then tildrakizumab were not included in the analysis.
Figure 3.
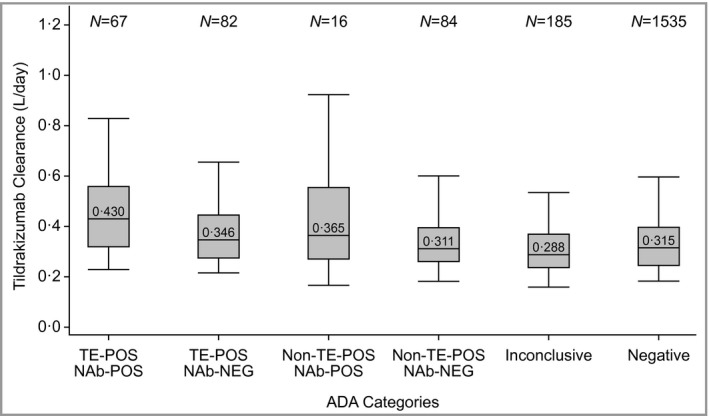
Distribution of clearance for all evaluable patients by antidrug antibody (ADA) patient category through the end of the trial in phase IIb and phase III. The boxes represent the interquartile range, the line in the box is the median and the whiskers are the 5th and 95th percentiles. TE‐POS, treatment emergent positive; NAb‐NEG, neutralizing antibody negative; NAb‐POS, neutralizing antibody positive.
Individual profiles from ADA‐positive patients showed that patients who had reduced PK often had detectable ADAs at multiple time points; in some patients these were identified as early as week 4. The effects of ADAs on PK through 12–16 weeks are shown in Figure S1 (see Supporting Information). The effects observed through 12–16 weeks were similar to the effects observed for the full duration of the trials, in that TE‐POS NAb‐POS patients showed a decrease in mean and individual tildrakizumab concentrations, whereas other categories were generally comparable with the ADA‐NEG patients.
Effect of antidrug antibodies on efficacy end points at week 12 for treatment‐emergent‐positive patients vs. negative and inconclusive patients (pooled analysis)
Two separate approaches were used to evaluate a potential effect of ADAs on efficacy. In the first analysis, the proportions of TE‐POS patients (TE‐POS NAb‐POS and TE‐POS NAb‐NEG combined) vs. ADA‐NEG and inconclusive patients (combined) were compared with the proportions of patients in each combined category achieving primary and secondary efficacy end points at week 12. This assessment included the largest number of patients (around 1300).
The proportions of patients who received tildrakizumab 100 mg or 200 mg in the first 12 weeks of the trials, who were categorized as TE‐POS for ADAs and who also achieved PASI 75, PASI 90, PASI 100 or PGA ‘clear’ or ‘minimal’ response at 12 weeks, were similar to the proportions of ADA‐NEG and inconclusive patients combined (Table 3).
Table 3.
Effect of antidrug antibodies (ADAs) on efficacy end points at week 12 in treatment‐emergent‐positive patients compared with negative and inconclusive (combined) patients
| Tildrakizumab treatment in part 1 | TE‐POS, N | Responders, n (%) | ADA‐NEG and inconclusive, N | Responders, n (%) |
|---|---|---|---|---|
| PASI 75 | ||||
| 100 mg | 30 | 18 (60.0) | 632 | 396 (62·7) |
| 200 mg | 29 | 20 (69.0) | 629 | 409 (65·0) |
| PASI 90 | ||||
| 100 mg | 30 | 11 (36.7) | 632 | 231 (36·6) |
| 200 mg | 29 | 9 (31.0) | 629 | 234 (37·2) |
| PASI 100 | ||||
| 100 mg | 30 | 3 (10.0) | 632 | 87 (13·8) |
| 200 mg | 29 | 3 (10.3) | 629 | 81 (12·9) |
| PGA ‘clear’ or ‘minimal’ | ||||
| 100 mg | 30 | 17 (56.7) | 632 | 364 (57·6) |
| 200 mg | 29 | 19 (65.5) | 629 | 378 (60·1) |
TE‐POS, treatment emergent ADA positive; NEG, negative; PASI, Psoriasis Area and Severity Index; PASI 75; ≥ 75% improvement in PASI; PGA, Physician's Global Assessment.
Effect of patients’ immunogenicity status on percentage Psoriasis Area and Severity Index improvement through 12–16 weeks and 52–64 weeks (nonpooled analysis)
In the second analysis examining potential effects of ADAs on efficacy, percentage PASI improvement by ADA patient category was summarized without pooling. At week 12, the percentage PASI improvements for TE‐POS NAb‐POS patients compared with ADA‐NEG patients were 62% (n = 4) vs. 75% (n = 603) for the 100‐mg group and 38% (n = 4) vs. 76% (n = 374) for the 200‐mg group (Fig. 4 and Table 4).
Figure 4.
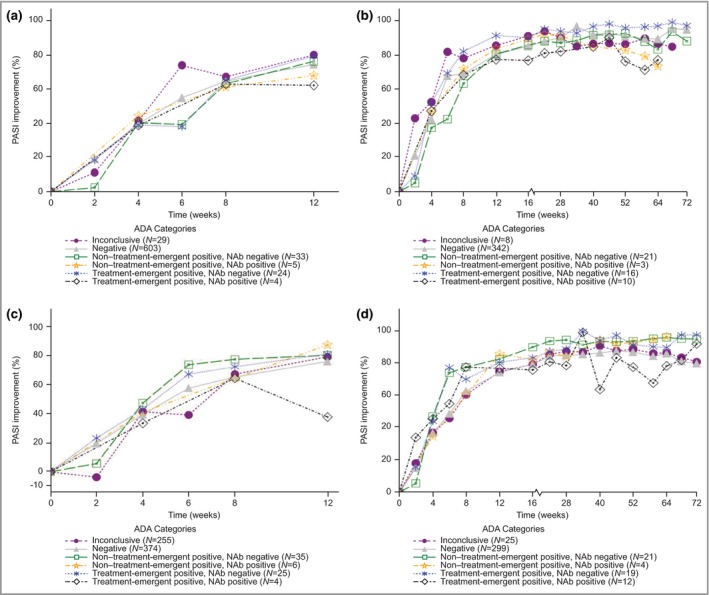
Effect of antidrug antibodies (ADAs) on percentage Psoriasis Area and Severity Index (PASI) improvement through week 12 or week 52. (a) Percentage PASI improvement through week 12 for patients treated with 100 mg in part 1. (b) Percentage PASI improvement through weeks 52–64 for patients treated with 100 mg continuously in parts 1, 2 and 3. (c) Percentage PASI improvement through week 12 for patients treated with 200 mg in part 1. (d) Percentage PASI improvement through weeks 52–64 for patients treated with 200 mg continuously in parts 1, 2 and 3. Continuous treatment with tildrakizumab means that patients treated with placebo or etanercept and then tildrakizumab were not included in the analysis. NAb, neutralizing antibodies.
Table 4.
Effect of antidrug antibodies (ADAs) on percentage Psoriasis Area and Severity Index (PASI) improvement at week 12 in patients on tildrakizumab 100 mg or 200 mg by ADA patient category
| Dose (mg) | ADA patient category | N | Mean % PASI improvement at week 12 |
|---|---|---|---|
| 100 | Inconclusive | 29 | 80 |
| ADA‐NEG | 603 | 75 | |
| Non‐TE‐POS NAb‐NEG | 33 | 76 | |
| Non‐TE‐POS NAb‐POS | 5 | 68 | |
| TE‐POS NAb‐NEG | 24 | 79 | |
| TE‐POS NAb‐POS | 4 | 62 | |
| 200 | Inconclusive | 255 | 79 |
| ADA‐NEG | 374 | 76 | |
| Non‐TE‐POS NAb‐NEG | 35 | 81 | |
| Non‐TE‐POS NAb‐POS | 6 | 88 | |
| TE‐POS NAb‐NEG | 25 | 81 | |
| TE‐POS NAb‐POS | 4 | 38 |
TE, treatment‐emergent ADA; NAb, neutralizing antibodies; POS, positive; NEG, negative.
To investigate the longer‐term effects of ADAs on PASI, patients who were on 100 mg or 200 mg continuously through weeks 52–64 were separated for analysis, as discussed in the methods section. At week 52, PASI improvement for TE‐POS NAb‐POS patients compared with ADA‐NEG patients was 76% (n = 10) vs. 91% (n = 342) for 100 mg continuously and 77% (n = 12) vs. 87% (n = 299) for 200 mg continuously (Fig. 4 and Table 5).
Table 5.
Effect of antidrug antibodies (ADAs) on percentage Psoriasis Area and Severity Index (PASI) improvement at week 52 in patients treated continuously with tildrakizumab 100 mg or 200 mg by ADA patient category
| Dose (mg)a | ADA patient category | N | Mean % PASI improvement at week 52 |
|---|---|---|---|
| 100 | Inconclusive | 8 | 86 |
| ADA‐NEG | 342 | 91 | |
| Non‐TE‐POS NAb‐NEG | 21 | 93 | |
| Non‐TE‐POS NAb‐POS | 3 | 83 | |
| TE‐POS NAb‐NEG | 16 | 96 | |
| TE‐POS NAb‐POS | 10 | 76 | |
| 200 | Inconclusive | 25 | 88 |
| ADA‐NEG | 299 | 87 | |
| Non‐TE‐POS NAb‐NEG | 21 | 94 | |
| Non‐TE‐POS NAb‐POS | 4 | 93 | |
| TE‐POS NAb‐NEG | 19 | 91 | |
| TE‐POS NAb‐POS | 12 | 77 |
TE, treatment‐emergent ADA; NAb, neutralizing antibodies; POS, positive; NEG, negative. aPatients were treated with the indicated dose continuously in parts 1, 2 and 3; does not include patients treated with placebo or etanercept and who were later treated with tildrakizumab.
At both week 12 and week 52, TE‐POS NAb‐POS patients trended towards lower PASI improvement. Other ADA‐positive patient categories and inconclusive patients were comparable with ADA‐NEG patients (Tables 4 and 5). Although inconclusive patients could potentially demonstrate false‐negative ADA samples, the observed mean percentage PASI improvement in this group was not reduced. In addition, testing positive for NAbs was not always related to decreased clinical response, as non‐TE‐POS NAb‐POS patients and ADA‐NEG patients showed similar tildrakizumab levels and PASI improvements.
Effect of immunogenicity on safety in part 1 (pooled analysis)
In the placebo‐controlled part 1 periods of each study, TE‐POS patients (TE‐POS NAb‐POS and TE‐POS NAb‐NEG combined) were compared with ADA‐NEG and inconclusive patients (combined). For the patients randomized to 100 mg in part 1, incidences of one or more AEs, one or more serious AEs and drug‐related AEs were similar in the TE‐POS group compared with the ADA‐NEG and inconclusive combined groups (Table 6). For patients randomized to 200 mg in part 1, incidences of patients with one or more AEs were somewhat higher for the TE‐POS group than for the ADA‐NEG and inconclusive combined group (62% vs. 48·3%), but were similar for both serious AEs (0% vs. 2·2%, respectively) and drug‐related AEs (14% vs.14·1%, respectively). No TE‐POS patients in either the 100‐mg or 200‐mg groups discontinued due to an AE.
Table 6.
Effect of antidrug antibodies (ADAs) on adverse events in treatment‐emergent‐positive patients compared with negative and inconclusive (combined) patients in the placebo‐controlled safety pool
| Tildrakizumab | |||
|---|---|---|---|
| 100 mg | 200 mg | 100 or 200 mg | |
| Treatment emergent positive | |||
| Patients in population | 30 (100) | 29 (100) | 59 (100) |
| With one or more adverse events | 15 (50) | 18 (62) | 33 (56) |
| With serious adverse events | 1 (3) | 0 | 1 (2) |
| With drug‐related adverse eventsa | 3 (10) | 4 (14) | 7 (12) |
| ADA‐negative and inconclusive | |||
| Patients in population | 632 (100) | 629 (100) | 1261 (100) |
| With one or more adverse events | 308 (49) | 304 (48) | 612 (48) |
| With serious adverse events | 9 (1) | 14 (2) | 23 (2) |
| With drug‐related adverse eventsa | 96 (15) | 89 (14) | 185 (15) |
Data are presented as n (%). aDetermined by the investigator to be related to the drug.
Effect of immunogenicity on immunogenicity‐related safety through 52–64 weeks (nonpooled analysis)
The potential of ADAs to affect specific immunogenicity‐related safety events in the phase IIb and phase III studies following continuous dosing with 100 mg or 200 mg through 52–64 weeks was additionally summarized for each ADA patient category without pooling. Drug‐related hypersensitivity was observed in one of 50 (2%) TE‐POS NAb‐NEG patients, in two of 812 (0·2%) ADA‐NEG patients and in one of 41 (2%) inconclusive patients. Anaphylaxis was not observed in any patient.
The incidence of injection‐site reactions was similar in the non‐TE‐POS NAb‐NEG, TE‐POS NAb‐POS and inconclusive subgroups: three of 47 (6%), two of 31 (6%) and two of 41 (5%), respectively, compared with the ADA‐NEG category: 49 of 812 (6·0%). The incidence in TE‐POS NAb‐NEG patients (five of 50, 10%) was somewhat higher than in ADA‐NEG patients. Overall, the incidence of potential immunogenicity‐related AEs showed no clear trend in any positive ADA patient category or the inconclusive category compared with ADA‐NEG patients (Table S2; see Supporting Information).
Discussion
In this integrated analysis of three randomized controlled clinical trials, we observed treatment‐emergent antibodies in around 4% of 1400 evaluable patients receiving tildrakizumab over 12–16 weeks and around 7% of 780 evaluable patients with continuous use for 52–64 weeks. ADA development was observed across all clinical trials, although most ADA‐positive patients did not have clinical effects. A small proportion of patients were TE‐POS NAb‐POS (0·6% at 12–16 weeks and around 3% at 52–64 weeks). This subgroup experienced a modest decrease in tildrakizumab PK and a reduction in clinical response as measured by PASI score mean reductions of 10–15% relative to ADA‐NEG patients at 52 weeks. No association was observed between tildrakizumab ADA development and the occurrence of AEs.
Biologic medications that target the IL‐23/IL‐17 axis demonstrate a wide range of immunogenicity. Although analyses of ADA development in individual studies are affected by factors such as sample handling, the assay platforms, characteristics of the assays and underlying disease, we observed that ADA development with tildrakizumab was in the range observed for other monoclonal antibodies targeting IL‐23/IL‐17. Among patients taking the IL‐12/23 and IL‐23 antagonists ustekinumab and guselkumab, approximately 6% were observed to develop ADAs.13, 14 For guselkumab, lower trough levels were observed in a portion of ADA‐positive patients.14 In the IL‐17 and IL‐17 receptor antagonists, a wider range was observed: less than 1% of patients on secukinumab developed antibodies in up to 52 weeks of treatment, 22% of patients on ixekizumab developed ADAs over 60 weeks of treatment, and 3% of patients on the IL‐17 receptor antagonist brodalumab developed ADAs.15, 16, 17 For patients treated with ixekizumab, higher antibody titres were associated with decreasing drug concentrations and clinical response.16
In our analysis, TE‐POS NAb‐POS patients showed the highest postdose titres. These patients demonstrated a decrease in mean tildrakizumab concentrations and a modest increase in median drug clearance compared with ADA‐NEG patients. Patients in other positive ADA categories were generally similar to ADA‐NEG patients with regard to mean tildrakizumab concentrations and drug clearance. We observed that ADAs were often detectable at multiple time points in patients with reduced tildrakizumab concentrations. Inconclusive patients had mean tildrakizumab concentration profiles and clearance values similar to ADA‐NEG patients, demonstrating no impact on PK in this subgroup (Fig. 2).
No effects on PASI or PGA responses at week 12 were observed for TE‐POS patients when NAb‐POS and NAb‐NEG patients were pooled. However, in the nonpooled analysis, TE‐POS NAb‐POS patients showed a reduction in mean PASI improvement compared with ADA‐NEG patients. These reductions were by 13% and 38% at week 12 and by 15% and 10% at week 52, for doses of 100 mg and 200 mg, respectively. Inconclusive patients showed a similar mean PASI improvement to ADA‐NEG patients at both week 12 and week 52 (Tables 4 and 5). Other studies have similarly shown the importance of looking at subgroups of TE‐POS patients to identify clinically relevant effects.18, 19, 20 However, only 3% of patients following long‐term treatment were TE‐POS NAb‐POS, which should be noted with regard to the interpretation of clinical effects from ADAs in this analysis.
The effects of ADAs were more pronounced on PK than clinical response. The reductions in exposure due to ADAs may not have been sufficiently extensive or prolonged to have a pronounced effect on efficacy. The exposure–response and PK–pharmacodynamic models for tildrakizumab estimated EC50 values at 0·36 μg mL−1 (90% confidence interval 0·22–0·61) and 0·25 μg mL−1, respectively, implying that at these exposure levels at least half of the maximum effect in PASI 75 can be expected.21 The finding that the effects of ADAs on PK are more sensitive than the effects on clinical response has also been observed with other biologics.22, 23, 24
ADA development was not associated with an increase in serious AEs or discontinuation from treatment. Overall, the incidence of potential immunogenicity‐related AEs showed no clear trend in inconclusive patients or any positive ADA patient category compared with ADA‐NEG patients (Table S2; see Supporting Information). This is similar to results with other IL‐23/IL‐17 biologics.
The high proportion of inconclusive patients on short‐term treatment with tildrakizumab 200 mg is a limitation related to the low drug tolerability level of the ADA assay (6 μg mL−1). A more drug‐tolerant ADA assay would have yielded a lower proportion of inconclusive patients. However, examples have shown that while sensitive ADA assays with high drug tolerance may detect a higher incidence of ADAs, the additional incidence is not generally correlated with additional effects on PK, efficacy and safety.20, 25 The inconclusive patients in the phase II–III programme consistently showed PK, efficacy and safety data that were similar to those in ADA‐NEG patients (Fig. 2, Tables 4 and 5 and Table S2; see Supporting Information). Thus, although the ADA assay performance was ambiguous for the inconclusive patients, no clinically relevant effects were observed. Another limitation is that effects of treatment interruptions on ADA development were not evaluated.
In summary, this analysis included a robust dataset of evaluable patients, particularly for the 100‐mg dose, which was recently approved by the FDA and EMA for treatment of chronic plaque psoriasis. TE‐POS NAb‐POS patients, who represented a small proportion (3%) of the patient population, experienced a modest decrease in tildrakizumab PK, correlating with a 10–15% reduction in clinical response at week 52. No apparent association between the development of antibodies to tildrakizumab and the occurrence of AEs was observed.
Supporting information
Fig S1. Effect of antidrug antibodies on tildrakizumab concentrations for patients treated with 100 mg or 200 mg for 12 weeks.
Table S1 Assays for detection of tildrakizumab, anti‐tildrakizumab antibodies and neutralizing antibodies in serum.
Table S2 Effect of antidrug antibodies (ADAs) on potential immunogenicity‐related adverse events in the base period safety pool by ADA patient category, all patients as treated.
Acknowledgments
The authors give sincere thanks and appreciation to the trial patients and staff who participated in these trials. The authors also wish to acknowledge medical writing and editorial support provided by Anish Mehta; submission assistance provided by Jennifer Pawlowski, MS; bioanalytical support by Lauren Nardini and Marina Ichetovkin; and immunogenicity dataset programming provided by Dan Xiao (all from Merck & Co., Inc., Kenilworth, NJ, U.S.A.).
Conflicts of interest A.B.K. is a consultant and investigator for Merck & Co., Amgen, AbbVie, Janssen, Novartis, Dermira and Pfizer; is a consultant for Sun Pharmaceuticals, Bristol‐Myers Squibb, Lilly and VBL; and has received fellowship funding from Janssen. A.B. has served as a scientific adviser and/or clinical study investigator for AbbVie, Aclaris, Akros, Allergan, Almirall, Amgen, Boehringer Ingelheim, Celgene, Dermavant, Dermira, Inc., Eli Lilly and Company, Galderma, Genentech/Roche, GlaxoSmithKline, Janssen, LEO Pharma, Meiji, Merck Sharp & Dohme, Novartis, Pfizer, Purdue Pharma, Regeneron, Revance, Sandoz, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, UCB Pharma, Valeant and Vidac; and as a paid speaker for Janssen, Regeneron and Sanofi Genzyme. K.A.P. has received honoraria for being a speaker, consultant and/or investigator for AbbVie, Active Biotech, Akesis, Allergan, Amgen, Anacor, Astellas, AstraZeneca, Basilea, Baxter, Bayer, Biogen, Boehringer Ingelheim, Bristol‐Myers Squibb, CanFite, Cato, Celgene, Centocor, Cepheid, Cipher, Coherus, Dow Pharma, Eli Lilly, Endocyte, Ferring Pharma, Forward Pharma, Galderma, Genentech, Gilead, GlaxoSmithKline, Janssen, Kyowa Hakko Kirin, Kythera, LEO Pharma, MedImmune, Meiji Seika Pharma, Merck Serono, Merck Sharp & Dohme, Mylan, Novartis, Pfizer, Regeneron Pharmaceuticals, Inc., Rigel, Roche, Sanofi‐Aventis, Sosei, Takeda, UBC and Vertex. K.R. has served as a consultant or paid speaker for, or participated in clinical trials sponsored by AbbVie, Amgen, Biogen, Celgene, Centocor, Covagen, Forward Pharma, GlaxoSmithKline, Janssen‐Cilag, LEO Pharma, Lilly, Medac, Merck & Co., Novartis, Pfizer, Takeda and Vertex. T.K. and P.K. are consultants paid for by Merck & Co., Inc., Kenilworth, NJ, U.S.A. Q.L. and D.M. are employees of Merck & Co., Inc., Kenilworth, NJ, U.S.A. F.vA. is a former employee of Merck & Co., Inc., Kenilworth, NJ, U.S.A.
Funding sources These analyses and the studies evaluated in this report were funded by Merck & Co., Inc., Kenilworth, NJ, U.S.A.
Conflicts of interest Conflicts of interest statements can be found in the Appendix.
https://doi.org/10.1111/bjd.18662 available online
References
- 1. Bendtzen K. Immunogenicity of anti‐TNF‐α biotherapies: I. Individualized medicine based on immunopharmacological evidence. Front Immunol 2015; 6:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther 2002; 24:1720–40. [DOI] [PubMed] [Google Scholar]
- 3. Carrascosa JM, van Doorn MB, Lahfa M et al Clinical relevance of immunogenicity of biologics in psoriasis: implications for treatment strategies. J Eur Acad Dermatol Venereol 2014; 28:1424–30. [DOI] [PubMed] [Google Scholar]
- 4. Muram TM, Sloan JM, Chain JS et al A highly sensitive and drug‐tolerant anti‐drug antibody screening assay for ixekizumab using affinity capture elution. J Invest Dermatol 2016; 136:1513–15. [DOI] [PubMed] [Google Scholar]
- 5. Reich K, Blauvelt A, Armstrong A et al Secukinumab, a fully human anti‐interleukin‐17A monoclonal antibody, exhibits minimal immunogenicity in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol 2017; 176:752–8. [DOI] [PubMed] [Google Scholar]
- 6. Reich K, Armstrong AW, Foley P et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76:418–31. [DOI] [PubMed] [Google Scholar]
- 7. Reich K, Papp KA, Baluvelt A et al Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 2017; 390:276–88. [DOI] [PubMed] [Google Scholar]
- 8. Papp K, Thaçi D, Reich K et al Tildrakizumab (MK‐3222), an anti‐interleukin‐23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo‐controlled trial. Br J Dermatol 2015; 173:930–9. [DOI] [PubMed] [Google Scholar]
- 9. Kopp T, Riedl E, Bangert C et al Clinical improvement in psoriasis with specific targeting of interleukin‐23. Nature 2015; 521:222–6. [DOI] [PubMed] [Google Scholar]
- 10. ILUMYA™ (tildrakizumab‐asmn) injection, for subcutaneous use. Merck Sharp & Dohme Corp., 2018. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761067s000lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 11. ILUMETRI (tildrakizumab). Almirall S.A., 2018. Available at: https://www.ema.europa.eu/en/documents/overview/ilumetri-epar-medicine-overview_en.pdf (last accessed 15 May 2019). [Google Scholar]
- 12. Jauslin P, Kulkarni P, Wada R et al Population pharmacokinetic modelling of tildrakizumab (MK‐3222), an anti‐interleukin‐23‐p19 monoclonal antibody, in healthy volunteers and patients with psoriasis. Br J Dermatol 2017; 177:e255. [DOI] [PubMed] [Google Scholar]
- 13. STELARA® (ustekinumab) injection, for subcutaneous or intravenous use. Janssen Biotech, Inc., 2009. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761044lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 14. TREMFYA™ (guselkumab) injection, for subcutaneous use. Janssen Biotech, Inc., 2017. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 15. COSENTYX® (secukinumab) injection, for subcutaneous use. Novartis Pharmaceuticals Corporation, 2015. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125504s001s002lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 16. TALTZ (ixekizumab) injection, for subcutaneous use. Eli Lilly and Company, 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125521s000lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 17. SILIQ™ (brodalumab) injection, for subcutaneous use. Valeant Pharmaceuticals North America LLC, 2017. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761032lbl.pdf (last accessed 15 May 2019). [Google Scholar]
- 18. Gordon KB, Blauvelt A, Papp KA et al Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016; 375:345–56. [DOI] [PubMed] [Google Scholar]
- 19. Ridker PM, Tardif JC, Amarenco P et al Lipid‐reduction variability and antidrug‐antibody formation with bococizumab. N Engl J Med 2017; 376:1517–26. [DOI] [PubMed] [Google Scholar]
- 20. Song S, Yang L, Trepicchio WL, Wyant T. Understanding the supersensitive anti‐drug antibody assay: unexpected high anti‐drug antibody incidence and its clinical relevance. J Immunol Res 2016; 2016:3072586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kerbusch T, Li H, Wada D, Wenning L. Exposure‐response analyses from a phase 2b and two phase 3 randomized controlled trials of tildrakizumab for the treatment of chronic plaque psoriasis. J Am Acad Dermatol 2018; 79 (Suppl. 1):AB139. [Google Scholar]
- 22. Wang YM, Wang J, Hon YY et al Evaluating and reporting the immunogenicity impacts for biological products – a clinical pharmacology perspective. AAPS J 2016; 18:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tham LS, Tang CC, Choi SL et al Population exposure‐response model to support dosing evaluation of ixekizumab in patients with chronic plaque psoriasis. J Clin Pharmacol 2014; 54:1117–24. [DOI] [PubMed] [Google Scholar]
- 24. Ungar B, Chowers Y, Yavzori M et al The temporal evolution of antidrug antibodies in patients with inflammatory bowel disease treated with infliximab. Gut 2014; 63:1258–64. [DOI] [PubMed] [Google Scholar]
- 25. Van Stappen T, Vande Casteele N, Van Assche G et al Clinical relevance of detecting anti‐infliximab antibodies with a drug‐tolerant assay: post hoc analysis of the TAXIT trial. Gut 2018; 67:818–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Effect of antidrug antibodies on tildrakizumab concentrations for patients treated with 100 mg or 200 mg for 12 weeks.
Table S1 Assays for detection of tildrakizumab, anti‐tildrakizumab antibodies and neutralizing antibodies in serum.
Table S2 Effect of antidrug antibodies (ADAs) on potential immunogenicity‐related adverse events in the base period safety pool by ADA patient category, all patients as treated.