

Abstract
Albicidin is a recently described natural product that strongly inhibits bacterial DNA gyrase. The pronounced activity, particularly against Gram‐negative bacteria, turns it into a promising lead structure for an antibacterial drug. Hence, structure–activity relationship studies are key for the in‐depth understanding of structural features/moieties affecting gyrase inhibition, antibacterial activity and overcoming resistance. The 27 newly synthesized albicidins give profound insights into possibilities for variations of the C‐terminus. Furthermore, in the present study, a novel derivative has been identified as overcoming resistance posed by the Klebsiella‐protease AlbD. Structural modifications include, for example, azahistidine replacing the previous instable cyanoalanine as the central amino acid, as well as a triazole amide bond isostere between building blocks D and E.
Keywords: albicidin, antibiotics, drug discovery, natural products, structure–activity relationships
27 newly synthesized albicidins give profound insights into possibilities for variations of the C‐terminus. In the present study a novel derivative has been identified as overcoming resistance posed by the Klebsiella‐protease AlbD. Structural modifications include, for example, azahistidine replacing the previous instable cyanoalanine as the central amino acid and a triazole amide bond isostere between building blocks D and E (see graphic).

The rise and spread of antimicrobial resistance poses one of the greatest threats to public health. According to a recent study, the number of annual deaths attributable to drug‐resistant infections could reach 10 million by the year 2050.1 The World Health Organization (WHO) and its member states have recognized the devastating prospect of a post‐antibiotic era and outlined the objectives to combat antimicrobial resistance (AMR) in a Global Action Plan. A Priority Pathogens List (PPL) was drawn up in a bid to promote research and development of new antibiotics, because the current clinical pipeline is insufficient to diminish the threat posed by AMRs.2 Since the majority of new drugs are merely modifications of existing ones, more investment is sorely needed to foster innovation, in particular, for therapeutics targeting the more challenging Gram‐negative pathogens. Novel antibiotic agents are classified innovative if they fulfil at least one of the following criteria: absence of cross‐resistance to existing antibiotics, new chemical class, new target or new mechanism of action.3
Exhibiting antibacterial activity at nanomolar concentrations against both Gram‐positive and ‐negative microorganisms, the oligoaromatic peptide antibiotic albicidin (1), which is produced by the sugarcane pathogenic bacterium Xanthomonas albilineans, represents a promising lead structure in the search for a new class of therapeutically useful anti‐infectives (Figure 1).4 The phytotoxin albicidin is a potent inhibitor of DNA gyrase, which has been similarly found for the structurally related cystobactamids5 and coralmycins.6 With a half maximum inhibitory concentration (IC50) of 40 nm, albicidin exhibits an inhibitory activity similar to that of DNA gyrase‐inhibiting quinolones and coumarins.4, 7 Its unique structure is composed of a cinnamoyl residue at the N‐terminus (building block A) and a dipeptidic moiety at the C‐terminus (building blocks E and F). The latter consists of two para‐aminobenzoic acids (pABAs), each decorated with adjacent methoxy and hydroxy groups. The only stereocenter of albicidin is featured in an unusual l‐cyanoalanine (building block C), which has been found to be prone to hydrolysis. Lastly, two unsubstituted pABAs (building blocks B and D) connect the central building block C with the two termini.8
Figure 1.
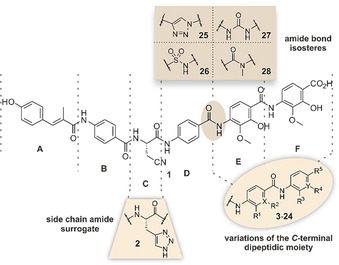
Structures of albicidin (1), azahistidine‐albicidin (2) and its novel derivatives 3–28. The individual fragments of the peptide are assigned a letter from A to F. For the identity of the residues investigated, see Table 1.
To maximize the bioactivity of albicidin while minimizing the effects of known resistance factors structure–activity relationship (SAR) studies are imperative. The first total synthesis of albicidin9 paved the way for initial SAR studies, which included variations of the cinnamoyl residue (building block A)10 and the incorporation of various α‐amino acids at the central building block C.11 We believe that albicidin's C‐terminal dipeptidic moiety (E–F) is a key pharmacophoric region. As of yet, there has been no extensive and systematic investigation of the SAR for this important part of the molecule. Herein, we report the synthesis of 26 new albicidin derivatives with variations of the C‐terminal dipeptidic group and their antimicrobial activities against a broad panel of pathogens, including members of the increasingly resistant ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp.) group. All analogues were additionally tested for their capacity to inhibit DNA gyrase and for their lack of sensitivity towards two of albicidin's most important resistance factors, the binding protein AlbA12 and the serine protease AlbD.13
Based on our previous investigations11 we first sought a viable replacement for the hydrolytically unstable β‐cyanoalanine as building block C. The noncanonical amino acid 2‐amino‐3‐(1H‐1,2,3‐triazol‐4‐yl)propanoic acid (azahistidine) appealed to us as a suitable substitute as it mimics the cyanoalanine and methoxy‐asparagine moieties present in the naturally occurring variants of albicidin (1).14 Indeed, the synthetic variant 2 exhibited superior antibacterial activity against the tested E. coli, S. typhimurium, B. subtilis and M. luteus strains, as well as an eight‐fold higher activity against ciprofloxacin (CIP) sensitive K. pneumoniae (Supporting Information, Table S1). Consequently, we used azahistidine‐albicidin 2 as a template structure for the subsequent SAR study: The first set of analogues (3–17) arises from sequential deletion of the methoxy and hydroxy groups present in the C‐terminal dipeptidic pABA moiety (Table 1, green). Removing any number of substituents leads to a varying spectrum of potency but generally reduces overall activity. The MIC values for trisubstituted variants 3–6 suggest that the methoxy group in E is the most critical one regarding single deletions. Its absence leads to a significant drop of activity for compound 3 against both CIP sensitive and resistant S. aureus, B. subtilis and M. luteus, while potency against the remaining panel, including all E. coli strains, is similar for 3–6. In general, potency against Gram‐positive pathogens gradually decreases with an increasing number of deletions of functional groups in E and F, leading to a loss of activity for unsubstituted variant 17 and monosubstituted derivatives 13–16. Despite the lack of all substituents, the overall activity against Gram‐negative pathogens only slightly decreases for 17. Contrary to the remaining analogues from the deletion sequence, compound 16—bearing a single methoxy group in E—shows an exceptionally low potency against all tested Gram‐negative pathogens.
Table 1.
Assignment of the residues for albicidin analogues with sequential deletion of the substituents of the C‐terminal dipeptidic moiety (3–17), sequential replacement of the methoxy groups with ethoxy groups (18–20), sequential replacement of the phenolic core structure with pyridines (21–23), and with replacement of the carboxylic acid group with a sulfonic acid group (24). All derivatives contain azahistidine as building block C.
|
|
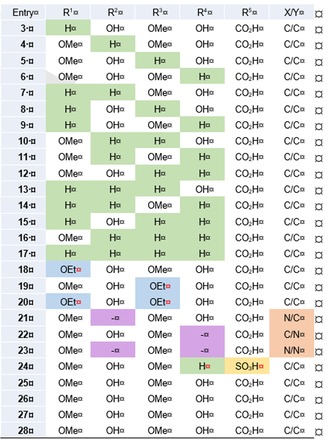
Previous findings have shown a tolerance for iso‐propoxy groups as methoxy‐substitutes.14, 15 Therefore, we hoped that more hydrophobic alkoxy groups in building blocks E and F might help boost activity. To test this hypothesis, the methoxy moieties of the parent compound 2 were successively replaced by ethoxy groups to produce synthetic analogues 18–20 (Table 1, blue). Although each of the three compounds inhibited DNA gyrase and showed high to very high activities throughout the series of tested pathogens—except for Klebsiella strains—the doubly substituted analogue 20 stands out (Figure 2). It displays an increased spectrum of activity and the highest potency against Gram‐positive B. subtilis, M. phlei and most importantly CIP sensitive and resistant S. aureus. Remarkably, variant 20 is highly potent on a CIP resistant strain of Gram‐negative A. baumannii, with MIC values below those of compound 2 by up to a factor of 65.
Figure 2.
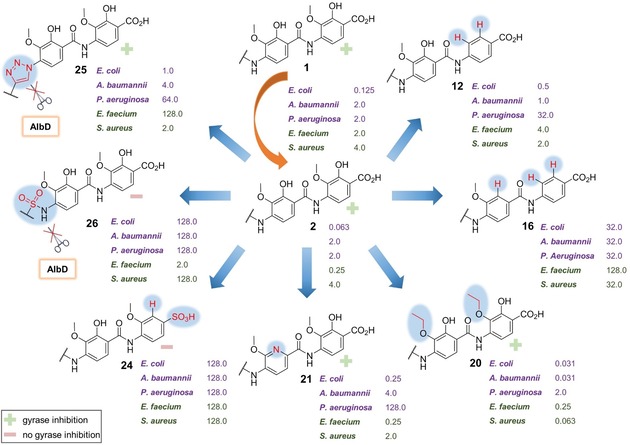
Minimum inhibitory concentrations (MICs) are given in μg mL−1 for selected albicidin derivatives. Values are shown for CIP resistant strains of E. coli, A. baumannii, P. aeruginosa, S. aureus, and a CIP sensitive strain of E. faecium. Gram‐negative bacteria are highlighted in purple and Gram‐positive ones in green. The capacity of the variants to inhibit bacterial DNA gyrase is indicated with a green (+) for active and a light red (−) for inactive. For a complete list of investigated compounds, see Supporting Information, Tables S1 and S2.
To expand the chemical space for the SAR study, heterocyclic derivatives 21–23, in which the phenolic core structures of E and F are replaced by pyridines, were also synthesized (Table 1, purple). While introducing a methoxypyridine as the building block E only had a minor effect on the overall activity of 21, a significant drop in activity—particularly against very important P. aeruginosa strains—and loss of the ability to inhibit gyrase was observed for variants 22 and 23 (Supporting Information Table S1 and S2). Possibly, the deleterious effect caused by the pyridine in building block F (compound 22) is predominant and explains the poor activity observed for compound 23. One could conclude that the hydroxy group in building block F is imperative, but a direct comparison of 22, for example, with the trisubstituted and still active analogue 6, negates that assumption and makes adverse electronic effects and H‐bonding interactions between the nitrogen atom of the pyridine and the adjacent carboxyl group more likely.
Since we have previously found that the amide bond between the C‐terminal dipeptide (E–F) and the pABA (D) is hydrolysed by the protease AlbD13 a series of analogues containing amide isosteres was synthesized in an effort to escape enzymatic cleavage. These included triazole 25, sulfonamide 26, urea derivative 27 and the N‐methyl amide 28 (Figure 1).16 All but the triazole‐containing analogue 25 were either poorly active or completely inactive in the MIC and supercoiling assays (Supporting Information, Tables S1 and S2). In contrast, compound 25 not only turned out to be highly potent against Gram‐negative strains of E.coli DSM 1116 and S. typhimurium TA100, with values in the range of the parent compound 2, but unlike the former it also maintained its activity in the presence of the serine protease AlbD as confirmed by agar diffusion assays (Supporting Information, Figure S4). The adverse results presumably stem from an altered geometry of the molecule upon introduction of an amide bond surrogate: The sulfonamide is considerably larger than an amide and likely induces a pronounced kink to compound 26. The three‐atom urea link is longer than the two‐atom amide link and possibly leads to a disfavoured conformation of 27 caused by an altered intramolecular H‐bonding network compared to 2. The latter reason might also hold true for analogue 28, because N‐methylation appears to disrupt the H‐bonding required to stabilize a favoured conformation. The same is true for the triazole‐containing analogue 25, which is much larger than the amide and lacks the ability to serve as an H‐bond donor. What appears to be a contradiction at first only strengthens the assumption that a certain degree of linearity is required for activity—the cyclic triazole moiety seems to confine a favoured geometry; however, to a lesser extent than intramolecular H‐bonding enabled by the amide. Additional MS‐cleavage experiments in the presence of AlbD were conducted for the biologically inactive compounds 26–28 to examine the stability of the respective surrogates (Supporting Information, Figures S8–S10). While the sulfonamide and urea linkers in analogues 26 and 27 proved to be stable towards AlbD, simple N‐methylation of the amide bond did not suffice to impede enzymatic cleavage of compound 28.
Due to favourable solubility characteristics offered by a sulfonic acid group,17 we intended to identify a derivative with a substituent in place of the C‐terminal carboxylic acid that would improve the poor aqueous solubility of albicidin. However, compound 24 did not exhibit any antibacterial activity or gyrase inhibition (Supporting Information, Tables S1 and S2). The reason for the lack of activity might be the higher acidity of the benzenesulfonic acid (pK a≈4) or simply the larger steric demand of a sulfonic acid group disfavouring interaction with the molecular target.18
With regard to the synthesis, our efforts were enormous, since the synthesis strategies for the preparation of the individual building blocks had to be adapted continuously. A [2+4]‐coupling strategy turned out to be the most viable for the convergent total syntheses of novel albicidin derivatives, combining an N‐terminal dipeptidic cinnamoyl residue (A–B) with a set of preassembled C‐terminal tetrapeptides (C–D–E–F).
As described in the literature,19 orthogonally protected azahistidine 30 was easily prepared in two steps from commercially available propargylglycine 29. Contrary to the cyano group present in the natural product, the robust triazole ring is stable towards basic conditions, allowing for convenient cleavage of the pivaloyloxymethyl (POM) protecting group of the side chain with aq. KOH after completed assembly of the albicidin analogues. A simple single‐step conversion of 30 afforded the dipeptidic aryl alkyne 31, which served as a substrate for a subsequent click‐reaction to form the triazole linker present in tetrapeptide 49 (Scheme 1 A,G). The synthesis of the tetrasubstituted pABA building block 36 from regioselectively protected dihydroxybenzoic acid 34 was achieved in a multigram scale following an alternative route compared to the original synthetic strategy. The key intermediate 35 served as a universal precursor for O‐alkylation, which enabled simple introduction of ethyl groups (Scheme 1 C). By omitting the hydroxy group in building block F, an acid‐labile sulfonate protecting group strategy could be adopted for the preparation of dipeptide 40, leading to the corresponding albicidin analogue 24 via tetrapeptide 47 (Scheme 1 D,G).20 Synthesis of N‐methyl amide 28 required selective monomethylation of aniline 41 in the presence of methyl iodide. This was achieved by installing a trifluoroacetyl protecting group prior to methylation to generate compound 42. The protecting group could easily be removed afterwards with K2CO3 in MeOH to afford the desired dipeptide 43 (Scheme 1 E). In the final step of the assembly, all tetrapeptides were coupled to the active ester 33, which had been prepared in two steps from previously reported cinnamoyl building block 32 (Scheme 1 B). Activation of the A–B dipeptide with pentachlorophenol (PCP), rather than previously used HATU, led to higher yields and considerably facilitated the final purification step by reversed‐phase preparative HPLC.
Scheme 1.
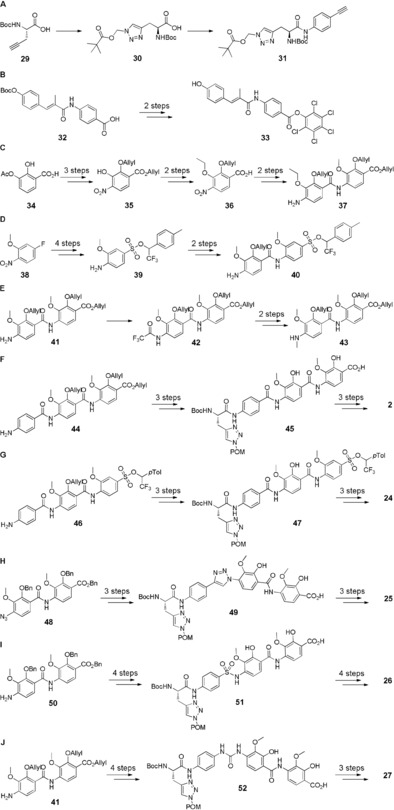
Synthetic pathways for the preparation of dipeptidic building blocks (A–E) and assembly of final albicidin derivatives (F–J). The C‐terminal dipeptidic building blocks inherent to compounds 3–15 and 21–23 have been prepared analogously to previously reported protocols. The assembly of the final albicidin derivatives was in line with that of azahistidine albicidin 2. For detailed schemes with reagents and conditions, see Supporting Information, Schemes S1 and S2.
In summary, we have carried out the first systematic SAR study for albicidin's C‐terminal dipeptidic pABA moiety. Initially, our extensive endeavour unearthed a new lead structure, azahistidine albicidin 2, exhibiting both superior antibacterial activity and chemical stability to the natural product 1. The doubly ethoxy‐substituted compound 20 exhibits outstanding potency and a broadened spectrum of activity, including a CIP‐resistant strain of A. baumannii. The triazole moiety in compound 25 was identified as a viable structural motif to overcome cleavage by the Klebsiella‐protease AlbD while preserving biological activity. Prospectively, our results will help improve a structure–activity guided drug design approach on the path to develop an urgently needed new clinical candidate.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SU 239/11‐1 and PE 2600/1‐1) and the BMBF (VIP grant: 03VP00030). The authors are grateful to Prof. Dr. Marcus Fulde (Freie Universität Berlin) for S2 assays. The authors also thank Dr. Andi Mainz for valuable discussions as well as Dr. Marius Morkunas, Lucas Härchen and Oliver Neumann for their contribution in the laboratory.
I. Behroz, P. Durkin, S. Grätz, M. Seidel, L. Rostock, M. Spinczyk, J. B. Weston, R. D. Süssmuth, Chem. Eur. J. 2019, 25, 16538.
References
- 1.J. O'Neill, Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, 2014.
- 2.World Health Organization, Global Action Plan on Antimicrobial Resistance, 2015. [DOI] [PubMed]
- 3.World Health Organization, Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis, 2017.
- 4. Birch R. G., Patil S. S., J. Gen. Microbiol. 1985, 131, 1069–1075. [DOI] [PubMed] [Google Scholar]
- 5. Baumann S., Herrmann J., Raju R., Steinmetz H., Mohr K. I., Hüttel S., Harmrolfs K., Stadler M., Müller R., Angew. Chem. Int. Ed. 2014, 53, 14605–14609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14835–14839. [Google Scholar]
- 6. Kim Y. J., Kim H.-J., Kim G.-W., Cho K., Takahashi S., Koshino H., Kim W.-G., J. Nat. Prod. 2016, 79, 2223–2228. [DOI] [PubMed] [Google Scholar]
- 7. Hashimi S. M., Wall M. K., Smith A. B., Maxwell A., Birch R. G., Antimicrob. Agents Chemother. 2007, 51, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cociancich S., Pesic A., Petras D., Uhlmann S., Kretz J., Schubert V., Vieweg L., Duplan S., Marguerettaz M., Noëll J., Pieretti I., Hugelland M., Kemper S., Mainz A., Rott P., Royer M., Sussmuth R. D., Nat. Chem. Biol. 2015, 11, 195–197. [DOI] [PubMed] [Google Scholar]
- 9. Kretz J., Kerwat D., Schubert V., Grätz S., Pesic A., Semsary S., Cociancich S., Royer M., Süssmuth R. D., Angew. Chem. Int. Ed. 2015, 54, 1969–1973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1992–1996. [Google Scholar]
- 10. Kerwat D., Grätz S., Kretz J., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1899–1903. [DOI] [PubMed] [Google Scholar]
- 11. Grätz S., Kerwat D., Kretz J., von Eckardstein L., Semsary S., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1499–1502. [DOI] [PubMed] [Google Scholar]
- 12. Rostock L., Driller R., Grätz S., Kerwat D., von Eckardstein L., Petras D., Kunert M., Alings C., Schmitt F.-J., Friedrich T., Wahl M. C., Loll B., Mainz A., Sussmuth R. D., Nat. Commun. 2018, 9, 3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vieweg L., Kretz J., Pesic A., Kerwat D., Grätz S., Royer M., Cociancich S., Mainz A., Süssmuth R. D., J. Am. Chem. Soc. 2015, 137, 7608–7611. [DOI] [PubMed] [Google Scholar]
- 14. von Eckardstein L., Petras D., Dang T., Cociancich S., Sabri S., Grätz S., Kerwat D., Seidel M., Pesic A., Dorrestein P. C., Royer M., Weston J. B., Sussmuth R. D., Chem. Eur. J. 2017, 23, 15316–15321. [DOI] [PubMed] [Google Scholar]
- 15. Hüttel S., Testolin G., Herrmann J., Planke T., Gille F., Moreno M., Stadler M., Brönstrup M., Kirschning A., Müller R., Angew. Chem. Int. Ed. 2017, 56, 12760–12764; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12934–12938. [Google Scholar]
- 16.
- 16a. Valverde I. E., Bauman A., Kluba C. A., Vomstein S., Walter M. A., Mindt T. L., Angew. Chem. Int. Ed. 2013, 52, 8957–8960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9126–9129; [Google Scholar]
- 16b. Brik A., Alexandratos J., Lin Y.-C., Elder J. H., Olson A. J., Wlodawer A., Goodsell D. S., Wong C.-H., ChemBioChem 2005, 6, 1167–1169; [DOI] [PubMed] [Google Scholar]
- 16c. Yang K.-W., Golich F. C., Sigdel T. K., Crowder M. W., Bioorg. Med. Chem. Lett. 2005, 15, 5150–5153; [DOI] [PubMed] [Google Scholar]
- 16d. Patani G. A., LaVoie E. J., Chem. Rev. 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- 17. Ballatore C., Huryn D. M., Smith A. B., ChemMedChem 2013, 8, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guthrie J. P., Can. J. Chem. 1978, 56, 2342–2354. [Google Scholar]
- 19. Roux S., Ligeti M., Buisson D.-A., Rousseau B., Cintrat J.-C., Amino Acids 2010, 38, 279–286. [DOI] [PubMed] [Google Scholar]
- 20. Pauff S. M., Miller S. C., J. Org. Chem. 2013, 78, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary