

Abstract
Group B Streptococcus serotypes Ia and Ib capsular polysaccharides are key targets for vaccine development. In spite of their immunospecifity these polysaccharides share high structural similarity. Both are composed of the same monosaccharide residues and differ only in the connection of the Neu5Acα2‐3Gal side chain to the GlcNAc unit, which is a β1‐4 linkage in serotype Ia and a β1‐3 linkage in serotype Ib. The development of efficient regioselective routes for GlcNAcβ1‐3[Glcβ1‐4]Gal synthons is described, which give access to different group B Streptococcus (GBS) Ia and Ib repeating unit frameshifts. These glycans were used to probe the conformation and molecular dynamics of the two polysaccharides, highlighting the different presentation of the protruding Neu5Acα2‐3Gal moieties on the polysaccharide backbones and a higher flexibility of Ib polymer relative to Ia, which can impact epitope exposure.
Keywords: carbohydrates, conformation analysis, glycosylation, regioselectivity, therapeutics
Glycosylation: A panel of GBS serotype Ia and Ib fragments was prepared by combined regioselective‐stereoselective approaches and used to unravel polysaccharide conformations responsible for immune specificity.
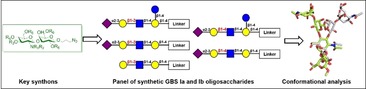
Introduction
Group B Streptococcus (GBS) is a leading cause of pneumonia, sepsis, meningitis, and death in neonates.1 It has also been associated with high rates of invasive diseases in the elderly.1 On the basis of variation in polysaccharide composition, the GBS sialic acid‐rich capsular polysaccharides (CPSs) are divided into ten serotypes (Ia, Ib, and II–IX).2 GBS CPSs are key virulence factors and considered the prime vaccine candidate to combat GBS infections.3 Monovalent conjugate vaccines prepared with GBS type‐specific polysaccharides representing the most frequent disease‐causing serotypes (Ia, Ib, II, III, and V), as well as a trivalent combination (Ia, Ib, III), have been tested in phase I/II clinical trials4 with the ultimate goal of developing a maternal vaccination strategy.1, 5 Multivalent formulations with six different serotypes are currently under clinical testing.6 GBS serotypes Ia, Ib, and III account for the majority of GBS related diseases.7 CPS Ia and Ib are structurally very similar. Both are composed of the same monosaccharide residues and differ only in the linkage between the Neu5Acα2‐3Gal side chain and the GlcNAc unit: a β1‐4 linkage in type Ia and a β1‐3 linkage in type Ib.8 This difference is critical in determining the immunospecificity (Figure 1).3, 9
Figure 1.
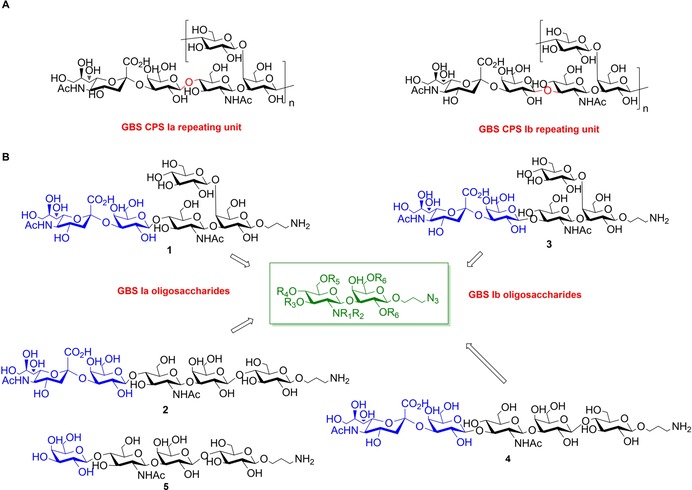
A) Chemical structure of GBS CPS Ia and Ib. B) Chemical structure of the target fragments from GBS CPS Ia (1 and 2) and CPS Ib (3 and 4). Synthesis of an unsialylated form of CPS Ia repeating units 5 was also envisaged.
The repeating units of CPS Ia and Ib can be described by the branched 1, 3 and linear 2, 4 frameshifts depicted in Figure 1. Intriguingly, the latter pentasaccharides 3 and 4 share identical monosaccharide composition with milk oligosaccharides, which have recently been proposed to inhibit GBS colonization.10 The availability of well‐defined GBS CPS glycans is key to explore interactions with serotype‐specific monoclonal antibodies in order to identify relevant glycoepitopes for elucidating the mechanism of action of the polysaccharide conjugates and for the development of synthetic carbohydrate‐based vaccines.11 The most studied of GBS polysaccharides is type III. This CPS is known to form a helical structure,12 and this feature has an impact on epitope exposure.13 Our group has recently synthesized CPSIII oligosaccharides that were used along with fragments obtained from CPS depolymerization to map a sialylated structural epitope spanning two repeating units.14 Considering that neither chemical nor enzymatic depolymerization reactions are available for CPS Ia and Ib, chemical synthesis is the only approach to obtain homogeneous oligosaccharides from the CPS.
Although the synthesis of the GBS CPS Ia repeating unit has been reported,15 the preparation of the pentasaccharide repeating unit of GBS CPS Ib has not been achieved. When approaching the synthesis of CPS Ia and Ib fragments, we envisaged the formation of the disaccharide GlcNAcβ1‐3Gal motif as a key step to enable convergent syntheses of a variety of structures depicted in Figure 1. Typically, installation of the GlcNAcβ1‐3Gal disaccharide within more complex glycans has been achieved with the 4‐hydroxyl group of the Gal acceptor either protected16, 17 or already engaged in a glycosidic linkage.18 Particularly, in the preparation of CPS Ia repeating units15 a 4,6‐O‐benzylidene‐protected Gal acceptor was used for glycosylation with a glucosamine trichloroacetimidate donor, and subsequent regioselective ring opening before further glycosylation of position 4 for the construction of the trisaccharide GlcNAcβ1‐3[Glcβ1‐4]Gal could take place. There is need of expeditious procedures for the construction of complex glycans, and regio‐ and stereoselective reactions distinguishing among diverse deprotected hydroxyls are highly desirable to simplify the oligosaccharide assembly.19 We reasoned that regioselective glycosylation of Gal 3‐OH would be the key for accelerating the synthesis of the GlcNAcβ1‐3Gal disaccharide and rendering the 4‐OH available for further glycosylation without the need of tedious protection/deprotection sequences.20
Herein, we report tactics to achieve regioselective syntheses of protected GlcNAcβ1‐3Gal building blocks and the use of these key synthons in convergent routes towards a series of fragments from CPS Ia and Ib repeating units with a built‐in aminopropyl linker amenable for future conjugation to carrier proteins (Figure 1). Furthermore, combination of NMR data from the synthetic GBS CPS Ia and Ib repeating units in their branched form 1 and 3, respectively, and molecular dynamics simulation allowed to shed light on how the variation of a single sugar connection dramatically affects the conformational properties of CPS Ia and Ib polysaccharides, and hence exposition of potential epitopes for antibody recognition.
Results and Discussion
Optimization of regioselective glycosylation of galactose
According to our retrosynthetic design (Figure 1), the target glycans 1–4 can be obtained through a [2+3] convergent strategy based on the glycosylation of a suitable trisaccharide acceptor with a Neu5Acα2‐3Gal donor. This approach envisages the challenging stereoselective α sialylation of the upstream galactose at an early stage of the synthesis.21 Alternative use of a Gal donor would enable the synthesis of 5. In this design, faster and efficient access to a GlcNAcβ1‐3Gal disaccharide building block plays a central role to obtain the trisaccharide acceptor without a temporary protection at position 4 for further assembly of GBS CPS Ia fragments. To achieve its regioselective synthesis, we investigated the effect of arming benzyl and disarming benzoyl groups22, 23 at position 2 and 6 of the Gal acceptors in tuning the reactivity of the 3‐ and 4‐OH, respectively, in combination with various protecting and leaving groups in the glucosamine donors. Despite the expected higher reactivity of the equatorial 3‐OH versus the axial 4‐OH, regioselective glycosylation of position 3 has been shown not to be trivial.16 Accordingly, we synthesized a series of glucosamine thioglycoside and trichloroacetimidate donors with the amine protected by the participating phthalimido (Phth) or trichloroethyl carbamate (Troc) group (experimental procedures are provided as Supporting Information).
Levulinoyl (Lev) and fluorenylmethyloxycarbonyl (Fmoc) were selected for temporary protection of either position 3 or 4. Alternatively, a 4,6‐O‐benzylidene was used to lock the 4‐ and 6‐hydroxyls to be subjected to regioselective ring opening delivering the 4‐OH at a later stage of the synthesis (Scheme 1). The prepared donors and acceptors were then coupled under several glycosylation conditions (Table 1 and Scheme 1) to optimize the synthesis of the GlcNAcβ1‐3Gal building block. The most efficient routes proved to be the combination of the 2,6‐di‐O‐benzoyl acceptor 11 with both donor 6 or 7 under N‐iodosuccinimide (NIS)/AgOTf‐mediated activation (Table 1, entries 7 and 8), which gave 14 a and 15 a in yields of 53 and 65 %, respectively, or the imidate 9 and acceptor 11 (Table 1, entry 9), which enabled the attainment of 14 a in 77 % yield.
Scheme 1.
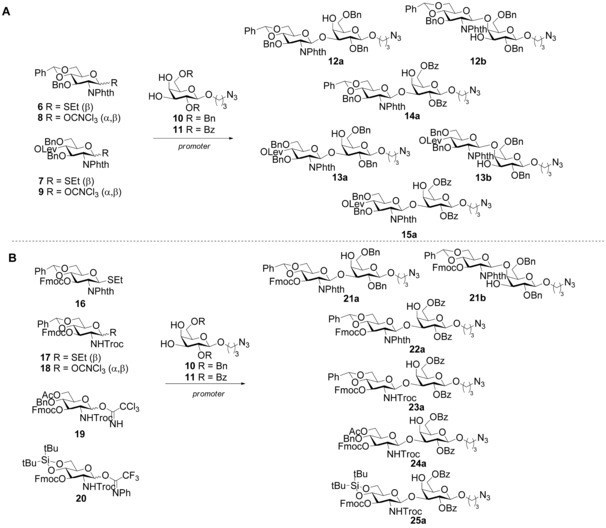
A) Preparation of disaccharide intermediates 12–15 for the synthesis of GBS CPS Ia repeating unit. Promoters and conditions are described in Table 1. B) Preparation of disaccharide intermediates 21–25 for the synthesis of GBS PSIb repeating unit. Promoters and conditions are described in Table 2.
Table 1.
Reaction of glucosamine donors 6–9 with Gal acceptors 10 and 11.
|
Entry |
Donor |
Acceptor |
Promoter, temperature [°C] |
Yield [%] |
|---|---|---|---|---|
|
1 |
6 |
10 |
NIS/TfOH, −30 |
n.d.[a] |
|
2 |
6 |
10 |
NIS/Ag(OTf), 30 |
12 a (43) |
|
12 b (26) | ||||
|
3 |
7 |
10 |
NIS/Ag(OTf), −30 |
13 a (40) |
|
13 b [b] (28) | ||||
|
4 |
8 |
10 |
TMSOTf, −10 |
12 a (31) |
|
5 |
9 |
10 |
TMSOTf, −10 |
13 a (45) |
|
6 |
6 |
11 |
NIS/TfOH, −30 |
n.d.[a] |
|
7 |
6 |
11 |
NIS/Ag(OTf), −30 |
14 a (53) |
|
8 |
7 |
11 |
NIS/Ag(OTf), −30 |
15 a (65) |
|
9 |
8 |
11 |
TMSOTf, −10 |
14 a (77) |
|
10 |
9 |
11 |
TMSOTf, −10 |
15 a (33) |
[a] CH2Cl2 was the solvent in all tested conditions; n.d.=not determined, product could not be detected; [b] The formation of the β1‐4 linkage was confirmed by acetylation of 13 b. In the 1H NMR spectrum a shift from 3.32 to 4.69 ppm of the H‐3 signal of Gal, appearing as a doublet of doublets with J 2,3=10.3 Hz and J 3,4=2.5 Hz was observed, confirming occurrence of glycosylation at position 4.
Similarly, conditions for the preparation of a GlcNAcβ1‐3Gal synthon with a temporary group at its C3′‐OH, to allow the ensuing assembly of GBS CPSIb fragments, were explored (Table 2 and Scheme 1). The glycosylation of di‐O‐benzyl acceptor 10 with donor 16 by using NIS with either TfOH or AgOTf as co‐promoters gave variable mixtures of the β1‐3 21 a and β1‐4 21 b disaccharides (Table 2, entries 1 and 2). Again, the di‐O‐benzoyl acceptor 11 in the presence of NIS/AgOTf activation at −30 °C allowed achieving a yield of 68 % (Table 2, entry 4), which confirms the improved capacity of the benzoyl substituents to govern the regioselectivity of the reaction compared with benzyl substituents. These conditions were also efficient for the GlcNTroc donor 17, which gave 23 a in 65 % yield (Table 2, entry 6). When the trichloroacetimidate 18 was used, the yield was increased up to 70 % (Table 2, entry 7), which corroborates the potential of this type of donor for the regioselective control of the reaction. Finally, trifluoroacetimidate glucosamine 20 bearing a 4,6‐O‐silylidene protection in the presence of TMSOTf as promoter afforded the target disaccharide 25 a in 62 % yield.24 The slighty higher flexibility or lower hindering effect of the silylidene relative to that of the benzylidene group favored the reaction. Overall, these results indicate that the regioselectivity of the glycosylation benefits from the decreased nucleophilicity of the axial 4‐hydroxyl, which is intrinsically less reactive than the 3‐hydroxyl group, induced by the electron‐withdrawing effect of the 2,6‐O‐benzoyl as compared with 2,6‐di‐O‐benzyl substituents in the Gal acceptor.
Table 2.
Reaction of glucosamine donors 16–20 with Gal acceptors 10 and 11.
|
Entry |
Donor |
Acceptor |
Promoter, temperature [°C] |
Yield [%] |
|---|---|---|---|---|
|
1 |
16 |
10 |
NIS/TfOH, −30 |
21 a (30) |
|
21 b (<5) | ||||
|
2 |
16 |
10 |
NIS/AgOTf, −30 |
21 a (38) |
|
21 b (26) | ||||
|
3 |
16 |
11 |
NIS/TfOH, −30 |
22 a (40) |
|
4 |
16 |
11 |
NIS/AgOTf, −30 |
22 a (68) |
|
5 |
17 |
11 |
NIS/TfOH, −30 |
n.d.[a] |
|
6 |
17 |
11 |
NIS/AgOTf, −30 |
23 a (65) |
|
7 |
18 |
11 |
TMSOTf, −10 |
23 a (70) |
|
8 |
19 |
11 |
TMSOTf, −10 |
24 a (50) |
|
9 |
20 |
11 |
TMSOTf, −10 |
25 a (62) |
[a] CH2Cl2 was the solvent in all tested conditions.
In addition, mild activation conditions (NIS/AgOTf) for the thioglycoside donor or the torsional disarming effect of the benzylidene/silylidene group for the imidate donors appears to favor the regioselectivity of glycosylation at position 3.
Synthesis of GBS CPS Ia linear and branched repeating units
Having identified the two glycosylation partners giving the GlcNAcβ1‐3Gal motif in a regioselective fashion, we elongated the disaccharide building block to assemble the pentasaccharide repeating unit of GBS CPS Ia. To this end, reactions of glucose donor 26 25 with disaccharide donors 12 a and 14 a were performed to furnish trisaccharides 27 a and 27 b in 75 and 68 % yield, respectively (Scheme 2). The newly formed glycosidic bond was in β configuration, as expected by the presence of a participating group.
Scheme 2.
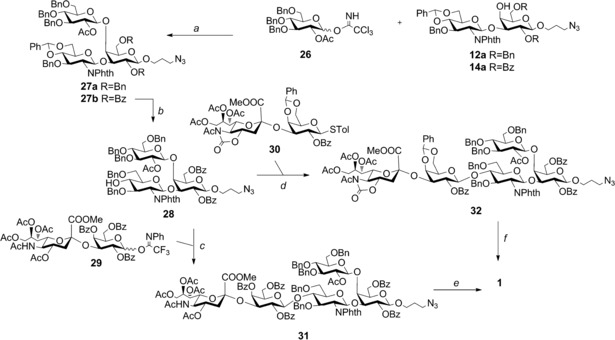
Assembly of GBS CPS Ia repeating unit. Reagents and conditions: a) TMSOTf, CH2Cl2 dry, −10 °C, (β‐) 75 % from 12 a, 68 % (β‐) from 14 a; b) Me3N⋅BH3, BF3 ⋅Et2O, MeCN, 0 °C, 70 %; c) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 75 %; d) TfOH, NIS, CH2Cl2 dry, −40 °C, (β‐) 73 %; e) LiI, Py, 120 °C; H2NCH2CH2NH2, EtOH, 90 °C; Ac2O, Py; MeONa, MeOH; H2, Pd‐C, 40 % (over five steps); f) 3 m NaOH, THF, reflux; Ac2O, MeOH; H2, Pd‐C, 45 % (over three steps).
Despite the deactivating effect of the 6‐O‐benzoyl ester relative to that of the 6‐O‐benzyl ether, the reaction proceeded with almost identical efficiency (Scheme 2), whereas a peracetylated trichloroacetimidate glucose donor with TMSOTf activation was ineffective for glycosylation of the 4‐OH. Considering the higher regioselectivity and yield achieved in synthesizing disaccharide 14 a, the resulting trisaccharide 27 b was advanced in the GBS CPS Ia repeating unit construction and subjected to regioselective opening of the 4,6‐O‐benzylidene acetal with BF3 ⋅Et2O and Me3N⋅BH3 to provide the acceptor 28 (70 %).
In order to complete the pentasaccharide construction, the sialo‐galactosyl trifluoroacetimidate donor 29 14a, 26 and thioglycoside 30 27 were tested. Of these two disaccharides, 30 can be prepared with a higher α stereoselectivity, whereas 29 is easily accessible from a commercial disaccharide precursor.14a
Glycosylation of trisaccharide 28 with 29 under TMSOTf activation gave the protected pentasaccharide 31 in 75 % yield, and the use of disaccharide 30 in the presence of NIS/TfOH led to the protected pentasaccharide 32 in a similar yield (73 %). Compound 30 was deprotected by a four‐step procedure,18 including 1) saponification of the methyl ester of Neu5Ac with lithium iodide in pyridine; 2) reaction with ethylenediamine in ethanol heated to reflux for concomitant removal of the O‐acetates and the NPhth protecting group; 3) reacetylation with acetic anhydride/pyridine to install the acetamide group of the GlcNAc residue along with acetyl esters; 4) methanolysis and final catalytic hydrogenation over Pd/charcoal to provide the target branched pentasaccharide 1.
Pentasaccharide 32 was first subjected to saponification with NaOH in THF heated to reflux, followed by amine reacetylation with a 2:3 acetic anhydride/methanol mixture.
Hydrogenation over Pd/charcoal afforded the target branched pentasaccharide 1 equipped with the aminopropyl linker suitable for conjugation. After purification by size exclusion chromatography, the final compound was obtained in 40 % overall yield from 31 and 45 % overall yield from 32, respectively (Scheme 2).
Next, we extended the same regioselective approach to the synthesis of the linear frameshift 2 of the serotype Ia repeating unit (Scheme 3). In this case, the benzoylated lactose 33 and the glucosamine donor 8 were chosen as glycosylation partners affording the linear trisaccharide acceptor 34 in 68 % yield with complete regioselectivity. Following benzylidene opening, the trisaccharide acceptor 35 was glycosylated with the two donors 29 and 30. The first glycosylation promoted by TMSOTf at 0 °C afforded the target linear pentasaccharide 36 in 65 % yield, with β stereo‐ and regioselectivity at C‐4 of GlcNAc over the C‐4 of Gal. The presence of the free galactose 4‐OH throughout all stages of the synthesis, from trisaccharide 34 to pentasaccharide 36, was monitored by following the signal of the Gal H‐4, which appeared at 3.97 ppm (d, J=2.7 Hz) in the 1H NMR and HSQC spectra of all synthetic intermediates. This confirmed the regioselectivity of the two glycosylations performed. Unexpectedly, reaction of 35 with the tolyl thioglycoside 30 under NIS/TfOH activation at −40 °C yielded only traces of the corresponding pentasaccharide, whereas mainly decomposition of the glycosyl donor was observed, as revealed by LC‐MS analysis. The linear pentasaccharide 36 was subjected to the five‐step deprotection protocol previously described for compound 31. The target oligosaccharide 2 was purified by size exclusion chromatography and obtained in 33 % overall yield (Scheme 3). NMR data of the synthesized fragments were in excellent agreement with those of the CPS Ia samples.8
Scheme 3.
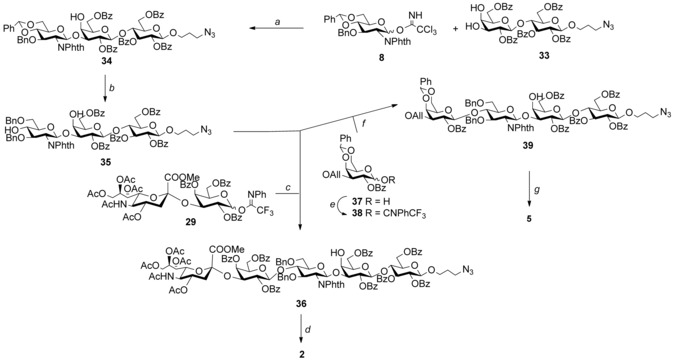
Assembly of linear GBS PS Ia fragments 2. Reagents and conditions: a) TMSOTf, CH2Cl2 dry, −10 °C, (β‐) 68 %; b) Me3N⋅BH3, BF3 ⋅Et2O, MeCN, 0 °C, 65 %; c) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 65 %; d) LiI, Py, 120 °C; H2NCH2CH2NH2, EtOH, 90 °C; Ac2O, Py; MeONa, MeOH; H2, Pd‐C, 33 % (over five steps); e) TFACl, Cs2CO3, CH2Cl2, 61 %; f) TMSOTf, −20 °C, CH2Cl2, (β‐) 72 %; g) PdCl2, MeOH; H2NCH2CH2NH2, EtOH, 90 °C; Ac2O, Py; MeONa, MeOH; H2, Pd‐C, 42 % (over five steps).
From acceptor 35 a desialylated CPS Ia linear fragment for future mapping studies was also obtained by glycosylation (72 % yield) with the trifluoroacetimidate 38, prepared from the known 1‐OH compound 37.18 After global deprotection tetrasaccharide 5 was obtained in 42 % yield (Scheme 3).
Synthesis of GBS CPS Ib linear and branched repeating unit
Differently than the GBS CPS Ia pentasaccharides, the two Ib frameshifts 3 and 4 required a glucosamine building block bearing a temporary protecting group at its C3‐OH and the creation of the Galβ1‐3GlcNAc linkage, which had a strong impact on our synthetic design. Initial attempts to prepare the branched pentasaccharide 3 by using a NPhth‐protected trisaccharide acceptor, similarly as done for the CPS Ia branched unit, were unsuccessful (Supporting Information, Scheme S9).
The C3‐OH of the glucosamine appeared significantly less reactive than the C4‐OH, which is likely due to the presence of the bulky NPhth group that could hinder the glycosylation reaction at the C3−OH. We anticipated that its replacement with a Troc protection would result in a higher nucleophilicity of the vicinal hydroxyl. Disaccharides 23 a and 25 a, which differ only in the cyclic protecting group blocking the glucosamine C4,6−OH groups, were selected to be elongated to the branched pentasaccharide 3 (Scheme 4). Glycosylation of the two acceptors with the armed Glc donor 26 under TMSOTf activation at 0 °C afforded the trisaccharides 40 and 41 in 63 and 70 % yield, respectively, as β anomers. After Fmoc removal by treatment with 10 % piperidine in CH2Cl2 (92 %), glycosylation with the sialogalactoside donor 29 of the two acceptors 42 and 43 was tested.
Scheme 4.
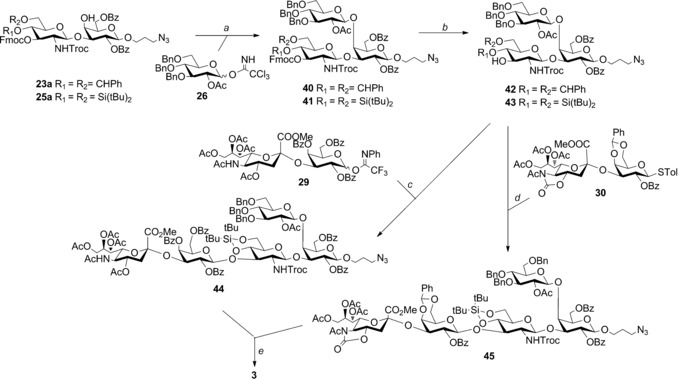
Assembly of GBS PSIb pentasaccharide branched unit 3. Reagents and conditions: a) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 63 % for 41, (β‐) 70 % or 42; b) piperidine, CH2Cl2 dry, 92 %; c) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 80 %; d) TfOH, NIS, CH2Cl2 dry, −40 °C, (β‐) 65 %; e) HF/pyridine, 0 °C; 3 m NaOH, THF, reflux; Ac2O/MeOH; H2/Pd‐C, 40 %.
Reaction of the 4,6‐O‐benzylidene trisaccharide 42 and 29 with TMSOTf as a promoter failed to afford the target pentasaccharide, leading to complete recovery of the unreacted acceptor. In contrast, reaction of acceptor 43, bearing the more flexible 4,6‐O‐silylidene ketal, with 29 in the presence of TMSOTf gave the target pentasaccharide 44 in 80 % yield (Scheme 4). This result suggests that the glycosylation of 42 was prevented by the steric and torsional constrain of the 4,6‐O‐benzylidene ring. Trisaccharide 43 was also efficiently β‐glycosylated with disaccharide donor 30 by NIS/TfOH activation, affording 45 in 65 % yield (Scheme 4). Despite a slightly lower yield in this step, the overall efficiency of the synthesis of the GBS serotype Ib branched repeating unit was superior when using the thioglycoside 30 with respect to the imidate 29 because of the better α stereoselectivity of the glycosylation leading to 30.26, 27 Pentasaccharides 44 and 45 were then deprotected by a four‐step protocol (Scheme 4): 1) desilylation by treatment with HF⋅pyridine, 2) saponification with NaOH in THF heated to reflux, for concomitant hydrolysis of the acyl esters, the Troc group, and the 5‐N,4‐O‐oxazolidinone protecting group and Neu5Ac methyl ester, 3) reacetylation of the amines by a 2:3 acetic anhydride/methanol mixture, 4) hydrogenation over Pd/charcoal. The target branched pentasaccharide 3 was obtained in 40 % yield.
Finally, we extended our regioselective approach to the synthesis of the linear frameshift 4 of the GBS serotype Ib repeating unit (Scheme 5). For this purpose, benzoylated lactose acceptor 33 was glycosylated with the 4,6‐O‐silylidene glucosamine imidate 20 under TMSOTf activation to give the target trisaccharide 46 with full β1‐3 stereo‐ and regioselectivity (55 %). Following Fmoc deprotection with piperidine in CH2Cl2, the obtained acceptor 47 was β‐glycosylated with imidate 29 to attain the linear protected pentasaccharide 48 (66 %). Reaction with thioglycoside 30 in TfOH and NIS reaction conditions also provided the analogous pentasaccharide 49 (40 %). The obtained pentasaccharides were deprotected and purified as described above. NMR spectroscopic data of the synthesized CPS Ib fragments were in excellent agreement with NMR spectroscopic data from samples of the bacterial polysaccharide.8a
Scheme 5.
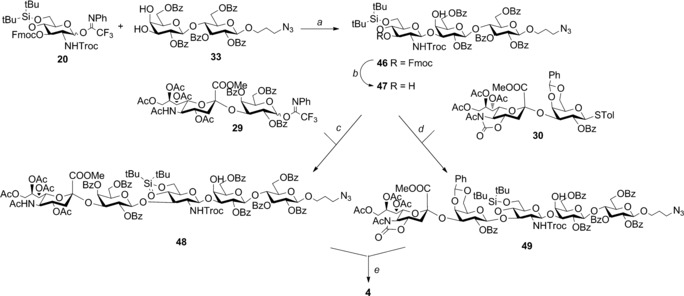
Synthetic route to type Ib linear repeating unit. Reagents and conditions: a) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 55 %; b) piperidine, CH2Cl2, 90 %; c) TMSOTf, CH2Cl2 dry, 0 °C, (β‐) 66 %; d) TfOH, NIS, CH2Cl2 dry, −40 °C, (β‐) 40 %; e) HF/pyridine; 3 m NaOH, THF, reflux; Ac2O/MeOH; H2/Pd‐C, 40 %.
Conformational analysis
The conformational properties of the CPS Ia and Ib branched repeating unit pentasaccharides 1 and 3 were studied by a combination of NMR spectroscopy and modeling tools,28 and compared with those of the corresponding polysaccharides. Interglycosidic interproton distances for 1 and 3 were estimated from ROESY spectra. The obtained experimental distances were compared with those derived from a 200 ns molecular dynamics (MD) simulation. Table 3 gathers the results for the CPS Ia pentasaccharide 1. The comparison reflects a good agreement between the NMR‐ and the MD‐derived distances for the glycosidic linkages GlcNAcβ1‐3Gal and Glcβ1‐4Gal (defined by the interproton distances H1GlcNAc‐H3Gal and H1Glc‐H4Gal, respectively). The Φ/Ψ population analysis from the MD simulation showed a single population for Φ fulfilling the exo‐anomeric effect (exo‐syn‐Φ),29, 30 and two populations around ψ for both linkages (Supporting Information, Figure S1).
Table 3.
Interglycosidic interproton [Å] distances for the CPS Ia pentasaccharide (1).
|
|
NMR |
MD |
|||
|---|---|---|---|---|---|
|
|
|
total average |
180/−30 |
−60/20 |
−60/−50 |
|
H3Gal‐H3eqNeuNAc |
none |
4.2 |
3.4 |
4.4 |
4.6 |
|
H3Gal‐H3axNeuNAc |
2.7 |
3.6 |
2.1 |
4.3 |
4.0 |
|
H3Gal‐H8NeuNAc |
very weak |
3.9 |
4.8 |
3.7 |
3.4 |
|
H1Gal‐H4GlcNAc |
2.4 |
2.4 |
|
|
|
|
H1Gal‐H6GlcNAc |
2.7 |
3.0 |
|
|
|
|
H1Gal‐H6'GlcNAc |
3.1 |
3.9 |
|
|
|
|
H1Glc‐H4Gal |
2.6 |
2.5 |
|
|
|
|
H1GlcNAc‐H3Gal |
2.5 |
2.4 |
|||
For the Galβ1‐4GlcNAc linkage, there is a perfect agreement for the H1Gal‐H4GlcNAc distance with slight discrepancies for the H1Gal‐H6,H6′GlcNAc ones, which is probably due to the MD bias around the GlcNAc ω torsion angle (Supporting Information, Figure S2). These data support the existence of a single population around the exo‐syn‐Φ/synΨ conformation, as predicted by the MD simulation.
For the Neu5Acα2‐3Gal linkage, MD simulations predict three different populations, 180°/−30°, −60°/−20°, and −60°/−50°. The interglycosidic interproton distances for each population are gathered in Table 3. There is a remarkable difference for the H3Gal‐H3axNeuNAc distance, being shorter according to NMR spectroscopy, which indicates that the MD simulation has a bias for the conformational ensemble towards exo‐syn‐Φ populations. Indeed, according the NOE‐derived distance, the exo‐anti‐Φ population should be the major one, representing around 75 % of the total ensemble. Two representative conformations for the CPS Ia pentasaccharide 1 are shown in the Supporting Information, differing in the Neu5Acα2‐3Gal linkage (Figure S3). The analysis for the CPS Ib pentasaccharide 3 yielded similar results (Supporting Information, Table S3), although the GlcNAcβ1‐3Gal linkage could not be fully characterized because of the overlapping between the H1GlcNAc and H3Gal protons. The linkage between Gal and GlcNAc, now β1‐3 instead of β1‐4, populates a minimum around the exo‐syn‐Φ/syn(−)‐Ψ conformation (Supporting Information, Figure S4). Two representative conformations for the CPS Ib pentasaccharide 3 are shown in the Supporting Information, which also differ in the orientation around the Neu5Acα2‐3Gal Φ torsion (Supporting Information, Figure S5). A superimposition of representative 3D structures for the CPS Ia and Ib pentasaccharides 1 and 3, with the major conformation exo‐anti‐Φ around the Neu5Acα2‐3Gal linkage is shown in Figure 2. The conformational behavior of the polysaccharides was then analyzed following a similar protocol. A model for the polysaccharide was built with 10 repeating units (50 monosaccharides) and MD simulations were run for 2.5 μs.
Figure 2.
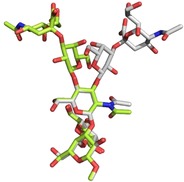
Superimposition of the major conformations for pentasaccharides 1 (lime) and 3 (grey), with the exo‐anti‐Φ geometry around the Neu5Acα2‐3Gal linkage.
The analysis of the glycosidic linkages was carried out for the 49 glycosidic bonds, revealing that the behavior for every glycosidic bond type is reproducible along the polysaccharide (Figure 3).
Figure 3.
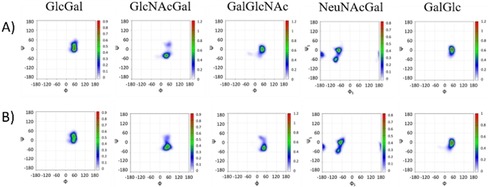
Glycosidic linkage analysis for GBS Ia (A) and Ib (B) polysaccharides: Φ/Ψ plots for representative glycosidic bonds of a 10 repeating unit model along the 2.5 μs MD simulation.
These populations are comparable to those of the corresponding pentasaccharide for every glycosidic linkage, and thus, the resulting interglycosidic interproton distances are very similar (Supporting Information, Table S1‐2). Remarkably, for both GBS serotype Ia and Ib, the HSQC spectra of the polysaccharide and the pentasaccharide were very similar, with the obvious exception for the Glcβ1‐4 linked moiety (E), which is not glycosylated at O4 in the pentasaccharides (Figure 4). The analysis of the interglycosidic NOE (from NOESY spectra at 20 ms mixing time) was consistent with the MD‐derived populations. The only discrepancies arose again for the Neu5Acα2‐3Gal linkages. Interestingly, for the Ia polysaccharide the NOE‐derived distance for H3axNeu5Ac‐H3Gal is 2.4 Å, shorter than that in the pentasaccharide. At the same time, there is a clear NOE between H3eqNeu5Ac‐H3Gal, not observed for the pentasaccharide. On the contrary, for the Ib polysaccharide the distance H3axNeu5Ac‐H3Gal is longer, 3.3 Å, whereas the NOE between H3eqNeu5Ac‐H3Gal does not exist (Figure 5 A, B). At the same time, the distance H8Neu5Ac‐H3Gal is slightly shorter for the Ib than that for the Ia polysaccharide (Figure 5 C, D). These data suggest that for the Ia polysaccharide, the major conformation around the Neu5Acα2‐3Gal fragment is the exo‐anti‐Φ (ca. 85 %), whereas for the Ib polysaccharide, there is a larger flexibility, with a major exo‐syn‐Φ form (ca. 55 %). The model structures for the polysaccharides with all Neu5Acα2‐3Gal linkages in exo‐anti‐Φ (Ia) and exo‐syn‐Φ (Ib) are displayed in Figure 6, showing different preferential shapes for the two polysaccharides. The Neu5Acα2‐3Gal branches of GBS PSIII have been shown to be strongly engaged in antibody recognition.14b The favored presentation of the different epitopes for the major conformation is rather different. However, given their intrinsic flexibility, especially for Ib, both molecules could be accommodated to interact with the monoclonal binding pockets without a major entropy penalty.31
Figure 4.
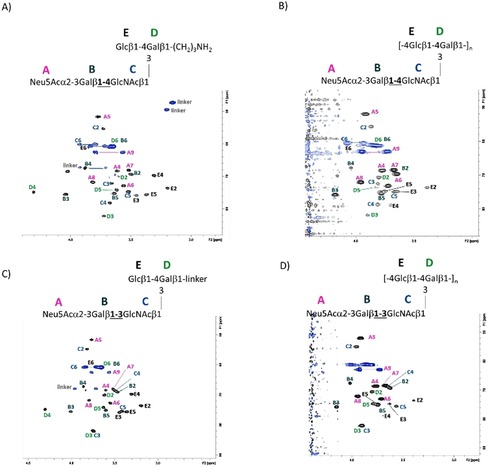
1H‐13C‐HSQC spectrum recorded for the pentasaccharide repeating unit of GBS Ia at 600 MHz, 298 K, D2O (A and B) and for GBS CPS Ib at 800 MHz, 318 K, D2O, showing the assignment of the 1H and 13C NMR signals. As expected, the matching is excellent except for some signals of residue E.
Figure 5.
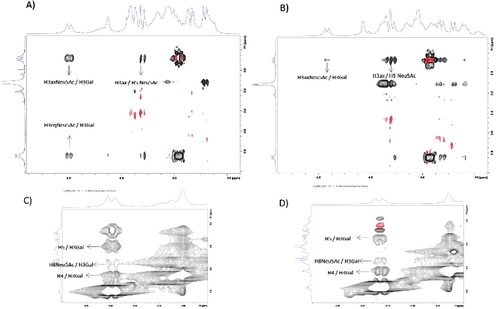
The key regions of the NOESY spectra recorded for the GBS CPS Ia (A and C) and GBS CPS Ib (B and D) polysaccharides showing the essential inter‐residue cross peaks.
Figure 6.
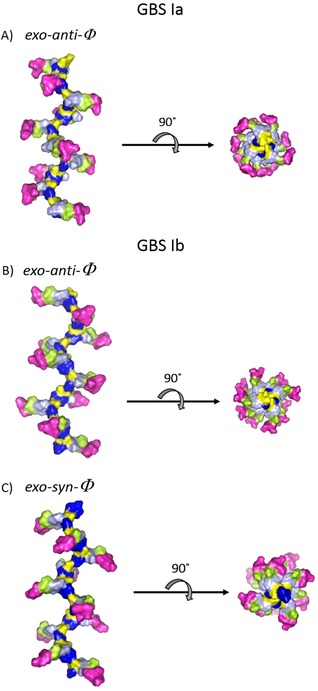
Model structures for the GBS Ia and Ib polysaccharides: A) Ia with the major exo‐anti‐Φ conformation around all the Neu5Acα2‐3Gal linkages B) Ib with exo‐anti‐Φ conformation around all Neu5Acα2‐3Gal linkages, and C) Ib with the major exo‐syn‐Φ conformation around all Neu5Acα2‐3Gal linkages.
Conclusions
To have fast access to homogeneous oligosaccharide antigens from GBS serotypes Ia and Ib and to gain insights into the conformational difference among these structurally similar polymers, we developed a highly convergent synthetic strategy based on the regioselective glycosylation of a galactose C3,4‐diol to obtain GlcNAcβ1‐3Gal disaccharide building blocks. Investigation of the different reactivities of the C3‐ and C4‐hydroxyls allowed us to reduce the number of protective group manipulations and synthetic steps to the final fragments, therefore simplifying the overall synthetic design.
Particularly, the use of a 2,6‐O‐benzoyl galactose diol resulted in improved regioselectively relative to that of the 2,6‐di‐O‐benzyl counterpart. In addition, mild activation conditions (NIS/AgOTf) for the glucosamine thiol donors or the torsional disarming effect of the benzylidene group for the trichloroacetimidate donors appear to favor the glycosylation reaction. The regioselective glucosamine incorporation was successfully applied to the synthesis of GBS CPS Ia and Ib branched repeating units (1 and 2). Their linear frameshifts (3 and 4) and a non‐sialylated CPS Ia form (5) were also synthesized to achieve an additional regioselective glycosylation of the Gal C3‐OH over the C4‐OH residue.
These results support the general applicability of the method to a variety of medically relevant glycans. Importantly, the structures synthesized through regioselective glycosylation appear extendible at the 4‐OH position of the Gal residue, thus potentially enabling the synthesis of longer and more complex GBS oligosaccharide structures.
Conformation analysis studies of the prepared oligosaccharides by NMR spectroscopy and MD simulations showed the impact of the GlcNAcβ1‐3Gal versus GlcNAcβ1‐4Gal connectivity in the orientation of the Neu5Acα2‐3Gal branching. The model, established from the single synthetic pentasaccharide repeating units, was used to study the conformational behavior of the GBS Ia and Ib polysaccharides, showing a different preferential shape for each polysaccharide with the Neu5Acα2‐3Gal linkages in exo‐anti‐Φ for Ia and exo‐syn‐Φ for Ib. These unique structural features are expected to influence antibody recognition and immunospecificity. Studies are ongoing to map the relevant glycoepitopes. Moreover, all glycans were designed with a chemical handle for conjugation to carrier proteins for immunological evaluation. Results on structural and immunogenic studies will be reported in due course.
Conflict of interest
L.D.B., D.O., M.M.R., R.C., and R.A. are employees of GSK groups companies. L.D.B. and R.A. are inventors of a patent related to the topic. R.A. is owner of GSK stocks.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Horizon 2020 research and innovation programme of the European Union for support through the Marie Skłodowska‐Curie grant agreement Glycovax No 675671.
L. Del Bino, I. Calloni, D. Oldrini, M. M. Raso, R. Cuffaro, A. Ardá, J. D. C. Codée, J. Jiménez-Barbero, R. Adamo, Chem. Eur. J. 2019, 25, 16277.
References
- 1. Nuccitelli A., Rinaudo C. D., Maione D., Ther. Adv. Vaccines 2015, 3, 76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Lin S. M., Zhi Y., Ahn K. B., Lim S., Seo H. S., Clin. Exp. Vaccine Res. 2018, 7, 76–81; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Paoletti L. C., Kasper D. L., Expert Opin. Biol. Ther. 2003, 3, 975–984; [DOI] [PubMed] [Google Scholar]
- 2c. Chaffin D. O., Mentele L. M., Rubens C. E., J. Bacteriol. 2005, 187, 4615–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paoletti L. C., Kasper D. L., Microbiol. Spectrum 2019, 7, GPP3-0001-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Madhi S. A., Cutland C. L., Jose L., Koen A., Govender N., Wittke F., Olugbosi M., Meulen A. S.-t., Baker S., Dull P. M., Narasimhan V., Slobod K., Lancet Infect. Dis. 2016, 16, 923–934; [DOI] [PubMed] [Google Scholar]
- 4b. Chen V. L., Avci F. Y., Kasper D. L., Vaccine 2013, 31, D13–D19. [Google Scholar]
- 5. Baker C. J., Carey V. J., Rench M. A., Edwards M. S., Hillier S. L., Kasper D. L., Platt R., J. Infect. Dis. 2014, 209, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. https://www.pfizer.com/news/press-release/press-release-detail/pfizer_awarded_grant_to_evaluate_vaccine_to_protect_newborns_against_group_b_streptococcus_infection-0.
- 7. Le Doare K., Heath P. T., Vaccine 2013, 31, D7–D12. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Pinto V., Berti F., J. Pharm. Biomed. Anal. 2014, 98, 9–15; [DOI] [PubMed] [Google Scholar]
- 8b. Jennings H. J., Katzenellenbogen E., Lugowski C., Kasper D. L., Biochemistry 1983, 22, 1258–1264. [DOI] [PubMed] [Google Scholar]
- 9. Schifferle R. E., Jennings H. J., Wessels M. R., Katzenellenbogen E., Roy R., Kasper D. L., J. Immunol. 1985, 135, 4164–4170. [PubMed] [Google Scholar]
- 10. Andreas N. J., Al-Khalidi A., Jaiteh M., Clarke E., Hyde M. J., Modi N., Holmes E., Kampmann B., Mehring Le Doare K., Clin. Transl. Immunol. 2016, 5, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Adamo R., Acc. Chem. Res. 2017, 50, 1270–1279; [DOI] [PubMed] [Google Scholar]
- 11b. Anish C., Schumann B., Pereira C. L., Seeberger P. H., Chem. Biol. 2014, 21, 38–50. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Kuttel M. M., Ravenscroft N., Pharmaceuticals 2019, 12, 28; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. González-Outeiriño J., Kadirvelraj R., Woods R. J., Carbohydr. Res. 2005, 340, 1007–1018. [DOI] [PubMed] [Google Scholar]
- 13. Jennings H. J., Lugowski C., Kasper D. L., Biochemistry 1981, 20, 4511–4518. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Cattaneo V., Carboni F., Oldrini D., De Ricco R., Donadio N., Ros I. M. Y., Berti F., Adamo R., Pure Appl. Chem. 2017, 89, 855–875; [Google Scholar]
- 14b. Carboni F., Adamo R., Fabbrini M., De Ricco R., Cattaneo V., Brogioni B., Veggi D., Pinto V., Passalacqua I., Oldrini D., Rappuoli R., Malito E., Margarit I. Y. R., Berti F., Proc. Natl. Acad. Sci. USA 2017, 114, 5017–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mondal P. K., Liao G., Mondal M. A., Guo Z., Org. Lett. 2015, 17, 1102–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Craft K. M., Townsend S. D., Carbohydr. Res. 2017, 440–441, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pozsgay V., Gaudino J., Paulson J. C., Jennings H. J., Bioorg. Med. Chem. Lett. 1991, 1, 391–394. [Google Scholar]
- 18. Demchenko A. V. B., Boons G.-J., J. Org. Chem. 2001, 66, 2547–2554. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Dimakos V., Taylor M. S., Chem. Rev. 2018, 118, 11457–11517; [DOI] [PubMed] [Google Scholar]
- 19b. Wang T., Demchenko A. V., Org. Biomol. Chem. 2019, 17, 4934–4950; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Niedbal D. A., Madsen R., Tetrahedron 2016, 72, 415–419. [Google Scholar]
- 20. Adamo R., Del Bino L., Int. Pat. Appl, 2018, WO2019012476A2019012471.
- 21. De Meo C. P., Priyadarshani U., Carbohydr. Res. 2008, 343, 1540–1552. [DOI] [PubMed] [Google Scholar]
- 22. Fraser-Reid B., López J. C. in Reactivity Tuning in Oligosaccharide Assembly, (Eds.: B. Fraser-Reid, J. Cristóbal López), Springer, Heidelberg, 2011, pp. 1–29. [Google Scholar]
- 23.
- 23a. van der Vorm S., van Hengst J. M. A., Bakker M., Overkleeft H. S., van der Marel G. A., Codée J. D. C., Angew. Chem. Int. Ed. 2018, 57, 8240–8244; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8372–8376; [Google Scholar]
- 23b. van der Vorm S., Hansen T., van Hengst J. M. A., Overkleeft H. S., van der Marel G. A., Codée J. D. C., Chem. Soc. Rev. 2019, 48, 4688–4706. [DOI] [PubMed] [Google Scholar]
- 24. Fraser-Reid B., Wu Z., Andrews C. W., Skowronski E., Bowen J. P., J. Am. Chem. Soc. 1991, 113, 1434–1435. [Google Scholar]
- 25. Zhang Z., Zong C., Song G., Lv G., Chun Y., Wang P., Ding N., Li Y., Carbohydr. Res. 2010, 345, 750–760. [DOI] [PubMed] [Google Scholar]
- 26. Ando T., Ishida H., Kiso M., Carbohydr. Res. 2003, 338, 503–514. [DOI] [PubMed] [Google Scholar]
- 27. Hsu C.-H., Chu K.-C., Lin Y.-S., Han J.-L., Peng Y.-S., Ren C.-T., Wu C.-Y., Wong C.-H., Chem. Eur. J. 2010, 16, 1754–1760. [DOI] [PubMed] [Google Scholar]
- 28. Lichtenthaler F. W., Oberthür M., Peters S., Eur. J. Org. Chem. 2001, 3849–3869. [Google Scholar]
- 29. Calloni I., Unione L., Jiménez-Osés G., Corzana F., Del Bino L., Corrado A., Pitirollo O., Colombo C., Lay L., Adamo R., Jiménez-Barbero J., Eur. J. Org. Chem. 2018, 4548–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu B., Unione L., Sardinha J., Wu S., Ethève-Quelquejeu M., Rauter A. P., Blériot Y., Zhang Y., Martín-Santamaría S., Díaz D., Jiménez-Barbero J., Sollogoub M., Angew. Chem. Int. Ed. 2014, 53, 9597–9602; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9751–9756. [Google Scholar]
- 31. Gimeno A., Delgado S., Valverde P., Bertuzzi S., Berbís M. A., Echavarren J., Lacetera A., Martín-Santamaría S., Surolia A., Cañada F. J., Jiménez-Barbero J., Ardá A., Angew. Chem. Int. Ed. 2019, 58, 7268–7272; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7346–7350. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary