

Abstract
Water‐splitting photoanodes based on semiconductor materials typically require a dopant in the structure and co‐catalysts on the surface to overcome the problems of charge recombination and high catalytic barrier. Unlike these conventional strategies, a simple treatment is reported that involves soaking a sample of pristine BiVO4 in a borate buffer solution. This modifies the catalytic local environment of BiVO4 by the introduction of a borate moiety at the molecular level. The self‐anchored borate plays the role of a passivator in reducing the surface charge recombination as well as that of a ligand in modifying the catalytic site to facilitate faster water oxidation. The modified BiVO4 photoanode, without typical doping or catalyst modification, achieved a photocurrent density of 3.5 mA cm−2 at 1.23 V and a cathodically shifted onset potential of 250 mV. This work provides an extremely simple method to improve the intrinsic photoelectrochemical performance of BiVO4 photoanodes.
Keywords: artificial photosynthesis, BiVO4, borate, photoelectrochemical cells, water oxidation
A BiVO4 photoanode was modified with borate at the molecular level (B‐BiVO4) by a simple immersion method, giving rise to increased photocurrent and decreased onset potential for water oxidation, which is comparable to loading a water‐oxidation co‐catalyst. The self‐anchored borate plays the role of passivator in decreasing the surface charge recombination and also a ligand in modifying the catalytic site to facilitate faster water oxidation.
Introduction
Water splitting by photoelectrochemical (PEC) cells is one of the most promising ways to obtain a renewable H2 fuel.1 Since electrochemical photolysis of water at a TiO2 photoanode was reported by Fujishima and Honda in 1972,2 metal oxide based semiconductors have become attractive materials for photocatalysis and PEC cells.3 An ideal semiconductor applicable for a PEC cell requires a suitable band gap to utilize a significant portion of the solar spectrum, an effective charge separation in the bulk, an efficient charge transfer at the semiconductor/electrolyte interface, and a long‐term stability in aqueous media.4 Among the metal oxide based semiconductors, monoclinic bismuth vanadate (BiVO4) is considered the most promising owing to its suitable band gap (ca. 2.4 eV) that enables it to absorb about 11 % of the visible light spectrum, its long carrier lifetime (ca. 40 ns), low cost, and good stability.5 Under the standard AM 1.5 G sunlight illumination, the theoretical photocurrent density of BiVO4 is estimated to reach a maximum of 7.5 mA cm−2, resulting in a solar‐to‐hydrogen conversion efficiency of close to 9.2 %.4b, 6
However, the PEC performance of pure BiVO4 photoanode is greatly limited by its low carrier mobility (ca. 4×10−2 cm2 V−1 s−1), short hole‐diffusion length (ca. 100 nm), and slow water oxidation kinetics.7 Plenty of approaches have been attempted to overcome these limitations, including element doping,8 morphology engineering,9 heterostructure formation,10 oxygen evolution catalysts (OECs)‐layer loading,11 crystal facet engineering,12 plasmonic enhancement,13 and combinations thereof. However, the efficiency of BiVO4 photoanodes is still far from an application level.5a Beside these well‐studied techniques, a series of postsynthetic treatments, a concept proposed by Smith and Stefik, have recently emerged as a simple and effective strategy to enhance the intrinsic photocatalytic activity of BiVO4 photoanodes.14 Instead of requiring the use of additional materials, such posttreatments stand out as methods to change the defect chemistry, both at the surface and in the bulk of BiVO4. It provides new mechanisms and opportunities to understand and enhance the intrinsic properties of BiVO4 photoanodes for higher PEC performance.
To date, a variety of postsynthetic modifications have been reported, including annealing under H2 or N2,15 illumination (that is, photocharging),16 UV curing,17 electrochemical treatment,18 acid vapor etching,12b Li/EDA (ethylenediamine) solution treatment,19 and so on. Herein, we found an extremely facile postsynthetic treatment for the improvement of BiVO4 photoanodes: modifying the BiVO4 electrodes with a borate species at the molecular level. The treated BiVO4 photoanodes (denoted as B‐BiVO4) consistently exhibit excellent PEC performance for water oxidation under AM 1.5 G illumination, with a near tenfold enhancement of photocurrent at 0.7 VRHE and a cathodic shift of the onset potential by 250 mV. A series of control experiments were performed; detailed physical characterizations, electrochemical impedance spectroscopy (EIS), and kinetic isotope effect (KIE) studies were conducted to reveal the significant role played by the addition of the borate moiety.
Results and Discussion
Nanoporous BiVO4 photoanodes were prepared according to an established method, with a few minor modifications.5c A typical worm‐like nanostructure of the resulting BiVO4 with a thickness of about 600 nm is shown in the SEM images (Supporting Information, Figure S1a,b). The monoclinic phase and a band gap of 2.42 eV are indicated by X‐ray diffraction and UV/Vis absorption spectra, respectively (Supporting Information, Figure S1c,d). Borate modification of the BiVO4 photoanode was performed by simply dipping the pristine BiVO4 in a 0.5 m borate buffer solution (pH 9.3) in a capped dark brown bottle (Figure 1 a). After 12 h, the treated BiVO4 electrode was removed from the borate solution and rinsed with Milli‐Q water to afford B‐BiVO4.
Figure 1.
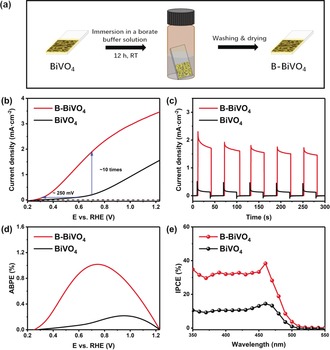
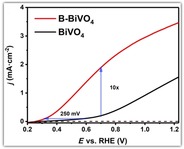
a) B‐BiVO4 photoanode preparation. b) Photocurrent–potential (J–V) curves of bare BiVO4 and B‐BiVO4 photoanodes under AM 1.5 G simulated sunlight at 100 mW cm−2 in a 0.5 m borate buffer (pH 9.3). Scan rate: 10 mV s−1. c) Transient photocurrents for BiVO4 and B‐BiVO4 photoanodes measured at 0.7 VRHE. d) Applied bias photon‐to‐current efficiencies (ABPEs) of BiVO4 and B‐BiVO4 photoanodes. e) Incident photon‐to‐current efficiencies (IPCEs) of BiVO4 and B‐BiVO4 photoanodes at 0.7 VRHE.
PEC performances of pristine BiVO4 and B‐BiVO4 were monitored in a three‐electrode cell, with 0.5 m borate buffer (pH 9.3) as electrolyte, under simulated sunlight illumination (AM 1.5 G, 100 mW cm−2). Pristine BiVO4 showed an onset potential of 0.57 V (defined at 0.1 mA cm−2 photocurrent density) and a maximum photocurrent density of only 1.6 mA cm−2 at 1.23 V vs. a reversible hydrogen electrode (RHE; Figure 1 b). Surprisingly, a highly improved photocurrent density was exhibited by B‐BiVO4, reaching 3.5 mA cm−2 at 1.23 V. The onset potential cathodically shifted to 0.32 V. Photocurrent density of B‐BiVO4 at 0.7 V is approximately ten times higher than that of the pristine BiVO4. The significantly enhanced PEC performance of B‐BiVO4 was further confirmed by the transient photocurrent (Figure 1 c), applied bias photon‐to‐current efficiency (ABPE, Figure 1 d), and incident photon‐to‐current conversion efficiency (IPCE) measurements (Figure 1 e). A maximum ABPE of 1.1 % was obtained by B‐BiVO4. IPCE of B‐BiVO4 at 0.7 V showed a universal double increment compared to the pristine BiVO4 and reached a maximum of 38 % at a wavelength of 460 nm.
The B‐BiVO4 photoanode, without the typical dopant or any co‐catalyst, displayed superior PEC performance even when compared to many doped and catalyst‐modified BiVO4 photoanodes (Supporting Information, Table S1). In general, state‐of‐the‐art performance of pristine BiVO4 is about 1.5 mA cm−2 at 1.23 V.5c For most of postsynthetically treated BiVO4 photoanodes, the photocurrent densities are only around 2.5 mA cm−2 (for example, 2.8, 2.4, and 2.5 mA cm−2 for N2,15a H2,15b and electrochemical treatments18 of BiVO4, respectively). Only two kinds of undoped and uncatalyzed BiVO4 photoanodes, reported recently, exhibited performances comparable to B‐BiVO4 (Supporting Information, Table S2). The photocharged BiVO4 photoanodes, investigated by Smith and co‐workers, achieved a photocurrent density of 4.3 mA cm−2 at 1.23 V.16a Cho and Zheng developed [001]‐oriented BiVO4 photoanodes with photocurrent density of 3.9 mA cm−2 at 1.23 V.12b The B‐BiVO4 displayed top level PEC performance among the undoped and uncatalyzed BiVO4 photoanodes. Additionally, the treatment method used in this case is more facile than other postsynthetic treatment methods.
Regarding the stability of B‐BiVO4 under an open‐circuit condition, when B‐BiVO4 was stored under air for 24 h, the PEC performance showed only a small decrease (Supporting Information, Figure S2); when B‐BiVO4 was stored in Milli‐Q water overnight, the PEC performance kept approximately 85 % of its incipient performance (Supporting Information, Figure S3). These observations distinguish B‐BiVO4 from the BiVO4 after photocharging treatment, where the photocharged BiVO4 totally lost its increment of PEC performance when stored in dark overnight in buffer solution,16b indicating that a different underlying mechanism is responsible for the improvement in the PEC performance of B‐BiVO4.
To investigate this underlying mechanism, we firstly established, by means of a series of control experiments on the immersion treatment, that the remarkable effect is indeed caused by the involvement of the borate species. The possibility that the improvement in PEC performance is due to the basic pH condition can be safely excluded as no obvious change in the photocurrent density is observed when the pristine BiVO4 is soaked in a NaOH aqueous solution (pH 9.3) instead of the borate solution (Figure 2 a). Regarding the effect of salt ions, a treatment with neither NaOAc nor NaClO4 solution brings in an improvement in the photocurrent of BiVO4. The bare BiVO4, treated with a phosphate buffer, showed some visible enhancement of PEC performance, but it was still far less than B‐BiVO4.
Figure 2.
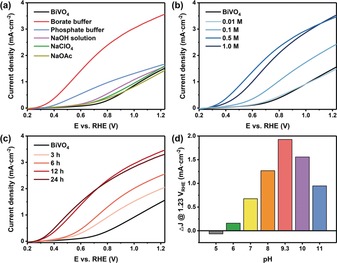
J–V curves for BiVO4 and B‐BiVO4 photoanodes treated a) with different salt solutions at pH 9.3; b) in different concentrations of borate buffer at pH 9.3; c) in a 0.5 m borate buffer at pH 9.3 for different durations. d) Increments of photocurrents at 1.23 VRHE of B‐BiVO4 photoanodes treated with a 0.5 m borate buffer at different pH values compared to the bare BiVO4.
Furthermore, the borate treatment itself was studied in greater detail by changing the borate concentration, immersion time, temperature, and pH value of the borate solution. PEC performances of the corresponding B‐BiVO4 photoanode markedly rose with the increase in the borate concentration under the same soaking duration (Figure 2 b). PEC performances of the resulting B‐BiVO4 treated in the same borate solution improved with respect to the immersion time (Figure 2 c) during the first 12 h. Extension of the immersion time over 12 h led to negligible improvement, indicating that the full transformation of the pristine BiVO4 to B‐BiVO4 was completed in the stipulated time. Interestingly, it was found that the borate treatment can be considerably accelerated by increasing the reaction temperature (Supporting Information, Figure S4); B‐BiVO4 with the best PEC performance can be generated after only 25 min of treatment at 100 °C. Especially noteworthy is the fact that the effect of the modification is highly dependent on the pH of the borate solution. The highest improvement was achieved by the treatment with a borate solution in the pH range of 9 to 10, approaching boric acid pK a of 9.24 (Figure 2 d; Supporting Information, Figure S5). The correlation between the enhancing effect and the pH of the borate solution suggests that [B(OH)4]−, the conjugate base of H3BO3, may be directly involved in modifying the BiVO4 sample and also plays a pivotal role in the PEC performance improvement. These results therefore confirm that the improvement of PEC performance resulted from the modification by the borate species, most likely to be [B(OH)4]− with a tetrahedral geometry.
To explore the structural changes of the BiVO4 film after the borate modification, physical characterizations were conducted for both the pristine and modified BiVO4. However, SEM images (Figure 3 a) and XRD patterns (Figure 3 b) of B‐BiVO4 show no noticeable differences compared to that of the bare BiVO4. The UV/Vis absorption spectra of both the modified and pristine BiVO4 also exhibit similar absorbance edges at approximately 520 nm, indicating the similar band gap of about 2.4 eV (Figure 3 c). These demonstrate that the nature of the bulk of B‐BiVO4, for example, structure and absorbance, remain unchanged.
Figure 3.
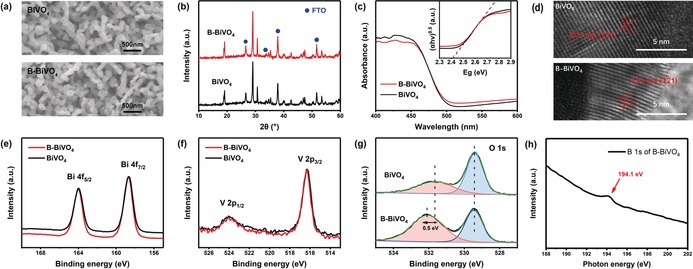
a) SEM images, b) X‐ray diffraction (XRD) spectra, and c) UV/Vis diffuse spectra of BiVO4 and B‐BiVO4 photoanodes. Inset: Tauc plots of BiVO4 and B‐BiVO4. d) HRTEM images of the bare BiVO4 and B‐BiVO4 photoanodes, respectively. e) Bi 4f, f) V 2p, and g) O 1s XPS spectra for bare BiVO4 and B‐BiVO4 photoanodes, respectively. h) B 1 s NEXAFS edge spectrum for the B‐treated BiVO4 sample.
Therefore, we can conclude that the alteration of the BiVO4 film, induced by the borate modification, happens owing to the changes on the surface. Raman spectroscopy, high‐resolution transmission electron microscopy (HRTEM), X‐ray photoelectron spectroscopy (XPS), and near‐edge X‐ray absorption fine structure (NEXAFS) spectroscopy, all of which are powerful techniques for surface characterization, were employed to explore structural details of the surface changes by the borate modification. Unfortunately, the Raman spectra of the pristine BiVO4 and B‐BiVO4 were found to be superimposable, showing no identifiable structural changes (Supporting Information, Figure S6). No obvious interface or newly generated nanolayer was observed from the HRTEM images either (Figure 3 d). The XPS spectra of both species exhibited typical O 1s, V 2p, and Bi 4f peaks (Supporting Information, Figure S7). The Bi 4f and V 2p peaks, and the O 1s peak at 529.4 eV, displayed negligible shifts before and after the borate treatment (Figures 3 e–g). The only obvious change is that the O 1s peak at 531.6 eV, which is commonly attributed to chemisorbed −OH groups, shifts to 532.1 eV, with an evidently higher density of such groups (Figure 3 g). This change can be a sign of an increase in chemisorbed ‐OH groups due to the absorption of [B(OH)4]− or −OH or both.20 However, it should be noted that surface contamination (−CO and −CO2) may also cause changes in the O 1s peak.21
It has been clearly demonstrated earlier that a borate moiety is involved in the modification of the surface of the BiVO4 film to afford an efficient B‐BiVO4 species. Unfortunately, most of the above characterization methods failed to show the nature of the exact changes. Even a boron signal could not be identified in the elemental analysis by XPS (Supporting Information, Figure S7) or HRTEM‐EDS (EDS corresponds to energy‐dispersive X‐ray spectroscopy; Supporting Information, Figure S8). However, this is not a factual contradiction, because boron is very light element. As it is in a system with heavy metal, the detection limits of both these techniques are very high.22 It is difficult to detect a B signal when its content is not abundant in the sample. Even for a typical boron‐doped BiVO4 with B‐compositions of 3 % and 10 %,20, 23 the observed B signals are very weak, indicating the level of a detection limit. In comparison, the amount of surface absorbed borate in this case can be orders of magnitude lower. This should explain the failure in detecting a B signal. The missing B signal in the regular physical characterization, in effect, indicates that the borate modification of the BiVO4 surface is at a molecular level with an extremely low borate concentration.
To display the presence of trace amount of borate on BiVO4 surface, we conducted a more sensitive characterization, the NEXAFS measurements by using low‐energy secondary electrons. The NEXAFS B 1s edge spectrum displayed that there may have been a trace of B at the surface of the B‐treated BiVO4 sample, as revealed by a small peak at approximately 194.1 eV owing to the boric acid/borate species in Figure 3 h.24 NEXAFS measurements were accomplished using low energy secondary electrons of about 14 eV (more precisely over a 13–15 eV range), noting that the inelastic mean free path (IMFP) of the detected secondary electrons, which is related to the escape depth and sampling depth of NEXAFS, is 3.6 nm at this electron energy with inorganic materials.25 Accordingly, the NEXAFS data pertain to the sample surface indicating the presence of B at a trace level. It is unsurprising that NEXAFS located a trace of B in the treated sample, although XPS were unable to detect B signal. Indeed, this is not a precedent in the NEXAFS detection of trace elements owing to the enhanced sensitivity of NEXAFS. For example, NEXAFS of the Fe L‐edge yielded high‐quality spectra with the detection of FeII/FeIII states at an Fe depleted iron chalcogenide surface since the photoabsorption cross‐section increased by several orders of magnitude, substantially boosting the analytical sensitivity of NEXAFS when the incident beam energy approached and resonated with the Fe L‐edge.26
To further reveal the underlying mechanism of the dramatic effect induced by the borate modification, we thoroughly investigated the photogenerated carrier transfer kinetics of BiVO4 before and after the borate treatment. Mott–Schottky curves of both samples show positive slopes, as expected, for the n‐type semiconductors (Figure 4 a). Based on the slope of the Mott–Schottky curves, carrier density increment of B‐BiVO4, compared to that of the pristine BiVO4, is negligible. The flat band potential (intercept on x axis) of the bare BiVO4 anodically shifts by only a small value of 25 mV. Moreover, an anodic shift cannot contribute to the negative shift of the photocurrent onset potential of B‐BiVO4 for water oxidation. The Mott–Schottky analysis again demonstrates that the bulk properties of BiVO4 are not affected by the borate modification. Electrochemical impedance spectroscopy (EIS) measurements show that the B‐BiVO4 photoanodes have the same series resistance R s but a much smaller interfacial charge transfer resistance R ct as that of the pristine BiVO4 (Figure 4 b), indicating that the improvement in photocurrent density of B‐BiVO4 can be attributed to the enhanced surface charge transfer rather than to the bulk charge transport.
Figure 4.
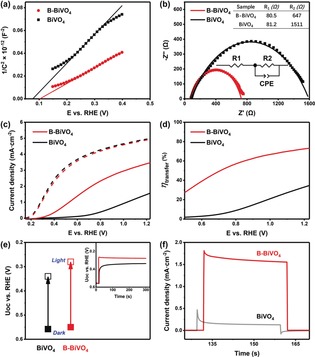
a) Mott–Schottky plots of BiVO4 and B‐BiVO4 photoanodes measured in a 0.5 m borate buffer at pH 9.3 in dark. b) Electrochemical impedance spectra (EIS) of BiVO4 and B‐BiVO4 photoanodes measured at 0.7 VRHE. c) J–V curves of BiVO4 and B‐BiVO4 photoanodes for sulfite oxidation measured in a 0.5 m borate buffer (pH 9.3) containing 0.5 m Na2SO3 (hole scavenger). d) Charge transfer efficiencies at the semiconductor/electrolyte interface (η transfer) of BiVO4 and B‐BiVO4 photoanodes. e) Open circuit potentials (UOC) of BiVO4 and B‐BiVO4 photoanodes under dark (solid) and illumination (hollow); inset: transient photovoltage response within immediate illumination. f) Transient photocurrents measured at 0.7 VRHE for BiVO4 and B‐BiVO4.
More precisely, the contributions of the increased photocurrent density (J), which is determined by three fundamental components [given by Eq. (1)], namely light absorption (represented as J abs), charge transport efficiency in the bulk (η transport), and charge transfer at the semiconductor/electrolyte interface for water oxidation (η transfer), were studied to confirm the identification of the key factors for the high PEC performance observed in the case of B‐BiVO4.
| (1) |
The borate treatment has trivial effect on J abs, because the unmodified BiVO4 and B‐BiVO4 have comparable light absorption properties, as shown by the similar UV/Vis absorption spectra for both. The η transport and η transfer were separately evaluated by employing a conventional hole‐scavenger method. Figure 4 c shows the J–V curves for the pristine BiVO4 and B‐BiVO4 photoanodes, determined in the electrolyte with and without a hole‐scavenger, Na2SO3. In contrary to the differences in PEC performances for water oxidation, the pristine BiVO4 and B‐BiVO4 exhibited comparable photocurrent density when sufficient Na2SO3 was introduced in the electrolyte. Considering that J abs is the same for both samples, it is rational to deduce that B‐BiVO4 has the same η transport as the pristine BiVO4. In contrast, η transfer of B‐BiVO4, as shown in Figure 4 d, is at least two‐fold higher than that of the pristine BiVO4, depending on the applied potential.
Finally, we found out that the immensely increased η transfer (that is, surface catalytic efficiency) is the key factor in the observed improvement in PEC performances of B‐BiVO4 after the borate treatment. Three factors can be responsible for an increase in η transfer, including a larger surface area, suppressed surface charge trapping, and an accelerated catalytic rate of water oxidation reaction. First, the surface areas of the BiVO4 photoanode before and after the borate treatment were evaluated by electrochemical capacitance measurements (Supporting Information, Figure S9). Electrochemically active surface areas (EASA) of the pristine BiVO4 and B‐BiVO4 were found to be similar, which rules out its contribution to the higher η transfer. The case of surface charge trapping was investigated by measuring the open‐circuit voltage (U oc).11d When BiVO4 is immersed in the electrolyte, the illumination induced increment in U oc depends on the photogenerated carrier density, which results in a new quasi‐Fermi level. The photovoltage for B‐BiVO4 was detected as 0.27 V, which was 50 mV higher than that detected for the bare BiVO4, indicating the suppression of surface charge trapping on B‐BiVO4 (Figure 4 e). The 50 mV of photovoltage difference between BiVO4 and B‐BiVO4 is much less than the 250 mV cathodic shift in the onset potential for water oxidation. Therefore, suppression of surface charge trapping is one of the factors that must have played a role in the enhancement of η transfer of B‐BiVO4.
Since the J–V curve for the B‐BiVO4 photoanode, determined with a hole‐scavenger, did not show any cathodic shift, while its J–V curve for water oxidation cathodically shifted 250 mV (Figure 4 c), it is obvious that the rate of water oxidation on the B‐BiVO4 photoanode was enhanced tremendously, which is the other important factor facilitating surface charge transfer in the case of B‐BiVO4. The faster water oxidation on the B‐BiVO4 surface can be further established by the study of photocurrent transients. Light on–off cycles in chopped light chronoamperometry is usually accompanied by photocurrent transient spikes, caused by the discrepancy between the fast carrier generation and slow surface reaction dynamics.8b, 11d, 27 The spikes for the B‐BiVO4 photoanodes are much smaller compared to that of the bare BiVO4 ones; moreover, no charge accumulation was found for B‐BiVO4, as shown by the damped‐current during light‐off (Figure 4 f). These observations demonstrate that the borate modification accelerated the catalytic rate of water oxidation on the modified BiVO4 surface. Indeed, the dramatic effect of surface modification on photocatalytic performance have been studied for other bismuth‐based semicondutors.28
Based on the control experiments, physical characterizations, and carrier transfer kinetics studies, we propose that the immersion treatment in borate buffer solution is indeed a spontaneous process in which the tetrahedral [B(OH)4]− gradually interacts with the active site (that is, defect) on the BiVO4 surface (Figure 5). The most likely sites for the tetrahedral [B(OH)4]− are the defects formed as a result of vanadium loss.23, 29 The adsorbed [B(OH)4]− may act as a passivator to reduce charge recombination22a and to facilitate extraction of holes to the surface.30 More importantly, the anchoring of the borate moiety at the catalytic active site significantly accelerated the catalytic rate of water oxidation. The role played by the self‐anchored borate can be considered as a ligand effect at the catalytic site on the BiVO4 surface. It can modify the electronic configuration of the bismuth catalytic site and consequently, accelerate the O−O bond formation rate. At the same time, the anchored borate, as an internal base, can also assist the concerted proton‐electron transfer, which has been shown to be essential for water oxidation by molecular catalysts,31 metal oxides,32 and semiconductor photoanodes.33 KIE studies of the pristine BiVO4 and B‐BiVO4 photoanodes indicated that proton transfer is involved in the rate determining step (RDS) because a KIE value of approximately 2.6 was observed for the pristine BiVO4 with low bias; the anchored borate evidently facilitated proton transfer in the RDS, with a much smaller KIE value of around 1.5 determined for the B‐BiVO4 photoanode (Supporting Information, Figure S10).
Figure 5.
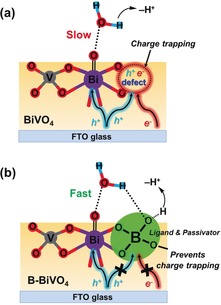
Illustration of the proposed mechanism for water oxidation on the surface of a) pristine BiVO4 and b) B‐BiVO4.
The stability of B‐BiVO4 under PEC test was evaluated by multiple cycles of linear sweep voltammetry (LSV) under illumination (Supporting Information, Figure S11) and photocurrent–time measurements (Supporting Information, Figure S12). PEC performance of B‐BiVO4 gradually decreased during 20 cycles of LSV. After 20 min of photoelectrolysis with 1.0 V bias, B‐BiVO4 lost approximately 35 % of the initial photocurrent. In a separate experiment, a Faradaic efficiency of 91 % for oxygen evolution by the B‐BiVO4 was calculated based on the record of the moles of electrons passing through and the determination of the amounts of evolved oxygen (Supporting Information, Figure S13). The deactivation of B‐BiVO4 can be induced by photocorrosion11b, 34 or desorption of the borate from the photocharged surface of B‐BiVO4 or both. Deactivation of the bare BiVO4, without a catalytic or passivating layer, has been widely observed under long‐term PEC tests.11b, 35 Interestingly, when the process of borate treatment was repeated on B‐BiVO4 after 20 cycles of LSV scanning, similar PEC performances as that from a freshly‐prepared B‐BiVO4 can be obtained again (Supporting Information, Figure S14). This self‐recovery process can be repeated several times and projects borate treatment as a possible strategy to produce self‐healing PEC cells, which can work during daytime and recover during the night (Supporting Information, Figure S15). Furthermore, modifying the B‐BiVO4 with co‐catalyst can further increase its photocurrent density for water oxidation and dramatically improve the stability. These related studies are ongoing in our group.
Conclusion
In summary, we reported the remarkable effect of modifying a BiVO4 surface with borate by a simple immersion method, leading to a significant increase in photocurrent as well as a decrease in the onset potential for water oxidation, which is comparable to the effect of loading a water‐oxidation co‐catalyst. Detailed characterizations and carrier transfer kinetics investigations indicated that the adsorption of tetrahedral [B(OH)4]− species near the active sites results in a molecular level modification. This acts as a regulating ligand and passivator, playing an important role in accelerating water‐oxidation rate and reducing charge trapping on the BiVO4 surface. The post‐synthetic borate treatment proposed in this work provides new opportunities to understand and improve the PEC performance of BiVO4 photoanodes. The method of small molecule modification can also be widely developed for improving the property of material‐based catalysts and photocatalysts.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support of this work by the Swedish Research Council (2017‐00935), Swedish Energy Agency, Knut and Alice Wallenberg Foundation (KAW 2016.0072), and the National Basic Research Program of China (973 Program, 2014CB239402). Q.M. and L.F. also thank the China Scholarship Council for a special scholarship award. We are grateful for the support of the European Community's Seventh Framework Programme (FP7/2007–2013, grant agreement no 312284) for the research at the Materials Science Beamline at the Elettra Synchrotron, the CERIC‐ERIC Consortium for access to experimental facilities and financial support of the Czech Ministry of Education (LM2015057). Special acknowledgement to Drs. Nataliya Tsud, Kevin C. Prince and J. Bradley for the assistance at the Elettra Synchrotron. M.C. and G.A.C. acknowledge the CERIC users’ grant, while RDM thanks the International Synchrotron Access Program of the Australian Synchrotron.
Q. Meng, B. Zhang, L. Fan, H. Liu, M. Valvo, K. Edström, M. Cuartero, R. de Marco, G. A. Crespo, L. Sun, Angew. Chem. Int. Ed. 2019, 58, 19027.
References
- 1.
- 1a. Tachibana Y., Vayssieres L., Durrant J. R., Nat. Photonics 2012, 6, 511–518; [Google Scholar]
- 1b. Walter M. G., Warren E. L., McKone J. R., Boettcher S. W., Mi Q., Santori E. A., Lewis N. S., Chem. Rev. 2010, 110, 6446–6473; [DOI] [PubMed] [Google Scholar]
- 1c. Gong J., Li C., Wasielewski M. R., Chem. Soc. Rev. 2019, 48, 1862–1864. [DOI] [PubMed] [Google Scholar]
- 2. Fujishima A., Honda K., Nature 1972, 238, 37–38. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Chen F., Huang H., Guo L., Zhang Y., Ma T., Angew. Chem. Int. Ed. 2019, 58, 10061–10073; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10164–10176; [Google Scholar]
- 3b. Yao T., An X., Han H., Chen J. Q., Li C., Adv. Energy Mater. 2018, 8, 1800210. [Google Scholar]
- 4.
- 4a. Park Y., McDonald K. J., Choi K.-S., Chem. Soc. Rev. 2013, 42, 2321–2337; [DOI] [PubMed] [Google Scholar]
- 4b. Xu X. T., Pan L., Zhang X., Wang L., Zou J. J., Adv. Sci. 2019, 6, 1801505; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Nellist M. R., Laskowski F. A. L., Lin F., Mills T. J., Boettcher S. W., Acc. Chem. Res. 2016, 49, 733–740. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Kim J. H., Lee J. S., Adv. Mater. 2019, 31, 1806938; [Google Scholar]
- 5b. Huang Z.-F., Pan L., Zou J.-J., Zhang X., Wang L., Nanoscale 2014, 6, 14044–14063; [DOI] [PubMed] [Google Scholar]
- 5c. Kim T. W., Choi K.-S., Science 2014, 343, 990–994. [DOI] [PubMed] [Google Scholar]
- 6. Seo J., Nishiyama H., Yamada T., Domen K., Angew. Chem. Int. Ed. 2018, 57, 8396–8415; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8530–8550. [Google Scholar]
- 7.
- 7a. Tan H. L., Amal R., Ng Y. H., J. Mater. Chem. A 2017, 5, 16498–16521; [Google Scholar]
- 7b. Abdi F. F., Savenije T. J., May M. M., Dam B., van de Krol R., J. Phys. Chem. Lett. 2013, 4, 2752–2757; [Google Scholar]
- 7c. Rettie A. J. E., Lee H. C., Marshall L. G., Lin J.-F., Capan C., Lindemuth J., McCloy J. S., Zhou J., Bard A. J., Mullins C. B., J. Am. Chem. Soc. 2013, 135, 11389–11396. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Abdi F. F., Han L., Smets A. H., Zeman M., Dam B., van de Krol R., Nat. Commun. 2013, 4, 2195; [DOI] [PubMed] [Google Scholar]
- 8b. Zhong D. K., Choi S., Gamelin D. R., J. Am. Chem. Soc. 2011, 133, 18370–18377; [DOI] [PubMed] [Google Scholar]
- 8c. Lee J. M., Baek J. H., Gill T. M., Shi X., Lee S., Cho I. S., Jung H. S., Zheng X., J. Mater. Chem. A 2019, 7, 9019–9024; [Google Scholar]
- 8d. Huang H., Tu S., Zeng C., Zhang T., Reshak A. H., Zhang Y., Angew. Chem. Int. Ed. 2017, 56, 11860–11864; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12022–12026. [Google Scholar]
- 9.
- 9a. Qiu Y., Liu W., Chen W., Chen W., Zhou G., Hsu P.-C., Zhang R., Liang Z., Fan S., Zhang Y., Cui Y., Sci. Adv. 2016, 2, e1501764; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Rao P. M., Cai L., Liu C., Cho I. S., Lee C. H., Weisse J. M., Yang P., Zheng X., Nano Lett. 2014, 14, 1099–1105; [DOI] [PubMed] [Google Scholar]
- 9c. Bielinski A. R., Lee S., Brancho J. J., Esarey S. L., Gayle A. J., Kazyak E., Sun K., Bartlett B. M., Dasgupta N. P., Chem. Mater. 2019, 31, 3221–3227. [Google Scholar]
- 10.
- 10a. Zhang K., Jin B., Park C., Cho Y., Song X., Shi X., Zhang S., Kim W., Zeng H., Park J. H., Nat. Commun. 2019, 10, 2001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Pihosh Y., Turkevych I., Mawatari K., Uemura J., Kazoe Y., Kosar S., Makita K., Sugaya T., Matsui T., Fujita D., Sci. Rep. 2015, 5, 11141; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Hong S. J., Lee S., Jang J. S., Lee J. S., Energy Environ. Sci. 2011, 4, 1781–1787. [Google Scholar]
- 11.
- 11a. Wang Y., Li F., Zhou X., Yu F., Du J., Bai L., Sun L., Angew. Chem. Int. Ed. 2017, 56, 6911–6915; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7015–7019; [Google Scholar]
- 11b. Lee D. K., Choi K.-S., Nat. Energy 2018, 3, 53–60; [Google Scholar]
- 11c. Ye S., Ding C., Chen R., Fan F., Fu P., Yin H., Wang X., Wang Z., Du P., Li C., J. Am. Chem. Soc. 2018, 140, 3250–3256; [DOI] [PubMed] [Google Scholar]
- 11d. Tang F., Cheng W., Su H., Zhao X., Liu Q., ACS Appl. Mater. Interfaces 2018, 10, 6228–6234; [DOI] [PubMed] [Google Scholar]
- 11e. Kuang Y., Jia Q., Ma G., Hisatomi T., Minegishi T., Nishiyama H., Nakabayashi M., Shibata N., Yamada T., Kudo A., Domen K., Nat. Energy 2016, 2, 16191; [Google Scholar]
- 11f. Zachäus C., Abdi F. F., Peter L. M., van de Krol R., Chem. Sci. 2017, 8, 3712–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Wang S., Liu G., Wang L., Chem. Rev. 2019, 119, 5192–5247; [DOI] [PubMed] [Google Scholar]
- 12b. Han H. S., Shin S., Kim D. H., Park I. J., Kim J. S., Huang P.-S., Lee J.-K., Cho I. S., Zheng X., Energy Environ. Sci. 2018, 11, 1299–1306; [Google Scholar]
- 12c. Li D., Liu Y., Shi W., Shao C., Wang S., Ding C., Liu T., Fan F., Shi J., Li C., ACS Energy Lett. 2019, 4, 825–831; [Google Scholar]
- 12d. Li M., Yu S., Huang H., Li X., Feng Y., Wang C., Wang Y., Ma T., Guo L., Zhang Y., Angew. Chem. Int. Ed. 2019, 58, 9517–9521; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9617–9621. [Google Scholar]
- 13.
- 13a. Abdi F. F., Dabirian A., Dam B., van de Krol R., Phys. Chem. Chem. Phys. 2014, 16, 15272–15277; [DOI] [PubMed] [Google Scholar]
- 13b. Gan J., Rajeeva B. B., Wu Z., Penley D., Liang C., Tong Y., Zheng Y., Nanotechnology 2016, 27, 235401; [DOI] [PubMed] [Google Scholar]
- 13c. Kim J. K., Shi X., Jeong M. J., Park J., Han H. S., Kim S. H., Guo Y., Heinz T. F., Fan S., Lee C.-L., Park J. H., Zheng X., Adv. Energy Mater. 2018, 8, 1701765. [Google Scholar]
- 14. Lamm B., Trześniewski B. J., Döscher H., Smith W. A., Stefik M., ACS Energy Lett. 2018, 3, 112–124. [Google Scholar]
- 15.
- 15a. Kim T. W., Ping Y., Galli G. A., Choi K.-S., Nat. Commun. 2015, 6, 8769; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Wang G., Ling Y., Lu X., Qian F., Tong Y., Zhang J. Z., Lordi V., Rocha Leao C., Li Y., J. Phys. Chem. C 2013, 117, 10957–10964. [Google Scholar]
- 16.
- 16a. Trześniewski B. J., Digdaya I. A., Nagaki T., Ravishankar S., Herraiz-Cardona I., Vermaas D. A., Longo A., Gimenez S., Smith W. A., Energy Environ. Sci. 2017, 10, 1517–1529; [Google Scholar]
- 16b. Trześniewski B. J., Smith W. A., J. Mater. Chem. A 2016, 4, 2919–2926. [Google Scholar]
- 17. Li T., He J., Peña B., Berlinguette C. P., Angew. Chem. Int. Ed. 2016, 55, 1769–1772; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1801–1804. [Google Scholar]
- 18. Wang S., Chen P., Yun J.-H., Hu Y., Wang L., Angew. Chem. Int. Ed. 2017, 56, 8500–8504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8620–8624. [Google Scholar]
- 19. Kim J. K., Cho Y., Jeong M. J., Levy-Wendt B., Shin D., Yi Y., Wang D. H., Zheng X., Park J. H., ChemSusChem 2018, 11, 933–940. [DOI] [PubMed] [Google Scholar]
- 20. Shan L.-W., Wang G.-L., Suriyaprakash J., Li D., Liu L.-Z., Dong L.-M., J. Alloys Compd. 2015, 636, 131–137. [Google Scholar]
- 21. Zhang B., Chen H., Daniel Q., Philippe B., Yu F., Valvo M., Li Y., Ambre R. B., Zhang P., Li F., ACS Catal. 2017, 7, 6311–6322. [Google Scholar]
- 22.
- 22a. Lan H., Wei A., Zheng H., Sun X., Zhong J., Nanoscale 2018, 10, 7033–7039; [DOI] [PubMed] [Google Scholar]
- 22b. Shard A. G., Surf. Interface Anal. 2014, 46, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y., Jing T., Liu Y., Huang B., Dai Y., Zhang X., Qin X., Whangbo M. H., ChemPlusChem 2015, 80, 1113–1118. [DOI] [PubMed] [Google Scholar]
- 24. Duffin A. M., Schwartz C. P., England A. H., Uejio J. S., Prendergast D., Saykally R. J., J. Chem. Phys. 2011, 134, 154503. [DOI] [PubMed] [Google Scholar]
- 25. Seah M. P., Dench W. A., Surf. Interface Anal. 1979, 1, 2–11. [Google Scholar]
- 26. Maric M., Sohail M., De Marco R., Electrochem. Commun. 2014, 41, 27–30. [Google Scholar]
- 27. Le Formal F., Sivula K., Grätzel M., J. Phys. Chem. C 2012, 116, 26707–26720. [Google Scholar]
- 28.
- 28a. Yu H., Li J., Zhang Y., Yang S., Han K., Dong F., Ma T., Huang H., Angew. Chem. Int. Ed. 2019, 58, 3880–3884; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3920–3924; [Google Scholar]
- 28b. Hao L., Kang L., Huang H., Ye L., Han K., Yang S., Yu H., Batmunkh M., Zhang Y., Ma T., Adv. Mater. 2019, 31, 1900546. [DOI] [PubMed] [Google Scholar]
- 29. Lamers M., Fiechter S., Friedrich D., Abdi F. F., van de Krol R., J. Mater. Chem. A 2018, 6, 18694–18700. [Google Scholar]
- 30. Kim J. Y., Jang J. W., Youn D. H., Magesh G., Lee J. S., Adv. Energy Mater. 2014, 4, 1400476. [Google Scholar]
- 31. Song N., Concepcion J. J., Binstead R. A., Rudd J. A., Vannucci A. K., Dares C. J., Coggins M. K., Meyer T. J., Proc. Natl. Acad. Sci. USA 2015, 112, 4935–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.
- 32a. Yamaguchi A., Inuzuka R., Takashima T., Hayashi T., Hashimoto K., Nakamura R., Nat. Commun. 2014, 5, 4256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Klingan K., Ringleb F., Zaharieva I., Heidkamp J., Chernev P., Gonzalez-Flores D., Risch M., Fischer A., Dau H., ChemSusChem 2014, 7, 1301–1310. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Zhang Y., Zhang H., Ji H., Ma W., Chen C., Zhao J., J. Am. Chem. Soc. 2016, 138, 2705–2711; [DOI] [PubMed] [Google Scholar]
- 33b. Takashima T., Ishikawa K., Irie H., Chem. Commun. 2016, 52, 14015–14018. [DOI] [PubMed] [Google Scholar]
- 34. Toma F. M., Cooper J. K., Kunzelmann V., McDowell M. T., Yu J., Larson D. M., Borys N. J., Abelyan C., Beeman J. W., Yu K. M., Nat. Commun. 2016, 7, 12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee D., Kvit A., Choi K.-S., Chem. Mater. 2018, 30, 4704–4712. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary