

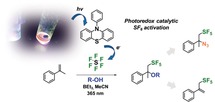
Abstract
SF6 was applied as pentafluorosulfanylation reagent to prepare ethers with a vicinal SF5 substituent through a one‐step method involving photoredox catalysis. This method shows a broad substrate scope with respect to applicable alcohols for the conversion of α‐methyl and α‐phenyl styrenes. The products bear a new structural motif with two functional groups installed in one step. The alkoxy group allows elimination and azidation as further transformations into valuable pentafluorosulfanylated compounds. These results confirm that non‐toxic SF6 is a useful SF5 transfer reagent if properly activated by photoredox catalysis, and toxic reagents are completely avoided. In combination with light as an energy source, a high level of sustainability is achieved. Through this method, the proposed potential of the SF5 substituent in medicinal chemistry, agrochemistry, and materials chemistry may be exploited in the future.
Keywords: addition reactions, electron transfer, phenothiazine, photocatalysis, photochemistry
Dual functionalization by light: Pentafluorosulfanyl and alkoxy substituents were introduced into styrenes with SF6. This method shows a broad substrate scope with respect to applicable alcohols for the conversion of α‐methyl‐ and α‐phenylstyrenes. The products bear a new structural motif with two functional groups installed in one step.
Pentafluorosulfanylation (SF5) chemistry has remained a challenging and difficult task since the initial report on CF3SF5 by Cady in 1950.1 This lack of modern methods is astonishing considering the proposed physicochemical profile of the SF5 substituent when added to small organic molecules.2, 3 For example, exchange of the widely used CF3 substituent that is bioisosteric to CH3 with a SF5 substituent in the anoretic norfenfluramine induces a dramatic change in the pharmacological profile.4 Further evidence for a benign profile of organic SF5 compounds has been reported.5 These features predict great potential for this functional group in chemistry.6 However, the accessibility of SF5 compounds is still rather difficult even though a mild synthesis starting from disulfides has been established by Umemoto and co‐workers in 20127 and further facilitated by Pitts, Togni, and co‐workers quite recently.8 However, formation of the C−S bond still requires the use of extraordinarily toxic reagents, like S2F10, and the mixed‐sulfur halogenides SF5Cl and SF5Br. In contrast, reports on non‐toxic SF6 in synthesis are rare although this would have strong environmental advantages.9, 10, 11, 12, 13, 14, 15 SF6 is still indispensable as an insulating gas in technical applications, like high‐voltage gears, and as a protecting gas in the production of metals. SF6 acts as an extremely potent greenhouse gas,16 so the use of SF6 as chemical reagent would be sustainable because the gas would be trapped and converted into potentially valuable chemical building blocks.
In general, the use of SF6 as an SF5 transfer reagent is difficult due to its alternating bond‐dissociation enthalpies.17 In particular the electron‐excess‐dependent fragmentation channels of the SF6 radical anion have hampered proper activation by photoinduced single‐electron transfer.18, 19, 20, 21, 22, 23 The dominant channel of activation at low electron excess energies is fragmentation into SF4 and a fluoride anion.18, 22, 23 This mode of reactivity was explored recently by Jamison and McTeague, as well as by Rueping and co‐workers, who reported deoxyfluorination‐type chemistry under photoredox conditions (Figure 1).12, 13 We unlocked the complementary mode of activation of SF6 for pentafluorosulfanylation of α‐substituted styrenes.11
Figure 1.
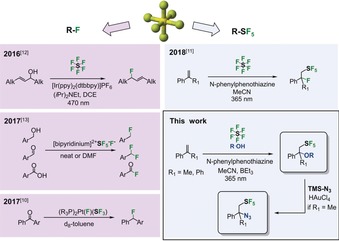
Overview of recent photochemical and chemical activation of SF6 for deoxyfluorinations (left) and pentafluorosulfanylations (right).
Photoredox catalysis applies light as an energy source for organic reactions.24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 Herein, we report an advanced photoredox catalytic method for the activation of SF6, which not only pentafluorosulfanylates α‐methyl‐ (1) and α‐phenyl‐ (2) styrenes but additionally forms a C−O bond, which significantly broadens the synthetic scope and opens the way for the functionalization of SF5 building blocks. In contrast to fluorination,12, 13 our approach precisely controls the local reductivity by N‐phenylphenothiazine (3) as a strong photoredox catalyst36 in order to transfer the SF5 group to α‐methyl‐ (1) and α‐phenyl‐ (2) styrenes to yield products 4 and 5.11 Mechanistic investigations revealed a two‐fold excitation process (Figure 2), similar to the conPET process reported by the König group.37 Quenching of the excited state of 3 by SF6 generates SF6 .−, which is fragmented by the electron excess energy into the SF5 radical. The second electron transfer activates the substrates through formation of the radical cations 1.+ and 2.+. This process seems to be critical for successful pentafluorosulfanylation due to the high oxidation power of the SF5 radical. The simple addition of MeOH to the reaction mixture consisting of 1 or 2, catalyst 3, and SF6 in MeCN yielded the SF5 methyl ethers 8 or 9. The previously observed dimerization of 2 11 was almost completely suppressed, which makes the fast trapping of 2.+ by MeOH very likely. The final step for pentafluorosulfanylation is the simple trapping of the resulting radicals 6. or 7., respectively, by the remaining SF5 radical. The competing nucleophilic attack by in situ generated fluoride anions could be reduced by the addition of a Lewis acid. The addition of 10–20 mol % BEt3 almost completely suppressed the formation of the vicinal fluoride 5 by trapping available fluoride anions in the solution. This is important for the preparation of a broader variety of SF5 compounds with alcohols as external nucleophiles. Further functional groups in the side chains of these alcohols give access to versatile SF5 building blocks.
Figure 2.
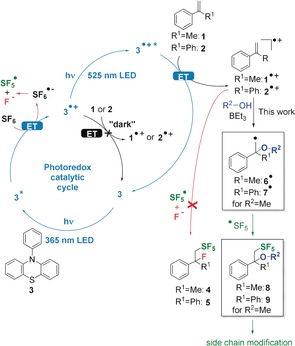
Proposed mechanism of photoredox catalytic activation of SF6 by N‐phenylphenothiazine (3) for pentafluorosulfanylation of α‐methyl (1) and α‐phenyl (2) styrene, and addition by fluoride as an internal nucleophile to give 4 and 5 or alcohols (R2‐OH) as external nucleophiles to give products 8 and 9 (shown for R2=Me).
We optimized the reaction conditions for 2 (Table 1). The initial yield of 29 % of 9 (determined by GC‐FID) was achieved with 10 mol % photocatalyst 3 and 5 equiv of MeOH in a 0.1 m solution of 2. The pressure of SF6 was adjusted to 2.8 bar (3.1 mmol) by a gas measure apparatus. A higher amount of MeOH (10 equiv) increased the yield to 44 %. Reducing the catalyst loading to 5 mol % 3 decreased the yield to 35 %. While dilution of the reaction mixture to 0.05 m also decreased the yield, an optimized yield of 53 % was observed using 0.2 m solution of 2. Higher concentrations did not further increase the yield. Additional control experiments were carried out before a broader substrate scope was investigated. The use of methoxide as a strongly basic nucleophile caused a collapse in reactivity and 9 was not observed. As expected, no product was observed during control reactions in the absence of light or catalyst 3, nor in the absence of MeOH. Finally, we explored the effect of BEt3. While the selectivity was dramatically increased by BEt3 (see above), the yield of 9 could not further be increased by the investigated range of 0–40 mol % BEt3. This indicated a passive interaction in the mechanism and deactivation of the generated fluoride anion by the Lewis acidic boron. The precise active species could not be identified although the formation of an intermediate alcohol coordination complex is likely based on previous observations by Renaud and co‐workers.38 The model reaction was also performed on a scale of 1.00 mmol of 2, which gave a yield of 45 % for 9 with a higher pressure of SF6 (5.5 bar), while the excess of SF6 could be reduced to 6.1 equiv. The preparative isolation of 9 in 40 % yield gave a pure product sample and allowed us to validate both the structure by NMR and XRD (Figure 3) and the applied 19F‐NMR quantification method. It is important to mention here that 8 or 9 are not produced by the reaction of the fluoride addition products 4 or 5 with methoxides, including Ca(OMe)2, KOMe and LiOMe, and with BEt3 (Figures S166–S173).
Table 1.
Photoredox catalytic pentafluorosulfanylations of 2 to the methoxylated 9.
|
Entry |
Conditions[a] |
[2] [m] |
MeOH [equiv] |
Yield [%] |
|---|---|---|---|---|
|
1 |
365 nm |
0.10 |
5 |
29 |
|
2 |
365 nm |
0.10 |
10 |
44 |
|
3 |
365 nm |
0.20 |
10 |
53 |
|
4 |
no light |
0.10 |
10 |
no reaction |
|
5 |
no catalyst |
0.10 |
10 |
no reaction |
[a] General reaction conditions: 20 mol % BEt3, 20 °C, 2.8 bar SF6, 368 nm, 22 h in MeCN. Yields determined by GC‐FID.
Figure 3.
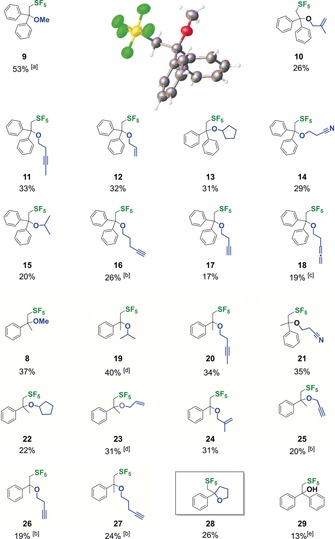
Substrate scope for the for the pentafluorosulfanylation of 1 and 2 and XRD structure of product 9. Yields were determined by 19F‐NMR spectroscopy in the crude reaction mixture. General reaction conditions: 0.20 mmol, 0.20 m in MeCN, 10 mol % 3, 10 mol % BEt3, 22 h, 20 °C, 2.8 bar (15 equiv) SF6, 368 nm. [a] Yield determined by GC‐FID, 20 mol % BEt3 used. [b] 3.0 equiv alkynol. [c] 0.15 m, 14 equiv. allene. [d] 1.00 mmol scale. [e] Prepared with 1‐ethinyl‐1‐cyclopentanol.
The substrate scope for the conversion of 1 and 2 is broad since a variety of functionalized alcohols, like branched alkohols, alkenols, internal and terminal alkynols, sterically demanding cyclopentanols, cyanoalcohols, and even allenes, were tolerated to give products 8, 10–18 and 19–27 (Figure 3).
The photoredox catalytic method is limited, of course, to the use of non‐oxidizable alcohols. Phenyl alcohols were not accepted, likely due to predominant oxidation by the catalyst 3. Even more complex molecules like spiroethers were obtained through an intramolecular addition, yielding 29 in 26 % yield. Full conversion of the starting materials, however, is problematic due to the aggressive reaction conditions and photocatalyst decomposition. Increased photocatalyst concentrations cause overreduction of the transients. Another competing reaction is the direct addition of alcohols to the substrates as well as in situ hydrolysis of the products probably due to the formation of oxophilic sulfur species. Nevertheless, we found a remarkably broad acceptance of various alcohols for the alkoxylation of 1 and 2, and the obtained yields between 13 % and 53 % should be viewed in the context of the fact that compounds 10–28 were not previously synthetically accessible and bear a new and doubly functionalized structural motif. Additionally, our results show an orthogonal reactivity by the SF5‐radical pathway, which allows the use a large excess (10.0 equiv) of alcohol without fully quenching of the reactive transient by deoxyfluorination.12, 13 While the use of water as a nucleophile shut down the reaction, the use of tertiary alcohols favored the formation of alcohol 29.
DSC experiments revealed a boiling point for 8 of about 18 °C (Figure S158) and a melting point for 9 of 125 °C (Figures S159 and S160). Compounds 8 and 9 were photochemically stable during irradiation (365 nm, 24 mm in DMSO, Figures S162 to S165) for at least 62 h. Compounds 8 and 9 were stable at 75 °C in DMSO (24 mm); at 125 °C 8 showed a half‐life of 68 min and 9 a half‐life of 84 min under air (Figures S161). This demonstrates sufficient stability for further chemical transformations, which we investigated: 1) Methoxylated 8 and 9 were successfully converted into the vinylic and allylic SF5 compounds 30 and 31 by the oxophilic Lewis acid BF3⋅Et2O (10 equiv) in CDCl3. The 19F‐NMR kinetic measurements showed remarkably fast conversion in both cases into the elimination products 30 and 31 with yields of more than 98 % in under 30 min (Figure 4, top). 2) Finally, we broadened the versatility of our method by conversion of the benzylic ether 8 into the corresponding azide 32. This reaction required HAuCl4 as the catalyst.39 Instead of the favored elimination reaction (by considering acidity), the 19F NMR spectra evidenced a clean an efficient conversion of 98 % after 5 h (Figure 4, bottom). Compound 32 showed the characteristic IR signatures of both the azide stretch mode at 2109 cm−1 and the SF5 signatures at around 813 cm−1 (Figure S156). It is important to mention here that such vicinal SF5 azides could potentially be used for click‐type cycloadditions or could serve as precursors for the corresponding amino acids.
Figure 4.
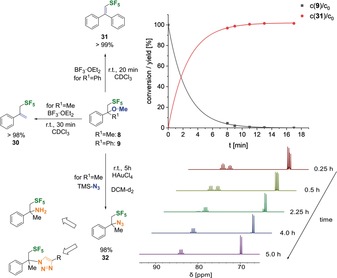
Top: Elimination of the methoxy substituent of 8 and 9 by 10.0 equiv BF3⋅Et2O, and representative 19F‐NMR kinetics for the conversion of 9 to 31. Bottom: Azidation of 8 to 32 (with potential following chemistry) and time‐resolved 19F‐NMR spectroscopy analysis for the conversion of 8 into 32.
In conclusion, we report herein a novel method to synthesize ethers with vicinal SF5 substituent through a one‐step method including photoredox catalysis. The products described herein bear a new structural motif with two functional groups, the SF5 and the alkoxy substituents, and thereby represent important new SF5 building blocks. Moreover, the alkoxy substituents allow further transformation by elimination and azidation. Our results complement the closed‐shell deoxyfluorination‐type photoredox chemistry of SF6 and pave the way to use SF6 as a highly valuable SF5‐transfer reagent if properly activated by highly reducing photoredox catalysts. Our method not only tolerates protic groups and high concentrations of alcohols, but uses them as nucleophiles. Unfortunately, the presence of water as a nucleophile is strictly prohibited by irreversible sulfoxidation of the photoredox catalyst. Despite this restriction, the corresponding SF5 alcohol can be prepared by the use of tertiary alcohols. Toxic reagents are completely avoided, and instead, non‐toxic SF6 is applied as a chemical reagent. Our vision is to reuse SF6 after technical applications for chemical synthesis of valuable SF5 molecules instead of simply destroying it, thereby enabling the proposed benign potential of the SF5 substituent in medicinal, agricultural, and materials chemistry to be exploited in the future. In combination with light as an energy source, the basis for a high level of sustainability is set.
Conflict of interest
D.R. and H.A.W. filed a patent application of the reported method.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/16‐2) and KIT is gratefully acknowledged. D.R. thanks the Landesgraduiertenstiftung Baden‐Württemberg for a doctoral fellowship and the GRK 1626 for their qualification program. We further thank Prof. Dr. Frank Breher and Prof. Dr. Michael A. R. Meier, as well as Prof. Dr. M. Wilhelm for sharing parts of their infrastructure. We thank M. Sc. Bernhard Birenheide for solving the XRD structure of 9, and Marie‐Christin Röpert for assistance with the DSC measurements.
D. Rombach, H.-A. Wagenknecht, Angew. Chem. Int. Ed. 2020, 59, 300.
References
- 1. Silvey G. A., Cady G. H., J. Am. Chem. Soc. 1950, 72, 3624–3626. [Google Scholar]
- 2. Savoie P. R., Welch J. T., Chem. Rev. 2015, 115, 1130–1190. [DOI] [PubMed] [Google Scholar]
- 3. Sowaileh M. F., Hazlitt R. A., Colby D. A., ChemMedChem 2017, 12, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 4. Welch J. T., Lim D. S., Bioorg. Med. Chem. 2007, 15, 6659–6666. [DOI] [PubMed] [Google Scholar]
- 5. Jackson D. A., Mabury S. A., Environ. Toxicol. Chem. 2009, 28, 1866. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Zhang Z. C., Chung T. C. M., Macromolecules 2006, 39, 5187–5189; [Google Scholar]
- 6b. Ollivier C., Renaud P., Chem. Rev. 2001, 101, 3415–3434; [DOI] [PubMed] [Google Scholar]
- 6c. Dolbier W. R., Aït-Mohand S., Schertz T. D., Sergeeva T. A., Cradlebaugh J. A., Mitani A., Gard G. L., Winter R. W., Thrasher J. S., J. Fluorine Chem. 2006, 127, 1302–1310. [Google Scholar]
- 7. Umemoto T., Garrick L. M., Saito N., Beilstein J. Org. Chem. 2012, 8, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pitts C. R., Bornemann D., Liebing P., Santschi N., Togni A., Angew. Chem. Int. Ed. 2019, 58, 1950–1954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1970–1974. [Google Scholar]
- 9. Zámostná L., Braun T., Angew. Chem. Int. Ed. 2015, 54, 10652–10656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10798–10802. [Google Scholar]
- 10. Berg C., Braun T., Ahrens M., Wittwer P., Herrmann R., Angew. Chem. Int. Ed. 2017, 56, 4300; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4364. [Google Scholar]
- 11. Rombach D., Wagenknecht H.-A., ChemCatChem 2018, 10, 2955–2961. [Google Scholar]
- 12. McTeague T. A., Jamison T. F., Angew. Chem. Int. Ed. 2016, 55, 15072–15075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15296–15299. [Google Scholar]
- 13. Rueping M., Nikolaienko P., Lebedev Y., Adams A., Green Chem. 2017, 19, 2571–2575. [Google Scholar]
- 14. Buß F., Mück-Lichtenfeld C., Mehlmann P., Dielmann F., Angew. Chem. Int. Ed. 2018, 57, 4951–4955; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5045–5049. [Google Scholar]
- 15.
- 15a. Harvey B. G., Arif A. M., Glöckner A., Ernst R. D., Organometallics 2007, 26, 2872–2879; [Google Scholar]
- 15b. Basta R., Harvey B. G., Arif A. M., Ernst R. D., J. Am. Chem. Soc. 2005, 127, 11924–11925. [DOI] [PubMed] [Google Scholar]
- 16.Intergovernmental Panel on Climate Change (IPCC). Green-house gases, aerosols and their radiative. InForcing Climate Change1995: The Science of Climate Change. Contribution of WGI to the SecondAssessment Report of the Intergovernmental Panel on Climate Change (Eds.: J. T. Houghton, L. G. Meira Filho, B. A. Callander, N. Harris, A. Kattenberg, K. Maskell), Cambridge University Press, Cambridge, 1996.
- 17. Kiang T., Zare R. N., J. Am. Chem. Soc. 1980, 102, 4024–4029. [Google Scholar]
- 18. Akhgarnusch A., Höckendorf R. F., Beyer M. K., J. Phys. Chem. A 2015, 119, 9978–9985. [DOI] [PubMed] [Google Scholar]
- 19. Drzaic P. S., Brauman J. I., J. Am. Chem. Soc. 1982, 104, 13–19. [Google Scholar]
- 20. Smardzewski R. R., Fox W. B., J. Chem. Phys. 1977, 67, 2309. [Google Scholar]
- 21. Menk S., Das S., Blaum K., Froese M. W., Lange M., Mukherjee M., Repnow R., Schwalm D., von Hahn R., Wolf A., Phys. Rev. A 2014, 89, 022502. [Google Scholar]
- 22. Christophorou L. G., Olthoff J. K., J. Phys. Chem. Ref. Data 2000, 29, 267. [Google Scholar]
- 23. Pelc A., Rapid Commun. Mass Spectrom. 2012, 26, 577–582. [DOI] [PubMed] [Google Scholar]
- 24. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marzo L., Paigre S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228. [Google Scholar]
- 26. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 27. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Staveness D., Bosque I., Stephenson C. R. J., Acc. Chem. Res. 2016, 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buzzetti L., Crisenza G. E. M., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 3730–3747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3768–3786. [Google Scholar]
- 30. Meggers E., Chem. Commun. 2015, 51, 3290–3301. [DOI] [PubMed] [Google Scholar]
- 31. Zou Y.-Q., Hörmann F. M., Bach T., Chem. Soc. Rev. 2018, 47, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravelli D., Fagnoni M., Albini A., Chem. Soc. Rev. 2013, 42, 97–113. [DOI] [PubMed] [Google Scholar]
- 33. Majek M., Jacobi von Wangelin A., Acc. Chem. Res. 2016, 49, 2316–2327. [DOI] [PubMed] [Google Scholar]
- 34. Hopkinson M. N., Tlahuext-Aca A., Glorius F., Acc. Chem. Res. 2016, 49, 2261–2272. [DOI] [PubMed] [Google Scholar]
- 35. Reckenthäler M., Griesbeck A. G., Adv. Synth. Catal. 2013, 355, 2727–2744. [Google Scholar]
- 36. Speck F., Rombach D., Wagenknecht H.-A., Beilstein J. Org. Chem. 2019, 15, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghosh I., Ghosh T., Bardagi J. I., König B., Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]
- 38. Povie G., Marzorati M., Bigler P., Renaud P., J. Org. Chem. 2013, 78, 1553–1558. [DOI] [PubMed] [Google Scholar]
- 39. Sawama Y., Goto R., Nagata S., Shishido Y., Monguchi Y., Sajiki H., Chem. Eur. J. 2014, 20, 2631–2636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary