

Abstract
Upadacitinib is a selective Janus kinase (JAK) 1 inhibitor being developed for treatment of rheumatoid arthritis. This study characterizes the relationships between upadacitinib exposure and interleukin (IL)‐6–induced signal transducer and activator of transcription proteins 3 (STAT3) phosphorylation (pSTAT3) and IL‐7–induced STAT5 phosphorylation (pSTAT5) in the ex vivo setting as measures for JAK1 and JAK1/JAK3 inhibition, respectively, with comparison to tofacitinib. Drug plasma concentrations and ex vivo IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 in blood from subjects evaluated in 2 phase 1 studies who received immediate‐release 1 mg to 48 mg upadacitinib, 5 mg twice daily (BID) tofacitinib, or placebo were determined. Exposure‐response models were developed, and the effects of different upadacitinib doses on ex vivo biomarker responses were simulated and compared to tofacitinib. Upadacitinib (and tofacitinib) reversibly inhibited IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 in a concentration‐dependent manner. Model‐estimated values of 50% of the maximum effect were 60.7 nM for upadacitinib and 119 nM for tofacitinib for IL‐6–induced pSTAT3 inhibition, and 125 nM for upadacitinib and 79.1 nM for tofacitinib for IL‐7–induced pSTAT5 inhibition. Tofacitinib 5 mg BID is estimated to have a similar magnitude of effect on IL‐6–induced pSTAT3 to ∼3 mg BID of upadacitinib (immediate‐release formulation), whereas a 4‐fold higher dose of upadacitinib (∼12 mg BID), is estimated to show a similar magnitude of inhibition on IL‐7–induced pSTAT5 as tofacitinb 5 mg BID. This study confirms that in humans, upadacitinib has greater selectivity for JAK1 vs JAK3 relative to the rheumatoid arthritis approved dose of tofacitinib, and results from these analyses informed the selection of upadacitinib IR doses evaluated in phase 2.
Keywords: IL‐6, IL‐7, JAK1, JAK3, Janus kinase, rheumatoid arthritis, STAT3, STAT5, target engagement, tofacitinib, upadacitinib
Rheumatoid arthritis (RA) is a chronic, progressive inflammatory disease of synovial joints that can lead to bone erosion, painful deformity, and disability. The treatment of RA was revolutionized by the introduction of biologic and conventional synthetic disease‐modifying antirheumatic drugs. Although these treatments have profound efficacy in some patients, there are a significant proportion of patients for whom these drugs have inadequate activity or no effect at all.1, 2
Cytokine signaling through Janus kinase (JAK) enzymes is critical for normal physiological functions, including immune regulation and erythropoiesis, but also plays an important role in the regulation of the immune system in autoimmune inflammatory diseases including RA.3, 4, 5, 6 Therefore, inhibition of JAK enzymes has been demonstrated to be an effective modality for the treatment of several autoimmune diseases.7 The JAK family comprises 4 members: JAK1, JAK2, JAK3, and tyrosine kinase 2; these kinases, coupled with phosphorylation of various isoforms of signal transducer and activator of transcription proteins (STATs), mediate signaling of cytokine and growth factor receptors.8 Enhancing potency for JAK1, while minimizing the inhibitory effects on other JAK isoforms, has been hypothesized as a potential approach to improve risk‐benefit profiles of JAK inhibitors in the treatment of RA compared to less selective JAK inhibitors.9, 10, 11, 12, 13
Upadacitinib is a selective JAK1 inhibitor that is currently in development for the treatment of several inflammatory diseases, including RA.14, 15, 16, 17, 18 In cellular assays, upadacitinib was ∼60‐fold selective for JAK1 over JAK2, and >100‐fold selective over JAK3.19 The phase 1 program of upadacitinib evaluated the pharmacokinetics and short‐term safety and tolerability of upadacitinib following the administration of single immediate‐release (IR) doses ranging from 1 mg to 48 mg in healthy subjects, multiple IR twice‐daily (BID) dosing ranging from 3 mg BID to 24 mg BID in healthy subjects, and multiple IR BID doses ranging from 6 mg BID to 24 mg BID in subjects with RA.20 Upadacitinib plasma exposure was approximately dose proportional over the evaluated dose range, with no significant accumulation with repeated BID dosing of the IR formulation. Upadacitinib terminal elimination half‐life was 6 to 16 hours, and the functional half‐life, calculated from maximum observed plasma concentration (Cmax) to trough plasma concentration ratio at steady state was 3 to 4 hours.20 Upadacitinib was administered in phase 2 trials in RA in the form of IR formulation. In order to enhance patient compliance and provide a more convenient once‐daily (QD) dosing, upadacitinib extended‐release (ER) formulation was developed and was used in phase 3 trials. Upadacitinib ER formulation has a relative bioavailability of 76% relative to the IR formulation.21, 22 A upadacitinib dose of 15 mg QD using the ER formulation provides equivalent daily area under the plasma concentration–time curve (AUC) and similar Cmax and minimum observed plasma concentration to 6 mg BID using the IR formulation.22
Tofacitinib is a non‐selective JAK inhibitor that was approved by the US Food and Drug Administration in 2012 for the treatment of subjects with moderate to severe RA at a dose of 5 mg BID.23 In vitro, tofacitinib potently inhibits JAK3, JAK1, and to a lower extent, JAK2.24 Upadacitinib demonstrated greater inhibitory potency in vitro to JAK1 and lower potency to JAK3 compared to tofacitinib.19, 25 To evaluate if the greater selectivity of upadacitinib for JAK1 over JAK3 compared to tofacitinib translates in human, the present analyses were conducted using ex vivo stimulation of STAT3 phosphorylation (pSTAT3) by IL‐6 (as a measure of JAK1 activity)26 and of STAT5 phosphorylation (pSTAT5) by IL‐7 (as a measure of JAK1/JAK3 activity)27 using blood samples collected from 2 upadacitinib phase 1 clinical studies in healthy subjects and in subjects with RA.20 The objectives of these analyses were to characterize the relationships between upadacitinib plasma exposures and IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5, respectively, and to compare upadacitinib and tofacitinib effects on these ex vivo pharmacodynamic biomarkers. This work benchmarked the activity of upadacitinib doses based on the biomarker effects relative to the approved RA dose of tofacitinib, which informed upadacitinib IR doses evaluated in phase 2 studies.
Methods
Data Sources and Subjects
Data from 2 phase 1 clinical studies were included in the analyses. Both phase 1 studies used in the analyses were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols and informed consent forms were approved by the institutional review boards, and participants provided written informed consent before any study‐related procedures were performed.
Overview of the design of the studies is presented in Table 1. Details of the design and eligibility criteria for the 2 studies were previously described (except for Part 3 of Study 2, described below).20 The IR formulation of upadacitinib was administered in both studies. In Study 1, healthy subjects (N = 56) were randomized to receive single doses of upadacitinib (1, 3, 6, 12, 24, 36, and 48 mg) or placebo. Study 2 had 3 parts: in Part 1, healthy subjects (N = 44) received multiple doses of upadacitinib (3, 6, 12, or 24 mg) or placebo BID for 14 days; in Part 2, subjects with mild to moderate RA (N = 14) received multiple doses of upadacitinib (6, 12, or 24 mg) or placebo BID for 26 days on background treatment of methotrexate (10‐25 mg/week).
Table 1.
Overview of the Studies Included in the Analysis
| Study | Population | N | Study Design Pharmacokinetic Sampling/Subject | Active Treatment | Dose(s)a | Reference |
|---|---|---|---|---|---|---|
| 1 | Healthy subjects | 56 | Single‐dose, randomized, placebo‐controlled | Upadacitinib | 1, 3, 6, 12, 24, 36, 48 mg | Mohamed et al20 |
| 2, Part 1 | Healthy subjects | 44 | Multiple‐dose, randomized, placebo‐controlled | Upadacitinib | 3, 6, 12, 24 mg BID | Mohamed et al20 |
| 2, Part 2 | Subjects with mild to moderate RA | 14 | Multiple dose, randomized, placebo‐controlled | Upadacitinib | 6, 12, 24 mg BID | Mohamed et al20 |
| 2, Part 3 | Healthy subjects | 9 | Multiple dose, open‐label | Tofacitinib | 5 mg BID | – |
BID, twice daily; RA, rheumatoid arthritis.
aUpadacitinib and tofacitinib were administered in the study as immediate‐release formulations.
Study 2, Part 3, was a single‐arm, open‐label study designed to characterize tofacitinib pharmacokinetics and its effects on ex vivo pharmacodynamic biomarkers in healthy subjects. Subjects received multiple doses of tofacitinib 5 mg BID for 14 days. This part of the study was conducted at PPD Development (Austin, Texas), and the study protocol and informed consent were approved by the RCRC Institutional Review Board (Austin, Texas). Adult male and female subjects (N = 9) between 18 and 55 years of age in general good health based on the results of medical history, laboratory profile, physical examination, chest x‐ray, and 12‐lead electrocardiogram, with a body mass index of 19 to 29 kg/m2 were selected to participate in Study 2, Part 3.
Ex Vivo Measurements of IL‐6–Induced Phosphorylation of Signal Transducer and Activator of Transcription Proteins 3 (pSTAT3) and IL‐7–Induced pSTAT5
For each subject, blood samples were collected by venipuncture into 2‐mL sodium heparin tubes before and at 1, 6, and 12 hours after administration of upadacitinib (day 1 in Study 1; days 1 and 14 in Study 2, Part 1; days 3 and 28 of Study 2, Part 2), tofacitinib (days 1 and 14 in Study 2, Part 3), or placebo (day 1 in Study 1; days 1 and 14 in Study 2, Part 1; days 3 and 28 of Study 2, Part 2). The blood samples were frozen and shipped to the AbbVie Bioresearch Center (Worcester, Massachusetts) for processing and analysis. IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 were assayed as previously described.19 Briefly, recombinant human IL‐6 or IL‐7 (R&D Systems, Minneapolis, Minnesota) was added to blood followed by addition of surface antibodies (CD14‐APC, CD3‐fluorescein isothiocyanate; BD Biosciences, San Jose, California), sample lysis, wash, and resuspension in BD Perm buffer III (BD Biosciences). Samples were then washed and stained with pSTAT5‐PE or pSTAT3‐PE (BD Biosciences) and analyzed on a FACSCalibur (BD Biosciences). The percent change was calculated as a change in pSTAT3 and pSTAT5 at 1, 6, and 12 hours following drug administration divided by the baseline measurement at t = 0 for each subject multiplied by 100.
Pharmacokinetic Assessments of Upadacitinib and Tofacitinib
In healthy subjects, serial blood samples for determination of upadacitinib or tofacitinib plasma concentrations were collected before dosing and for 72 hours after single‐dose administration of upadacitinib in Study 1 or for 12 hours after the first dose and for 72 hours after the last dose administration in Study 2, Part 1 (for upadacitinib), and Study 2, Part 3 (for tofacitinib). In subjects with RA (Study 2, Part 2), serial blood samples for upadacitinib assay were collected over 12 hours following the first study drug dose on study day 3, over 12 hours following the morning study drug dose on study day 28, and over 48 hours following the last study drug dose on study day 29 as previously described.20 Upadacitinib plasma concentrations were measured using a validated high‐performance liquid chromatography–tandem mass spectrometry as described previously.20 Tofacitinib plasma concentrations were measured using a validated high‐performance liquid chromatography–tandem mass spectrometry method at AbbVie (North Chicago, Illinois) with lower limit of quantification of 0.6 ng/mL; the mean bias of the analytical assay was ≤3.2%, and the percent coefficient of variation (as a measure of precision) was ≤6.2%.
Pharmacokinetic Models for Upadacitinib and Tofacitinib
Plasma concentrations vs time data for upadacitinib and tofacitinib were analyzed separately using the nonlinear mixed‐effects modeling software NONMEM (Version 7.3; ICON Development Solutions, Ellicott City, Maryland) to characterize upadacitinib and tofacitinib pharmacokinetics with data from subjects in Study 1 and Study 2. The pharmacokinetic models were fitted to the data using the first‐order conditional estimation method with INTERACTION employed within NONMEM. The population pharmacokinetic analyses followed the same methodology we previously reported elsewhere.21, 28, 29 The developed population pharmacokinetic models used as input in the exposure‐response analyses were a 2‐compartment model with absorption lag time for upadacitinib (apparent oral clearance of 45 L/h in healthy subjects and 36 L/h in subjects with RA; steady‐state apparent volume of 218 L) and a 1‐compartment model for tofacitinib (apparent oral clearance of 34.9 L/h and steady‐state apparent volume of 110 L). These parameter estimates were consistent with estimates from more extensive analyses including larger data sets and data from later‐phase trials.21, 28, 29
Exposure‐Response Analyses for the Effect of Upadacitinib and Tofacitinib on IL‐6–Induced pSTAT3 and IL‐7–Induced pSTAT5
IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 models were built in a stepwise manner. The empirical Bayesian individual pharmacokinetic parameters from the pharmacokinetic models were used to build exposure‐response models and the data for upadacitinib and tofacitinib were fit simultaneously. First, a model describing the placebo effect was developed considering only the IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 time course in subjects who received placebo. Different placebo models (no placebo response, linear placebo response, exponential placebo response) were evaluated.
The no placebo response model (intercept only) was defined as:
| (1) |
with θ1 representing the intercept. If needed, a slope was added to the model to describe a linear time course of the placebo response:
| (2) |
with θ2 representing the slope. Alternatively, an exponential model was defined as:
| (3) |
Second, the active treatment (upadacitinib and tofacitinib) data were modeled along with the placebo data to describe the IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 time course under each treatment group. Different effect models were evaluated to estimate the time course of the dose‐dependent effects of upadacitinib and tofacitinib on IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5.
The direct drug effect model was defined as a combination of the placebo effect and a maximum effect (Emax) model:
| (4) |
with θ3 representing the Emax and θ4 the drug concentration producing 50% of Emax (EC50). Cp is the drug plasma concentration generated from the empirical Bayesian individual pharmacokinetic parameters. A common Emax parameter was used for upadacitinib and tofacitinib with different EC50 values.
Delayed effect and indirect effect models were also evaluated to estimate IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 following upadacitinib or tofacitinib administration.
Intersubject variability was evaluated using additive error models for the placebo effect (or intercept) and Emax, and exponential error models for the EC50 of each drug. Residual variability was modeled using the additive, proportional, or a combination of additive and proportional error model and relevant covariate‐parameter relationships were investigated using forward inclusion/backward elimination procedures.
Model selection was performed based on achieving physiologically reasonable, precise, and statistically significant parameter estimates (95% confidence intervals do not include reference values). In addition, the likelihood ratio test was used for hypothesis testing to discriminate among alternative nested models. Because the difference between 2 objective function values (OFVs) provided by NONMEM is approximately chi‐squared distributed, this statistic was used to guide model building. When comparing nested models, 1 additional model parameter, corresponding to 1 degree of freedom in the higher‐order model, was considered significant if it lowered the OFV by >6.63, corresponding to P < .01. For 2 degrees of freedom, the required reduction in OFV was 9.21. All statistical tests were conducted at the .01 significance level, except tests in the backward elimination step of the covariate selection procedure that were conducted at the .001 significance level (changes in OFV of 10.83 and 13.82 for 1 and 2 degrees of freedom, respectively). Model adequacy was evaluated using goodness‐of‐fit plots, visual predictive checks, and bootstrap evaluation. For the visual predictive checks, final model parameters were used to simulate 1000 replicates of the original data set. Model evaluation was performed by superimposing the median with 5th and 95th percentiles of observed data on median with 5th and 95th percentiles of the simulated data. For bootstrap evaluation, N participants were randomly sampled with replacement from the original data set to form 1000 bootstrap replicates, where N is the number of participants in the original data set. The final model was used to estimate population parameters for each bootstrap replicate, and medians and 95% confidence intervals for each parameter were calculated across the successfully converging runs. Final model parameter estimates based on the original data set were compared against the bootstrap results.
Covariates evaluated included RA population, age, weight, body mass index, body surface area, baseline IL‐6–induced pSTAT3, sex, and race on placebo effect and on EC50.
The final exposure‐response models were used to simulate the effects of different BID doses of upadacitinib using the IR formulation as well as of the RA approved dose of tofacitinib 5 mg BID and placebo on IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5, which informed the selection of upadacitinib IR doses evaluated in phase 2.
Results
Upadacitinib pharmacokinetic parameters after single and multiple doses in these studies have been previously reported.20 Following the administration of 5 mg BID doses of tofacitinib for 14 days, tofacitinib mean Cmax was 40.9 ng/mL (130 nM), Ctrough was 1.23 ng/mL [3.9 nM], AUC0‐12 was 135 ng • h/mL (432 nM • h), and harmonic mean half‐life was 2.3 hours. Tofacitinib Cmax was reached with a median time of 1 hour after oral administration. These results for tofacitinib pharmacokinetics are in agreement with previously reported results from other studies.30
Observed Changes in IL‐6–Induced pSTAT3 and IL‐7–Induced pSTAT5
Blood samples from 90 healthy volunteers and 10 RA patients were used in the ex vivo pharmacodynamic analyses. Of these, 68 received upadacitinib, 23 received placebo, and 9 received tofacitinib. The time course (observed and model‐predicted) for the percent changes from baseline in IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 over a dosing interval are presented in Figures 1 and 2, respectively, after administration of single and multiple doses of upadacitinib, placebo, or tofacitinib. Following the administration of upadacitinib, IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 were reduced in a dose‐dependent manner, with maximum inhibition observed at 1 hour after dosing. The effects of upadacitinib and tofacitinib at steady state (day 14) were consistent with the effects after a single dose for the corresponding doses. The mean observed percent decrease from baseline in IL‐6–induced pSTAT3 levels after 1 hour following upadacitinib administration were 18% to ∼100%, and mean percent decreases for IL‐7–induced pSTAT5 levels were 21% to 93% across the full upadacitinib single‐ and multiple‐dose range. Following administration of 5 mg of tofacitinib (after the first dose and at steady state), mean IL‐6–induced pSTAT3 levels after 1 hour (also corresponds to maximum observed effect of tofacitinib on IL‐6 and IL‐7 ex vivo biomarkers) were 45% to 49% lower compared to baseline, and IL‐7–induced pSTAT5 levels were 61% to 71% lower compared to baseline. The inhibitory effects of upadacitinib and tofacitinib on IL‐6–induced pSTAT3 and of IL‐7–induced pSTAT5 were reversible, with levels returning close to baseline at the end of the 12‐hour dosing interval.
Figure 1.
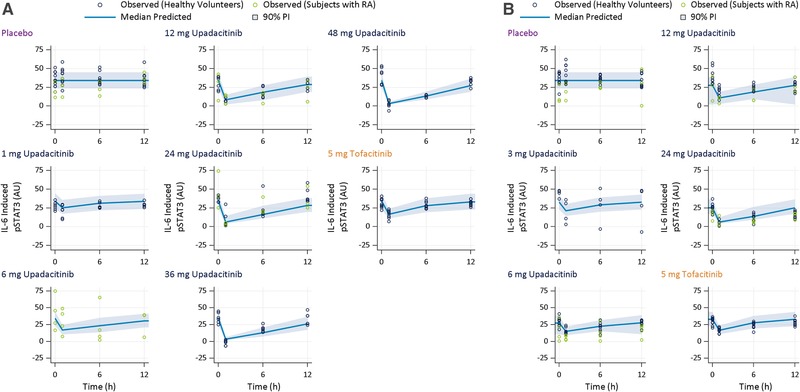
Observed and model‐predicted IL‐6–induced pSTAT3 over a dosing interval after (A) the first/single dose and (B) multiple twice‐daily doses of placebo, upadacitinib, or tofacitinib. AU, arbitrary units of fluorescence; IL, interleukin; PI, prediction interval; pSTAT3, phosphorylation of signal transducer and activator of transcription proteins 3; RA, rheumatoid arthritis.
Figure 2.
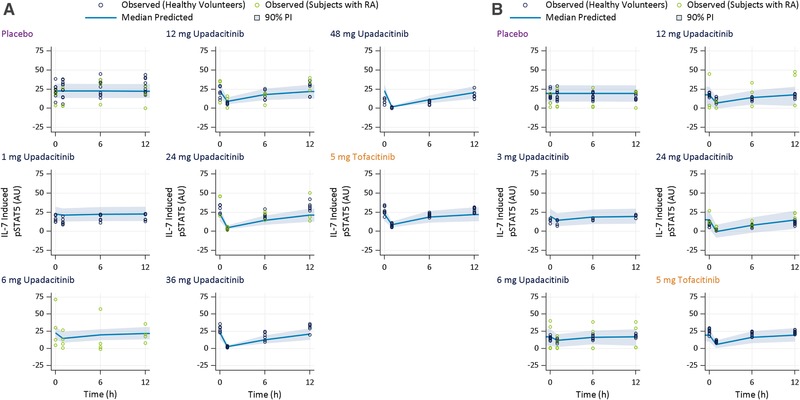
Observed and model‐predicted IL‐7–induced pSTAT5 over a dosing interval after (A) the first/single dose and (B) multiple twice‐daily doses of placebo, upadacitinib, or tofacitinib. AU, arbitrary units of fluorescence; IL, interleukin; PI, prediction interval; pSTAT3, phosphorylation of signal transducer and activator of transcription proteins 3; RA, rheumatoid arthritis.
Relationships Between Upadacitinib and Tofacitinib Plasma Exposures and Changes in IL‐6–Induced pSTAT3 and IL‐7–Induced pSTAT5
IL‐6–induced STAT phosphorylation following oral administration of upadacitinib or tofacitinib to healthy subjects, and oral administration of upadacitinib to subjects with RA was described by a combination of an intercept for baseline and an additive direct drug effect model. The drug (upadacitinib or tofacitinib) effect was described by an Emax model. Emax was fixed in the model to the negative value of the estimated intercept, which corresponds to 100% inhibition of IL‐6–induced pSTAT3. Different EC50 values were estimated for upadacitinib and tofacitinib. The combined error model was found to be most appropriate for explaining the residual error, with the same standard deviation assumed as the additive intersubject error on intercept. Observed baseline IL‐6–induced pSTAT3 value was included as a covariate on EC50 for both upadacitinib and tofacitinib (higher EC50 with higher baseline IL‐6–induced pSTAT3).
IL‐7–induced STAT phosphorylation following oral administration of upadacitinib and tofacitinib to healthy subjects and subjects with RA was best described by a combination of a linear placebo response model and Emax direct drug effect model. The model included intersubject variability on the slope of the placebo response. Different EC50 values were estimated for upadacitinib and tofacitinib and a sigmoidity parameter (Hill coefficient) was found to be significant (ΔOFV = –7.73, P < .01) in the IL‐7 model. No covariates were found to be significant in the IL‐7 exposure‐response model. The additive error model was found to be most appropriate for explaining the residual error, with the same standard deviation assumed as the additive intersubject variability of intercept.
The model‐estimated EC50 values for inhibition of IL‐6–induced pSTAT3 were 61 nM (23.1 ng/mL) for upadacitinib and 119 nM (37.1 ng/mL) for tofacitinib, while the model‐estimated EC50 values for inhibition of IL‐7–induced pSTAT5 were 125 nM (47.7 ng/mL) for upadacitinib and 79 nM (24.7 ng/mL) for tofacitinib (Table 2). The mode‐estimated and observed change from baseline in IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 for different upadacitinib doses and for tofacitinib 5 mg BID are presented in Figures 1 and 2, which demonstrate that the models adequately describes the observed effects of upadacitinib and tofacitinib on ex vivo biomarkers.
Table 2.
Parameter Estimates and Variability for the Final Models Describing the Effects of Upadacitinib and Tofacitinib on Ex Vivo IL‐6–Induced pSTAT3 and IL‐7–Induced pSTAT5
| Bootstrap Results (N = 1000) | |||
|---|---|---|---|
| Parameter | Population Estimate (%RSE) | Median | 95% Confidence Interval |
| IL‐6–induced pSTAT3 | |||
| EC50 for upadacitinib (nM) | 61 (11) | 60 | 48‐77 |
| EC50 for tofacitinib (nM) | 119 (24) | 117 | 87‐160 |
| Intercept (AU) | 34 (3) | 34 | 32‐37 |
| Emax for drug effect (AU) | −34 (fixed) | −34 (fixed) | – |
| Standard deviation of additive ISV of intercept and residual error (AU) | 6.5 (7.8) | 6.4 | 5.2‐7.9 |
| Exponent of the nonlinear relationship between observed baseline IL‐6–induced phosphorylation and EC50 a | 1.2 (20) | 1.2 | 0.5‐1.8 |
| Proportional residual error (%) | 24 (12) | 23 | 4.5‐32 |
| IL‐7–induced pSTAT5 | |||
| EC50 for upadacitinib (nM) | 125 (9.4) | 124 | 106‐143 |
| EC50 for tofacitinib (nM) | 79 (16) | 80 | 67‐91 |
| Slope of placebo effect (AU/h) | −0.009 (25) | −0.009 | −0.013 to −0.004 |
| Intercept (AU) | 23 (3.4) | 23 | 21‐25 |
| Emax for drug effect (AU) | −23 (fixed) | −23 (fixed) | – |
| Hill factor | 1.3 (10) | 1.3 | 1.1‐1.5 |
| Standard deviation of additive ISV of intercept and residual error (AU) | 5.6 (3.3) | 5.5 | 4.7‐6.3 |
| Standard deviation of additive ISV of slope of placebo effect (AU/h) | 0.008 (20) | 0.008 | 0.0001‐0.01 |
AU, arbitrary units of fluorescence; EC50, 50% of the maximum effect; Emax, maximum effect; IL, interleukin; ISV, intersubject variability; pSTAT3, phosphorylation of signal transducer and activator of transcription proteins 3; RSE, relative standard error.
Proportional residual error = proportional residual error calculated as σprop *100.
aEC50 in IL‐6–induced pSTAT3 model was defined as EC50 = θ4* (BLINDUC6/32.95)θ6, where θ4 is the estimate of typical EC50 for an individual with baseline IL‐6–induced phosphorylation equivalent to the data median (32.95 AU), BLINDUC6 is the observed IL‐6–induced pSTAT3 at baseline, and θ6 is the exponent of the nonlinear relationship between the observed baseline IL‐6–induced phosphorylation and EC50.
Estimating the Effects of Different Regimens of Upadacitinib Immediate‐Release Doses Compared to Tofacitinib 5 mg BID on IL‐6–Induced pSTAT3 and IL‐7–Induced pSTAT5 Using the Exposure‐Response Models
The developed models were used to simulate the effects of different regimens using the IR formulation of upadacitinib as well as tofacitinib 5 mg BID at steady state. Model‐estimated effects of different IR regimens of upadacitinib or tofacitinib are presented in Figure 3. Tofacitinib 5 mg BID is estimated to have a similar magnitude of effect on IL‐6–induced pSTAT3 to ∼3 mg BID of upadacitinib (IR formulation), whereas on IL‐7–induced pSTAT5 a much higher dose of upadacitinib, ∼12 mg BID, is estimated to show a similar magnitude of inhibition as tofacitinb 5 mg BID.
Figure 3.
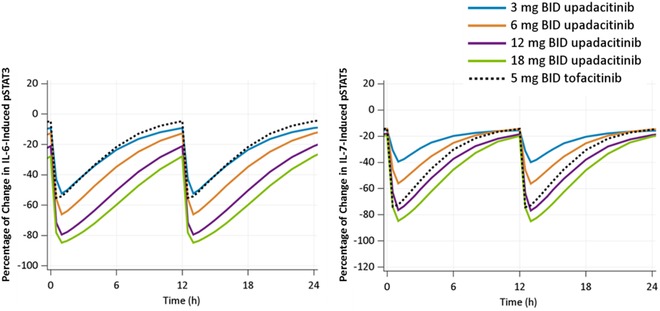
Model‐predicted median percent inhibition of ex vivo IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 by immediate‐release regimens of upadacitinib and 5 mg of tofacitinib after 14 days of twice‐daily administration. IL, interleukin; pSTAT3, phosphorylation of signal transducer and activator of transcription proteins 3.
Discussion
Results from the current study demonstrated that upadacitinib, a selective JAK1 inhibitor, rapidly inhibited IL‐6–induced STAT3 phosphorylation and IL‐7–induced STAT5 phosphorylation in vivo in a reversible and concentration‐dependent manner. The time course for these signaling events by upadacitinib and tofacitinib appeared to follow the plasma concentration levels of both JAK inhibitors with maximum inhibition coinciding with the maximum plasma concentration for each (∼1 hour) and STAT phosphorylation levels returning close to baseline level by the end of the dosing interval for each compound (Figure 1). IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 measurements returned to near their baseline levels by the end of the upadacitinib or tofacitinib dosing interval, which confirms that both compounds are reversible inhibitors. The analyses demonstrated greater potency of inhibition of IL‐6–induced pSTAT3 (as a measure of JAK1 activity) and lower potency of inhibition of IL‐7–induced pSTAT5 (as a measure of JAK1/3 activity) for upadacitinib compared to tofacitinib using blood samples from subjects who received upadacitinib or tofacitinib (Table 2 and Figure 3). The potency ratio for inhibition of IL‐6–induced pSTAT3 relative to IL‐7–induced pSTAT5 (based on the ratio of estimated EC50 values; Table 2) is 2.0 for upadacitinib and 0.67 for tofacitinib, indicating ∼3‐fold higher selectivity for upadacitinib over tofacitinib for inhibition of IL‐6–induced pSTAT3 than IL‐7–induced pSTAT5. These results are in agreement with the in vitro results, which demonstrated greater JAK1 to JAK3 relative potency of upadacitinib compared to tofacitinib.19, 25
The involvement of IL‐6 in RA pathogenesis is well established, and targeting IL‐6 or IL‐6 receptors was demonstrated to be efficacious in the treatment of RA.31, 32, 33 Therefore, by comparing the effects of different doses of upadacitinib to the approved tofacitinib dose on IL‐6–induced pSTAT3 through these analyses, it was possible to inform upadacitinib IR dose selection in the 2 phase 2 studies (BALANCE I and II).27 The analyses demonstrated that the approved dose of tofacitinib in RA (5 mg BID) provides comparable effects on IL‐6–induced pSTAT3 to ∼3 mg BID of upadacitinib (IR formulation). As such, upadacitinib doses of 3 mg BID and higher were predicted to be efficacious, with the potential of 6‐ and 12‐mg BID doses to provide greater efficacy benefit than a tofacitinib 5‐mg BID dose. Upadacitinib doses of 12 mg BID appeared to result in comparable effects on IL‐7–induced pSTAT5 phosphorylation to tofacitinib 5 mg BID, suggesting that upadacitinib doses of 12 mg BID and higher start to lose the JAK1 selectivity. Informed with these analyses, a dose range of 3 mg BID to 18 mg BID of upadacitinib IR formulation was selected for evaluation in the BALANCE I and II phase 2 trials to characterize the plateau of efficacy or the maximal therapeutic benefit of upadacitinib in the different RA subpopulations, and to better understand the selectivity profile of upadacitinib in the target patient population with a longer treatment duration.27
Results from upadacitinib BALANCE I and BALANCE II phase 2b trials were consistent with the pharmacodynamic biomarker analyses presented in this paper. Exposure‐response analyses characterized the relationships between upadacitinib exposures and efficacy (assessed as the percentage of subjects achieving ACR20/50/70) using data from the 2 dose‐ranging phase 2 studies, and indicated that upadacitinib plasma exposures associated with 6 mg BID to 12 mg BID using the IR formulation may maximize efficacy in patients with moderately to severely active RA who are on background treatment of methotrexate. The exposure‐response analyses indicated the upadacitinib dose of 3 mg BID using the immediate‐release formulation may provide suboptimal efficacy compared to higher doses, especially in the more refractory anti‐TNF‐IR population.27 The exposure‐response analyses also indicated that upadacitinib doses higher than 12 mg BID using the IR formulation may result in greater effects on the natural killer cells (believed to be mediated through JAK1/JAK3 inhibition) as well as some other laboratory parameters, suggesting progressive loss of JAK1 selectivity with higher doses.
To enhance patient compliance and provide a more convenient QD dosing in phase 3 studies, the ER formulation of upadacitinib was developed at the transition between phase 2 and phase 3 development of upadacitinib for treatment of RA. We have demonstrated that doses of 15 mg and 30 mg QD using the ER formulation provide similar plasma exposures over day (AUC, maximum and minimum plasma concentrations) to 6 mg BID and 12 mg BID using the IR formulation.22 While upadacitinib doses >6 mg BID using the IR formulation result in greater inhibition of ex vivo IL‐6–induced pSTAT3 (Figure 3), this greater inhibition does not translate into greater clinical efficacy in the treatment of RA based on results as demonstrated by the clinical end‐point results from the phase 2 dose‐ranging studies and from the phase 3 trials (which evaluated 15‐ and 30‐mg ER QD doses of upadacitinib that are equivalent to 6 mg and 12 mg IR BID).27, 28, 34 This indicates that only partial inhibition of IL‐6 signaling is adequate to maximize efficacy of JAK inhibitors in RA. Doses (or equivalent exposures) >12 mg BID using the IR formulation (or 30 mg QD using the ER formulation) have provided clinically meaningful additional efficacy benefit in Crohn disease and ulcerative colitis.35, 36, 37 This difference in the doses that maximize efficacy between different autoimmune inflammatory diseases is likely attributed to the different patterns of cytokine involvement as well as the different sites of inflammation.
The blood samples used in the ex vivo analyses were collected from mostly healthy subjects in addition to 10 subjects with mild to moderate RA. To explore whether the effects of upadacitinib on IL‐6–induced pSTAT3 and IL‐7–induced pSTAT5 are different between healthy subjects and subjects with RA, subject population was evaluated as a covariate on upadacitinib EC50, which demonstrated lack of statistically significant difference in upadacitinib EC50 for inhibition of ex vivo STAT phosphorylation between healthy subjects and subjects with RA. This assessment is limited, however, by the small number of subjects with RA included in the analyses.
Conclusion
In summary, upadacitinib demonstrated reversible and concentration‐dependent inhibition of IL‐6–induced pSTAT3 (as a measure of JAK1 activity) and IL‐7–induced pSTAT5 (as a measure of JAK1/3 activity) in samples from healthy subjects and subjects with RA treated with different upadacitinib doses. Ex vivo pharmacodynamic assay results showed a greater selectivity of upadacitinib on JAK1 versus JAK3 compared to tofacitinib, confirming higher in vitro potency of upadacitinib against JAK1 compared to JAK3. Maximizing efficacy of JAK inhibitors in the treatment of RA does not necessarily require maximizing inhibition of IL‐6 signaling in vivo. The biomarker analyses presented in this paper informed the selection of upadacitinib IR doses evaluated in phase 2 studies, supporting an overall successful development program for upadacitinib in RA.
Conflicts of Interest
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that the submitted work was supported by AbbVie Inc. AbbVie contributed to the study design, research, and interpretation of data, and the writing, review, and approval of the publication. All authors are employees of AbbVie and may hold AbbVie stock or stock options.
Acknowledgments
The authors thank AbbVie employees Ben Kluender for helping with simulations, Wesley Wayman for medical writing support, and Jonathan George for supporting the ex‐vivo stimulation blood sample analyses.
Data Sharing
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan and execution of a Data Sharing Agreement. Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html
Dr. Ahmed A Othman is a Fellow of the American College of Clinical Pharmacology
[Correction added on 7 October 2019, after first online publication: “FCP” has been attributed to Ahmed A. Othman in the author byline, and Jonathan George has been added to the acknowledgments.]
References
- 1. Wilkie WS, Schwieterman P. Strategies for the management of rheumatoid arthritis. Orthopedics. 2012;35(2):125‐130. [DOI] [PubMed] [Google Scholar]
- 2. Rindfleisch JA, Muller D. Diagnosis and management of rheumatoid arthritis. Am Fam Physician. 2005;72(6):1037‐1047. [PubMed] [Google Scholar]
- 3. Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol. 2001;73(3):271‐277. [DOI] [PubMed] [Google Scholar]
- 4. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune‐mediated disease. Immunity. 2012;36(4):542‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vaddi K, Luchi M. JAK inhibition for the treatment of rheumatoid arthritis: a new era in oral DMARD therapy. Expert Opin Investig Drugs. 2012;21(7):961‐973. [DOI] [PubMed] [Google Scholar]
- 6. Isomaki P, Junttila I, Vidqvist KL, Korpela M, Silvennoinen O. The activity of JAK‐STAT pathways in rheumatoid arthritis: constitutive activation of STAT3 correlates with interleukin 6 levels. Rheumatology (Oxford). 2015;54(6):1103‐1113. [DOI] [PubMed] [Google Scholar]
- 7. Fragoulis GE, McInnes IB, Siebert S. JAK‐inhibitors: new players in the field of immune‐mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford). 2019;58(suppl. 1):i43‐i54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aaronson DS, Horvath CM. A road map for those who don't know JAK‐STAT. Science. 2002;296(5573):1653‐1655. [DOI] [PubMed] [Google Scholar]
- 9. Ghaffari S, Kitidis C, Fleming MD, Neubauer H, Pfeffer K, Lodish HF. Erythropoiesis in the absence of Janus‐kinase 2: BCR‐ABL induces red cell formation in JAK2(‐/‐) hematopoietic progenitors. Blood. 2001;98(10):2948‐2957. [DOI] [PubMed] [Google Scholar]
- 10. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23(8):1067‐1077. [DOI] [PubMed] [Google Scholar]
- 11. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397‐409. [DOI] [PubMed] [Google Scholar]
- 12. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234‐243. [DOI] [PubMed] [Google Scholar]
- 13. Kontzias A, Kotlyar A, Laurence A, Changelian P, O'Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12(4):464‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Vollenhoven R, Takeuchi T, Pangan AL, et al. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX‐naïve patients with active rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). https://acrabstracts.org/abstract/a-phase-3-randomized-controlled-trial-comparing-upadacitinib-monotherapy-to-mtx-monotherapy-in-mtx-naive-patients-with-active-rheumatoid-arthritis/. Accessed August 15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smolen JS, Pangan AL, Emery P, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT‐MONOTHERAPY): a randomised, placebo‐controlled, double‐blind phase 3 study. Lancet. 2019;393(10188):2303‐2311. [DOI] [PubMed] [Google Scholar]
- 16. Fleischmann R, Pangan AL, Song IH, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase 3, double‐blind, randomized controlled trial [published online ahead of print 2019]. Arthritis Rheumatol. 10.1002/art.41032. [DOI] [PubMed] [Google Scholar]
- 17. Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10139):2503‐2512. [DOI] [PubMed] [Google Scholar]
- 18. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease‐modifying anti‐rheumatic drugs (SELECT‐BEYOND): a double‐blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513‐2524. [DOI] [PubMed] [Google Scholar]
- 19. Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT‐494). BMC Rheumatol. 2018;2(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55(12):1547‐1558. [DOI] [PubMed] [Google Scholar]
- 21. Klünder B, Mittapalli RK, Mohamed MF, Friedel A, Noertersheuser P, Othman AA. Population pharmacokinetics of upadacitinib using the immediate‐release and extended‐release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I‐III clinical trials. Clin Pharmacokinet. 2019;58(8):1045‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohamed MF, Zeng J, Marroum PJ, Song IH, Othman AA. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin Pharmacol Drug Dev. 2019;8(2):208‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaur K, Kalra S, Kaushal S. Systematic review of tofacitinib: a new drug for the management of rheumatoid arthritis. Clin Ther. 2014;36(7):1074‐1086. [DOI] [PubMed] [Google Scholar]
- 24. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voss J, Graff C, Schwartz A, et al. Pharmacodynamics of a novel Jak1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects. Arthritis Rheum. 2013;65(10):S1015. [Google Scholar]
- 26. Guschin D, Rogers N, Briscoe J, et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin‐6. Embo J. 1995;14(7):1421‐1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohamed MF, Klunder B, Camp HS, Othman AA. Exposure‐response analyses of upadacitinib efficacy in phase 2 trials in rheumatoid arthritis and basis for phase 3 dose selection [published online ahead of print 2019]. Clin Pharmacol Ther. 10.1002/cpt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klunder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin Pharmacokinet. 2018;57(8):977‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.US Food and Drug Administration . Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review(s): tofacitinib. Application number 203214Orig1s000. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203214Orig1s000ClinPharmR.pdf. Accessed August 14, 2019.
- 30. Krishnaswami S, Boy M, Chow V, Chan G. Safety, tolerability, and pharmacokinetics of single oral doses of tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev. 2015;4(2):83‐88. [DOI] [PubMed] [Google Scholar]
- 31. Scott LJ. Tocilizumab: a review in rheumatoid arthritis. Drugs. 2017;77(17):1865‐1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lamb YN, Deeks ED. Sarilumab: a review in moderate to severe rheumatoid arthritis. Drugs. 2018;78(9):929‐940. [DOI] [PubMed] [Google Scholar]
- 33. Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2010;2(5):247‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Genovese MC, Smolen JS, Weinblatt ME, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68(12):2857‐2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sandborn W, Feagan B, Panes J, et al. Safety and efficacy of ABT‐494 (upadacitinib), an oral Jak1 inhibitor, as induction therapy in patients with Crohn's disease: results from Celest. Gastroenterology. 2017;152( 5):S1308‐1309. [Google Scholar]
- 36. Panaccione R, D'Haens G, Sandborn W, et al. Efficacy of upadacitinib as an induction therapy for patients with moderately to severely active ulcerative colitis, with or without previous treatment failure of biologic therapy: data from the dose‐ranging phase 2B study U‐Achieve. Gastroenterology. 2019;156(6):S‐170. [Google Scholar]
- 37. Mohamed MF, Klünder B, Lacerda AP, Othman AA. Exposure‐response relationships for the effect of upadacitinib on clinical and endoscopic efficacy endpoints in subjects with moderately to severely active Crohn's disease—analysis of CELEST study. United Eur Gastroenterol J. 2017;5(5S). [Google Scholar]