

Abstract
Objective
To test the hypothesis that Alzheimer's disease and related neuropathologies contribute to the association between hospitalization and cognitive decline in old age.
Methods
As part of a longitudinal clinical–pathologic cohort study, 526 older persons (mean age at death = 90.9 years, 71% female) without dementia at baseline completed annual cognitive testing and were autopsied at death. Hospitalization information was obtained from linked Medicare claims records. Neuropathologic examination assessed β‐amyloid burden, tau tangle density, neocortical Lewy bodies, hippocampal sclerosis, chronic gross and microscopic cerebral infarcts, and transactive response DNA binding protein 43 kDa.
Results
Over a mean of 5.1 years, a total of 1,383 hospitalizations occurred, and the mean annual rate of hospitalization was 0.5 (standard deviation = 0.6, median = 0.4). Higher rate of hospitalization was not directly related to higher burden for any of the neuropathologic markers. Higher rate of hospitalization was associated with more rapid cognitive decline (estimate = −0.042, standard error [SE] = 0.012, p < 0.001), and after controlling for all 7 neuropathologic markers, the association was essentially the same (estimate = −0.040, SE = 0.013, p = 0.002). In a multivariable model with 3‐way interactions of neuropathologic markers with hospitalization rate and time, the association between hospitalization rate and faster cognitive decline was greater in persons with more tangle pathology (estimate for interaction = −0.007, SE = 0.002, p = 0.002) and in persons with neocortical Lewy bodies (estimate for interaction = −0.117, SE = 0.042, p = 0.005).
Interpretation
Older persons with more hospitalizations experienced faster rates of cognitive decline, and this association was more pronounced in persons with more tau tangle density and with neocortical Lewy body pathologies. ANN NEUROL 2019;86:844–852
Hospitalization of older persons is associated with subsequent increases in rate of cognitive decline1, 2, 3, 4, 5 and risk of dementia.6 The association increases with advancing age,7, 8 but its basis is uncertain. In particular, it is not clear whether neuropathologic processes linked to late life cognitive decline and dementia are somehow contributing to the association between hospitalization and cognitive dysfunction. One possibility is that hospitalization in old age is directly related to level of neuropathology as either a cause or consequence, which could explain why cognitive impairment and decline are associated with higher subsequent rate of hospitalization.9 Alternatively, if hospitalization were not directly related to neuropathology, then level of neuropathologic burden might modify the association of hospitalization with cognitive decline, as hypothesized for delirium.10, 11 Finally, the association of hospitalization with late‐life cognitive dysfunction might be completely independent of common neuropathologies.
In the present study, we test the hypothesis that common neurodegenerative and cerebrovascular lesions contribute to the association of hospitalization with cognitive decline in old age. Participants are from a longitudinal clinical–pathologic cohort study,12 with annual evaluations including detailed cognitive testing linked to data on hospitalization from Medicare records. After death, participants underwent a uniform neuropathologic examination to quantify pathologic markers linked to cognitive impairment and dementia. We tested 3 hypotheses: (1) whether hospitalizations were related to greater burden of dementia‐related neuropathologies, (2) whether hospitalizations were related to greater cognitive decline independent of neuropathological markers, or (3) whether neuropathologic markers modified the relationship of hospitalizations with cognitive decline.
Patients and Methods
Participants
Analyses are based on participants in the Rush memory and aging project (MAP), an ongoing longitudinal clinical–pathologic cohort study that began in 1997.12 Eligibility required agreement to annual clinical evaluations and brain autopsy at death. Individuals were recruited from retirement communities, social service agencies, subsidized housing facilities, and churches. After a presentation about the project, interested persons had detailed discussions with staff who obtained written informed consent. The study was approved by the institutional review board of Rush University Medical Center.
At the time that data were requested from the Centers on Medicare and Medicaid Services (CMS) in 2012, 1,593 participants had enrolled, and Medicare records were obtained for 1,389 (87%) of them. As described previously,5 we excluded persons who did not have valid overlapping Medicare and Memory and Aging Project data, who had dementia at first year of Medicare records, and who did not have at least 2 cognitive assessments, leaving 930 persons available for these analyses. Of these 930 persons, 625 died by the time of these analyses, and 526 (84%) were autopsied and had complete neuropathological data available; pathologic analyses are based on this group. Compared to the 404 participants who were not included in the pathologic analyses, they were more likely to be male (29% vs 21%, χ2 = 7.43, p = 0.006), were older (82.9 vs 78.1 at baseline, t = −11.49, p < 0.001) and had lower Mini‐Mental State Examination (MMSE; 27.9 vs 28.4, t = 3.79, p < 0.001) and global cognition scores (0.00 vs 0.14, t = 4.12, p < 0.001); there was no significant difference by education (t = 0.29, p = 0.77).
Assessment of Cognitive Function and Clinical Diagnosis
A battery of 21 cognitive performance tests was administered in person annually, as the MMSE is used only for descriptive purposes, and scores from the Complex Ideational Material test were highly skewed, the remaining 19 tests were used to create a composite measure of cognition. It included 7 tests of episodic memory, 3 tests of semantic memory, 3 tests of working memory, 4 tests of perceptual speed, and 2 tests of visuospatial ability. A composite measure of global cognition based on all 19 tests was used as the outcome of this analysis. Raw test scores were converted to z scores, using the baseline mean and standard deviation (SD) for the entire cohort. The z scores of individual tests were averaged to yield the composite score as previously described.13 Clinical diagnosis of dementia was conducted at each annual evaluation following the criteria of the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association.14
Assessment of Hospitalization
Data on hospitalizations came from Medicare claims records for the years 1999 to 2010 obtained from CMS through the Research Data Assistance Center research identifiable files request system (https://www.resdac.org/research-identifiable-files-rif-requests). Information on years of eligible Medicare Part A coverage, which covers inpatient care, was included in the Master Beneficiary Summary File, whereas hospitalization data came from the Medicare provider analysis and review (MedPAR) file, which concatenates all claims filed for a hospitalization into a single record. Length of hospital stay in days was available in the MedPAR file. The Charlson Comorbidity Index, a measure of severity of illness during hospitalization based on number and seriousness of diseases,15 was created from International Classification of Diseases, Ninth Revision (ICD‐9) codes in the MedPAR file. Because participants had different lengths of Medicare coverage, burden of hospitalization was characterized as annual rate of hospitalization, calculated as the total number of hospitalization divided by years of follow‐up. Hospitalization rate was truncated at 3 for 4 individuals with extreme values.
Neuropathologic Examination
Neuropathologic markers were derived from postmortem autopsy and characterized in the fashion most strongly related to cognitive decline in this cohort.16, 17, 18 The brain was removed a median of 6.9 hours after death (interquartile range = 5.7–8.9). One cerebral hemisphere, 1 cerebellar hemisphere, and the brainstem were fixed in 4% paraformaldehyde for at least 3 days. The brain was cut coronally into 1cm slabs, and all slabs were examined for gross infarcts. A standard protocol was followed for tissue preservation, tissue sectioning, and quantification of pathologic data by examiners blinded to all clinical data.19 We used hematoxylin and eosin to identify microinfarcts (ie, visible on microscopic but not gross inspection) in at least 9 regions in 1 hemisphere.20 In analyses, chronic gross and microscopic infarcts were each treated as present or absent.
β‐amyloid–immunoreactive plaques were assessed in 8 limbic and neocortical regions (entorhinal cortex, CA1/subiculum, anterior cingulate cortex, dorsolateral prefrontal cortex, superior frontal cortex, inferior temporal cortex, inferior parietal cortex, and primary visual cortex) using a monoclonal antibody (4G8; Covance Labs, Madison, WI; 1:9000 or 6F/3D; Dako North America, Carpinteria, CA; 1:50 or 10D5; Elan Pharmaceuticals, San Francisco, CA; 1:600) with diaminobenzidine as the reporter with 2.5% nickel sulphate to enhance contrast. The percent of each area occupied by β‐amyloid–immunoreactive pixels was estimated with computer‐assisted sampling and image analysis. Regional measures were averaged to form a composite measure of β‐amyloid burden.21
An anti–paired helical filaments–tau antibody clone AT8 (ThermoScientific, Rockford, IL; 1:2,000) and computer‐assisted sampling21 were used to assess density of tau‐immunoreactive neurofibrillary tangles from the same 8 regions assessed for amyloid. Raw scores in each section and region were averaged to yield a composite measure of tangle density/millimeter squared.21
Lewy bodies in the substantia nigra, 2 limbic sites (entorhinal cortex, anterior cingulate cortex), and 3 neocortical sites (midfrontal cortex, superior or middle temporal cortex, inferior parietal cortex) were identified with a monoclonal antibody to α‐synuclein (LB509; Zymed Laboratories, San Francisco, CA; 1:100; or phosphorylated anti–α‐synuclein antibody; Wako Chemicals USA, Richmond, VA; 1:20,000). We classified Lewy body disease as nigral, limbic, or neocortical using a modified version22 of the staging criteria of McKeith et al.23 Neocortical disease required Lewy bodies in frontal, temporal, or parietal cortex and was accompanied by nigral and/or limbic Lewy bodies. A dichotomous variable indicating presence of neocortical Lewy bodies was used in this analysis, as it has been shown to be most predictive of dementia.24
Hippocampal sclerosis was evaluated unilaterally in a coronal section of midhippocampus at the level of the lateral geniculate body. It was graded as present or absent based on severe neuronal loss and gliosis typically in the CA1 sector and/or subiculum with or without other hippocampal subfields affected.25, 26
Transactive response DNA binding protein 43 kDa (TDP‐43) pathology was assessed in 8 brain regions (amygdala, entorhinal cortex, hippocampus CA1 and subiculum, dentate gyrus, anterior temporal pole, interior frontal, middle temporal cortex, and midfrontal cortex), an expansion beyond the 6 described in previous publications25, 27 using monoclonal antibodies (pS409/410;1:100), which stain pathologically phosphorylated TDP‐43 proteins but not normal nuclear TDP‐43.28 A dichotomous variable indicating TDP‐43 positivity beyond the amygdala was used in this analysis.
Statistical Analysis
First, we tested whether neuropathologic markers were directly related to hospitalization rate using linear regression models for continuous neuropathologic markers and logistic regression for binary neuropathologic markers. Separate models were fit with each neuropathological marker as the outcome, adjusted for age at death, sex, and education.
We then analyzed whether change in cognitive function was associated with annual rate of hospitalization using mixed‐effects regression models adjusted for age at baseline, sex, and education. All models included terms for time (since baseline), hospitalization rate, and the interaction of hospitalization rate with time; the interaction represented the association between rate of hospitalization and change in cognitive function. Follow‐up time for this analysis was defined as the minimum interval between (1) first available year of Medicare records or MAP baseline and (2) last available year of Medicare records or most recent MAP assessment.
To this initial model, we added a term for each of the neuropathologic markers to examine if the relationship between hospitalization rate and cognitive change was attenuated by adjusting for neuropathology. Finally, we added a term for the 2‐way interactions of each neuropathologic marker with time, the 2‐way interactions of each neuropathologic marker with hospitalization rate, and the 3‐way interactions of each neuropathologic marker with hospitalization and time. The 3‐way interaction tested whether the relationship of hospitalization rate and cognitive change was modified by presence/level of a particular neuropathologic marker. As a secondary analysis, we repeated this final model after removing any participants who developed dementia at any cognitive assessment before the first hospitalization. We also repeated the analysis using only the rate of hospitalization before dementia in persons who developed dementia. Finally, to assure the results were not driven by hospitalizations for neurovascular events, we repeated the analyses after removing all persons who experienced a hospitalization with primary diagnosis of stroke. For all analyses involving skewed continuous variables, the influence of high leverage values was checked and alternate transformations were tested. All analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC).
Results
During a mean of 5.1 (SD = 2.6) years of observation, a total of 1,383 hospitalizations occurred during the study period for the 526 participants who received postmortem autopsy, ranging from 0 (23.1%) to 17 hospitalizations (median = 2, interquartile range = 1–5). To quantify individual differences in exposure to multiple hospitalizations accounting to differences in length of observation time, we also calculated each person's annual rate of hospitalization. To suppress the influence of potential outliers, the annual rate of hospitalization was truncated at 3 for these analyses (4 participants had rate >3, ranging from 3.7 to 7.7). After truncation, the mean rate was 0.53 hospitalizations per year (SD = 0.59, skewness = 1.8, median = 0.36). Basic descriptive statistics of the cohort and hospitalization characteristics are provided in Table 1.
Table 1.
Description of Cohort and Hospitalization Characteristics (N = 524)
| Characteristic | Value |
|---|---|
| Age at baseline, yr, mean (SD), range | 82.9 (5.7), 65.7 to 100.5 |
| Age at death, yr, mean (SD), range | 90.9 (5.9), 72.7 to 108.3 |
| Women, n (%) | 374 (71.0%) |
| Education, yr, mean (SD), range | 14.6 (2.8), 4 to 23 |
| Baseline MMSE, mean (SD), range | 27.9 (2.1), 18 to 30 |
| Baseline global cognitive function, mean (SD), range | 0.00 (0.5), −1.85 to 1.25 |
| Rate of hospitalization per year, mean (SD), rangea | 0.53 (0.59), 0 to 3 |
| Developed dementia between baseline and death, n (%) | 208 (40%) |
| Hospitalization characteristics | |
| Length of stay, d, mean (SD), range | 3.3 (2.8), 0 to 18 |
| Charlson Comorbidity Index score, mean (SD), range | 0.8 (0.9), 0 to 4 |
| Neuropathologic markersb | |
| Amyloid burden, n, mean (SD), range | 511, 5.0 (4.6), 0 to 22.9 |
| Tau tangle density, n, mean (SD), range | 519, 7.0 (7.7), 0.0 to 43.9 |
| Gross infarcts, presence of, n (%) | 526 (35.0%) |
| Microinfarcts, presence of, n (%) | 526 (30.4%) |
| Neocortical Lewy bodies, presence of, n (%) | 511 (12.5%) |
| Hippocampal sclerosis, presence of, n (%) | 525 (8.4%) |
| TDP‐43, presence of, n (%)c | 521 (33.0%) |
Rate was truncated at 3 to suppress influence of potential outliers (n = 4).
Complete data on all neurological markers were available for 491 participants.
TDP‐43 stage 2 (extension to hippocampus or entorhinal cortex) or stage 3 (extension to the neocortex).
SD = standard deviation.
Hospitalization and Neuropathologic Burden
Distributions of the neuropathological markers are listed in Table 1. We tested whether each neuropathologic marker was related to hospitalization rate in a series of models adjusted for age at death, sex, and education (Table 2). There was no evidence of a relationship between any of the neuropathologic markers and hospitalizations except for an inverse association between tau density and hospitalization rate. Model checking indicated this was driven by high leverage individuals in the highest decile of tangle density with few hospitalizations. When the model was repeated with the highest decile of tangles removed or with decile rank as the dependent variable, there was no significant association with hospitalizations. Hospitalization rate was not related to a higher burden of any neuropathology.
Table 2.
Association of Neuropathologic Markers with Rate of Hospitalization
| Neuropathologic Marker | Estimatea | SE | p |
|---|---|---|---|
| Amyloid burden | 0.21 | 0.35 | 0.54 |
| Tau tangle density | −1.30 | 0.57 | 0.024 |
| OR b | 95% CI | ||
| Gross infarcts | 1.08 | 0.79–1.47) | |
| Microinfarcts | 1.12 | 0.81–1.54) | |
| Neocortical Lewy bodies | 0.86 | 0.52–1.40) | |
| Hippocampal sclerosis | 0.57 | 0.27–1.22) | |
| TDP‐43 | 0.80 | 0.56–1.14) | |
Estimated from separate linear regression models for each neuropathologic marker, adjusted for age at death, sex, and education.
Estimated from separate logistic regression models for each neuropathologic marker, adjusted for age at death, sex, and education.
CI = confidence interval; OR = odds ratio; SE = standard error; TDP‐43 = transactive response DNA binding protein 43 kDa.
Hospitalization, Neuropathologic Burden, and Cognitive Decline
A higher annual rate of hospitalization was associated with more rapid cognitive decline after adjusting for age, sex, education, and the interaction of age at baseline and time since baseline (Table 3, Model A). When we repeated the analysis with terms to control for the neuropathologic markers, the association was essentially the same (see Table 3, Model B).
Table 3.
Association of Annual Hospitalization Rate with Global Cognitive Decline, before and after Adjustment for Neuropathologic Burden
| Model Term | Model A, n = 526a | Model B, n = 491b | ||||
|---|---|---|---|---|---|---|
| Estimate | SE | p | Estimate | SE | p | |
| Timec | −0.088 | 0.009 | <0.001 | −0.088 | 0.009 | <0.001 |
| Hospitalization rate | −0.109 | 0.035 | 0.002 | −0.127 | 0.036 | <0.001 |
| Hospitalization rate × time | −0.042 | 0.012 | <0.001 | −0.040 | 0.013 | 0.002 |
| Aged | −0.028 | 0.004 | <0.001 | −0.027 | 0.004 | <0.001 |
| Sex | −0.140 | 0.045 | 0.002 | −0.161 | 0.047 | <0.001 |
| Education | 0.048 | 0.007 | <0.001 | 0.047 | 0.008 | <0.001 |
| Time × age | −0.003 | 0.001 | 0.007 | −0.003 | 0.001 | 0.040 |
| Amyloid burden | −0.002 | 0.005 | 0.72 | |||
| Tau tangle density | −0.014 | 0.003 | <0.001 | |||
| Gross infarcts | −0.019 | 0.044 | 0.67 | |||
| Microinfarcts | −0.033 | 0.045 | 0.47 | |||
| Neocortical Lewy bodies | −0.024 | 0.062 | 0.70 | |||
| Hippocampal sclerosis | −0.222 | 0.082 | 0.007 | |||
| TDP‐43 | 0.011 | 0.048 | 0.82 | |||
Estimated from mixed‐effects model.
Estimated from mixed‐effects model with nonmissing data for all 7 neuropathological measures.
Time = time since baseline.
Age = age at baseline.
SE = standard error; TDP‐43 = transactive response DNA binding protein 43 kDa.
To determine whether neuropathologic markers modified the association of hospitalization with cognitive decline, we included terms for the 3‐way interactions of hospitalization rate, time since baseline, and each neuropathologic marker. As shown in Table 4, tau tangle density modified the relationship of hospitalization and cognitive decline such that the rate of decline was steeper in persons who had more tangle pathology. This is displayed in Figure 1, in which the rate of decline for persons with the mean rate of 0.5 hospitalizations per year versus persons with no hospitalizations are displayed for the 75th percentile versus 25th percentile of tau tangle density. Persons with 0.5 hospitalizations per year in the 75th percentile of tau tangle density had the steepest rate of cognitive decline. Inferences were similar in sensitivity analyses repeating the models with decile rank of tau tangle density replacing the continuous measure. The presence of neocortical Lewy bodies also modified the relationship of hospitalization and cognitive decline such that the rate of decline was steeper in persons who had neocortical Lewy bodies (Fig 2).
Table 4.
Modification of Association of Hospitalization Rate with Global Cognitive Decline by Neuropathologic Markersa
| Interaction Term | Estimate | SE | p |
|---|---|---|---|
| Hospitalization × amyloid burden × time | 0.004 | 0.003 | 0.12 |
| Hospitalization × tau tangle density × time | −0.007 | 0.002 | 0.002 |
| Hospitalization × gross infarcts × time | −0.038 | 0.028 | 0.17 |
| Hospitalization × microinfarcts × time | 0.024 | 0.028 | 0.39 |
| Hospitalization × neocortical Lewy bodies × time | −0.117 | 0.042 | 0.005 |
| Hospitalization × hippocampal sclerosis × time | 0.059 | 0.069 | 0.39 |
| Hospitalization × TDP‐43 × time | −0.016 | 0.030 | 0.60 |
Estimated from a mixed‐effects model including terms for age at baseline, sex, education, time (since baseline), the interaction of age with time, hospitalization rate, the interaction of hospitalization rate with time, each neuropathology, the interactions of each neuropathological marker with time, the interactions of each neuropathological marker with hospitalization rate, and the 3‐way interactions of each neuropathological marker with hospitalization rate and time in all participants with nonmissing data for all 7 neuropathological measures (n = 491).
SE = standard error; TDP‐43 = transactive response DNA binding protein 43 kDa.
Figure 1.
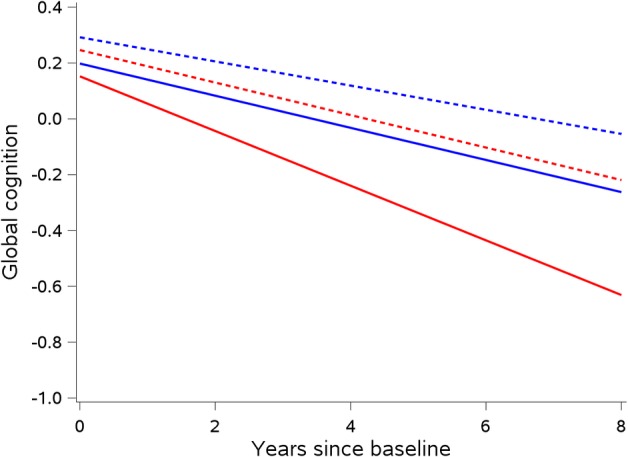
Interaction of hospitalization and tau tangle pathology on rate of cognitive decline. Predicted change in a global cognitive function comparing persons who were not hospitalized (blue) versus mean hospitalization rate of 0.5 per year (red), and persons with 25th percentile tau tangle density (dotted) versus 75th tangle density (solid), for a female of average age and education, with no amyloid burden, no gross or microinfarcts, no Lewy bodies, no hippocampal sclerosis, and no transactive response DNA binding protein 43 kDa pathology (n = 491).
Figure 2.
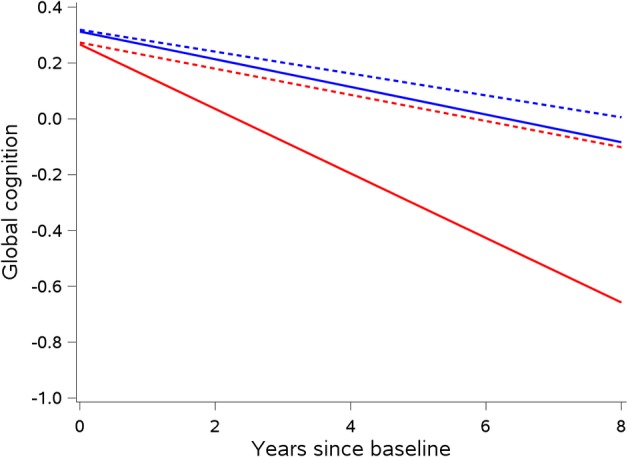
Interaction of hospitalization and Lewy body pathology on rate of cognitive decline. Predicted change in a global cognitive function comparing persons who were not hospitalized (blue) versus mean hospitalization rate of 0.5 per year (red), and persons without neocortical Lewy bodies (dotted) versus with neocortical Lewy bodies (solid), for a female of average age and education, with no amyloid or tangle burden, no gross or microinfarcts, no hippocampal sclerosis, and no transactive response DNA binding protein 43 kDa pathology (n = 491).
We conducted secondary analyses to determine if persons who developed dementia were driving these findings. First, we repeated the final interaction model after removing any participants who developed dementia prior to any hospitalization (n = 24), and the findings were similar. In the 208 persons who developed dementia at any point over follow‐up, the rate of hospitalization was higher before incident dementia (mean = 0.57, SD = 0.74) than after (mean = 0.24, SD = 0.50; t = 5.83, p < 0.001), and slightly higher than the rate in those who never developed dementia (n = 318; mean = 0.54, SD = 0.75), though this difference was not significant (t = −0.52, p = 0.60). When we repeated the analyses using only hospitalization rate before the development of dementia in the persons who developed dementia, the inferences were the same for the 3‐way interaction with tangles (estimate = −0.004, standard error [SE] = 0.002, p = 0.041) and Lewy bodies (estimate = −0.102, SE = 0.034, p = 0.002). Finally, when we repeated the analyses after removing all persons who were hospitalized for stroke (n = 71), the inferences were the same for the 3‐way interaction with tangles (estimate = −0.010, SE = 0.003, p < 0.001) and Lewy bodies (estimate = −0.083, SE = 0.038, p = 0.030).
Discussion
In a cohort of >500 older persons who had come to autopsy with Medicare records linked to annual cognitive assessment over a mean of 5 years, a higher rate of hospitalization was associated with more rapid cognitive decline. Although greater burden of neuropathology was not related to more hospitalizations, certain neuropathologies appeared to modify the relationship, such that persons with more tau tangle pathology and persons with neocortical Lewy bodies had steeper rates of decline as hospitalization rates increased. This is the first study that we are aware of to provide evidence compatible with the claim that older adults with greater burden of Alzheimer's disease and other dementia‐related pathologies may experience worse cognitive outcomes after hospitalization.
The association of hospitalization with faster cognitive decline is consistent with previous research,1–4,6 although 1 study indicated that most decline occurs before hospitalization.29 The basis of the association is uncertain. A novel feature of the present study is that these participants died and had a uniform neuropathologic examination, allowing us to test the hypothesis that common neurodegenerative and cerebrovascular lesions play a role in hospitalization‐related cognitive decline. We found no evidence that higher rate of hospitalization was directly related to greater neuropathologic burden. We also found no evidence that degree of neuropathology accounted for the relationship of hospitalization rate and cognitive decline, as the association did not change after adjustment for neuropathology. We did, however, find evidence that certain pathologies modify the relationship of hospitalization rate and cognitive decline. One way to interpret this interaction is that persons with higher tau tangle density or Lewy bodies experience faster rates of cognitive decline compared to persons without these pathologies given the same rate of hospitalization. Another interpretation is that given the same level of tau or Lewy body pathology, persons who are hospitalized more will experience faster cognitive decline. Therefore, hospitalizations may lower an older person's resilience to negative cognitive consequences for a given level of underlying pathology.30 It has been postulated that the events surrounding hospitalization may “unmask” preclinical Alzheimer's disease,31, 32 and these findings provide support to that notion, though more research is needed, especially with incident dementia outcomes.
The specific mechanisms that underlie this interaction are unknown at this time. From these data, it seems most likely that hospitalization is making existing neuropathologies have a more pronounced effect on the brain rather than having a direct relationship with neuropathology. However, hospitalization did not interact with all neuropathologies; therefore, it does not appear that hospitalization simply acts as another “hit” to the brain, but may work to potentiate the effects of Alzheimer's disease, and perhaps Lewy bodies. Exploring this interaction could provide insights into the mechanisms of Alzheimer's disease, as well as shed light on how hospitalization leads to negative cognitive outcomes in older adults. Specific mechanisms for the association between hospitalization and cognitive impairment in old age have been posited, including the use of surgery and anesthesia, stressors of the hospital experience, delirium, exposure to sedatives and mechanical ventilation, polypharmacy, and sleep fragmentation. Examining these potential mechanisms in the context of underlying neuropathology in patients through the use of neuroimaging and other burgeoning biomarkers33 could help to reveal specific etiologies, and eventually lead to opportunities for personalized medicine by identifying persons at highest risk for hospital‐related cognitive problems due to their biomarker profile.
We are not aware of any published data that directly address the intersecting roles of hospitalization, neuropathologies, and cognition in late‐life cognitive decline. One study indirectly examined this association using delirium, a common acute consequence of hospitalization in older patients, with cognitive decline.11 Similar to our findings, the study reported an interaction such that persons with a combination of delirium and higher neuropathological burden displayed the greatest rate of cognitive decline. This raises the possibility that the relationship between neuropathology and dementia is modified by delirium.10 We were not able to explore this mechanism, because only 5% of the cohort had ICD‐9 codes indicating delirium, reflecting underreporting of delirium in claims records.34 Another study examined the association of hospitalization with long‐term cognitive decline and brain magnetic resonance imaging (MRI) changes; they found hospitalization was associated with higher odds of increasing ventricular size, and there was modest evidence of mediation of the association of hospitalization and cognitive decline when ventricular change was added to the model (there was little evidence of mediation by white matter hyperintensities).2 Finally, a recent MRI study demonstrated a relationship between more frequent hospitalizations and smaller brain volumes and lower white matter integrity.33 Taken together, these studies provide compelling evidence that hospital‐related events intersect with brain pathology to promote cognitive decline, providing a potential avenue for dementia prevention in the absence of interventions to directly target neuropathology.30, 35 This could potentially fit more broadly into the cognitive reserve theory, with hospitalization acting to lower reserve capacity to deal with existing pathology, though other mechanisms could be at play, and more research is needed to explore this claim.
This study has important strengths and limitations. Rates of participation in clinical follow‐up and brain autopsy were high, reducing likelihood of bias due to selective attrition. Hospital admission was assessed using Medicare claims rather than participant reports, leading to more accurate assessment of hospitalization burden. Cognitive function was assessed with a previously established psychometrically sound index, minimizing floor and ceiling artifacts and other sources of measurement error. Cognitive testing took place at annual intervals for a mean of about 5 years, enhancing ability to model trajectories of change. A series of secondary analyses indicated that the main association of interest was not due to reverse causation (ie, the development of dementia leading to more hospitalizations). The main limitation is that participants were selected, and so results may not generalize to other groups, though it should be noted that findings are consistent with previous population‐based studies.1, 6 Additionally, this analysis is predicated on the assumption that postmortem neuropathologic burden reflects the level of burden in years prior to death. This assumption is supported by evidence that Alzheimer's disease neuropathologic burden progresses slowly over decades,36 and that in vivo measures are highly correlated with postmortem Alzheimer neuropathology irrespective of time to death,37 but these findings must be confirmed in future work when in vivo biomarkers for all neuropathologies are available. Delirium was not able to be evaluated, and no measures of direct insults to the brain at the time of hospitalization, such as inflammation, stress response, or hypoxia, were available. Annual cognitive testing did not allow for assessment of acute onset of impairment immediately after hospitalization. Though related to change in cognitive function, hospitalization rate was not significantly higher in persons who developed clinical dementia; the relationship of hospitalization with incident dementia needs to be tested formally in future analyses with more data, taking into account competing risk of mortality for rapid decliners. Medicare claims data cannot be used to observe hospitalizations at Veterans Affairs (VA) facilities; however, only 2 participants in our cohort indicated that they utilize VA hospitals (inferences were unchanged when they were removed from analyses). Finally, although our laboratory has standard protocols to establish reliability across changes in methods and raters, some variation in pathological assessment methods over time may be unavoidable. Certain pathologies, such as amyloid and τ, may be measured with more precision than others in this analysis. More research is needed to understand the intersecting roles of hospitalization and Alzheimer's disease and related pathologies in the development of cognitive impairment. Future work will also include incident dementia outcomes and the incorporation of biomarkers.
Author Contributions
All authors contributed to the conception and design of the study. B.D.J., A.W.C., P.A.B., R.C.S., D.A.B., and J.A.S. contributed to the acquisition and analysis of data. B.D.J., R.S.W., and J.A.S. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This work was funded by the National Institute on Aging grants K01AG050823 (B.D.J.), R01AG17917 (D.A.B., B.D.J.), R01AG033678 (P.A.B., B.D.J.), R01AG034374 (P.A.B.), R01AG035117 (E.W.E.), R01AG027472 (E.W.E.), P30AG010161 (J.A.S.), and the Illinois Department of Public Health.
We thank the Illinois residents who participated in the Rush Memory and Aging Project; T. Colvin and K. Skish for study coordination; J. Gibbons and G. Klein for data management; and W. Bang for statistical programming.
Data for participants of the Rush Memory and Aging Project are available upon request through the Rush Alzheimer's Disease Center Research Resource Sharing Hub: https://www.radc.rush.edu.
References
- 1. Wilson RS, Hebert LE, Scherr PA, et al. Cognitive decline after hospitalization in a community population of older persons. Neurology 2012;78:950–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown CH, Sharrett AR, Coresh J, et al. Association of hospitalization with long‐term cognitive and brain MRI changes in the ARIC cohort. Neurology 2015;84:1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pandharipande PP, Girard TD, Jackson JC, et al. Long‐term cognitive impairment after critical illness. N Engl J Med 2013;369:1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mathews SB, Arnold SE, Epperson CN. Hospitalization and cognitive decline: can the nature of the relationship be deciphered? Am J Geriatr Psychiatry 2014;22:465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. James BD, Wilson RS, Capuano AW, et al. Cognitive decline after elective and nonelective hospitalizations in older adults. Neurology 2019;92:e690–e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA 2010;303:763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen CC, Chiu MJ, Chen SP, et al. Patterns of cognitive change in elderly patients during and 6 months after hospitalisation: a prospective cohort study. Int J Nurs Stud 2011;48:338–346. [DOI] [PubMed] [Google Scholar]
- 8. Moller JT, Cluitmans P, Rasmussen LS, et al. Long‐term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post‐Operative Cognitive Dysfunction. Lancet 1998;351:857–861. [DOI] [PubMed] [Google Scholar]
- 9. Wilson RS, Rajan KB, Barnes LL, et al. Cognitive aging and rate of hospitalization in an urban population of older people. J Gerontol A Biol Sci Med Sci 2014;69:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis DH, Muniz Terrera G, Keage H, et al. Delirium is a strong risk factor for dementia in the oldest‐old: a population‐based cohort study. Brain 2012;135(pt 9):2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis DH, Muniz‐Terrera G, Keage HA, et al. Association of delirium with cognitive decline in late life: a neuropathologic study of 3 population‐based cohort studies. JAMA Psychiatry 2017;74:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bennett DA, Buchman AS, Boyle PA, et al. Religious orders study and rush memory and aging project. J Alzheimers Dis 2018;64(suppl 1):S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilson RS, Barnes LL, Krueger KR, et al. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc 2005;11:400–407. [PubMed] [Google Scholar]
- 14. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis 1987;40:373–383. [DOI] [PubMed] [Google Scholar]
- 16. Jansen WJ, Wilson RS, Visser PJ, et al. Age and the association of dementia‐related pathology with trajectories of cognitive decline. Neurobiol Aging 2018;61:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boyle PA, Yu L, Wilson RS, et al. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018;83:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boyle PA, Wilson RS, Yu L, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 2013;74:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology 2007;69:2197–2204. [DOI] [PubMed] [Google Scholar]
- 20. Arvanitakis Z, Leurgans SE, Barnes LL, et al. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011;42:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bennett DA, Schneider JA, Wilson RS, et al. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 2004;61:378–384. [DOI] [PubMed] [Google Scholar]
- 22. Wilson RS, Yu L, Schneider JA, et al. Lewy bodies and olfactory dysfunction in old age. Chem Senses 2011;36:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology 1996;47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 24. Schneider JA, Arvanitakis Z, Yu L, et al. Cognitive impairment, decline and fluctuations in older community‐dwelling subjects with Lewy bodies. Brain 2012;135(pt 10):3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson RS, Yu L, Trojanowski JQ, et al. TDP‐43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013;70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP‐43 pathology in aging and Alzheimer disease. Ann Neurol 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. James BD, Wilson RS, Boyle PA, et al. TDP‐43 stage, mixed pathologies, and clinical Alzheimer's‐type dementia. Brain 2016;139:2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP‐43 is a consistent feature in all sporadic and familial forms of TDP‐43 proteinopathies. Acta Neuropathol 2009;117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hallgren J, Fransson EI, Reynolds CA, et al. Cognitive trajectories in relation to hospitalization among older Swedish adults. Arch Gerontol Geriatr 2018;74:9–14. [DOI] [PubMed] [Google Scholar]
- 30. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: clarifying terminology for preclinical studies. Neurology 2018;90:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fong TG, Davis D, Growdon ME, et al. The interface between delirium and dementia in elderly adults. Lancet Neurol 2015;14:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guerra C, Linde‐Zwirble WT, Wunsch H. Risk factors for dementia after critical illness in elderly Medicare beneficiaries. Crit Care 2012;16:R233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walker KA, Gottesman RF, Wu A, et al. Association of hospitalization, critical illness, and infection with brain structure in older adults. J Am Geriatr Soc 2018;66:1919–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCoy TH, Snapper L, Stern TA, Perlis RH. Underreporting of delirium in statewide claims data: implications for clinical care and predictive modeling. Psychosomatics 2016;57:480–488. [DOI] [PubMed] [Google Scholar]
- 35. Bennett DA. Mixed pathologies and neural reserve: Implications of complexity for Alzheimer disease drug discovery. PLoS Med 2017;14:e1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367. [DOI] [PubMed] [Google Scholar]
- 37. La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB‐PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement 2019;15:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]