

Abstract
Radiotherapy is one of the most used treatment approaches for head and neck squamous cell carcinoma (HNSCC). Targeted inhibition of DNA repair machinery has the potential to improve treatment response by tailoring treatment to cancer cells lacking specific DNA repair pathways. Human papillomavirus (HPV)‐negative and HPV‐positive HNSCCs respond differently to radiotherapy treatment, suggesting that different approaches of DNA repair inhibition should be employed for these HNSCC groups. Here, we searched for optimal radiosensitization approaches for HPV‐positive and HPV‐negative HNSCCs by performing a targeted CRISPR‐Cas9 screen. We found that inhibition of base excision repair resulted in a better radiotherapy response in HPV‐positive HNSCC, which is correlated with upregulation of genes involved in base excision repair. In contrast, inhibition of nonhomologous end‐joining and mismatch repair showed strong effects in both HNSCC groups. We validated the screen results by combining radiotherapy with targeted inhibition of DNA repair in several preclinical models including primary and recurrent patient‐derived HNSCC xenografts. These findings underline the importance of stratifying HNSCC patients for combination treatments.
Keywords: CRISPR‐cas9, DDR, HNSCC, HPV, radiotherapy
Short abstract
What's new?
The combination of radiotherapy and targeted inhibition of DNA repair pathways can potentially improve therapeutic response in patients with head and neck squamous cell carcinoma (HNSCC). Here, a targeted CRISPR‐Cas9 screen was used to identify optimal radiosensitization approaches for human papillomavirus (HPV)‐positive and HPV‐negative HNSCC. Inhibition of base excision repair was associated with improved radiotherapy response in HPV‐positive HNSCC cells. By comparison, inhibition of non‐homologous end‐joining and mismatch repair was effective in both HPV‐positive and HPV‐negative cells. The screen results were validated in patient‐derived xenograft models, suggesting that stratification of HNSCC patients by HPV status may benefit therapeutic outcome.
Abbreviations
- BER
Base excision repair
- CNV
copy number variations
- DDR
DNA damage response
- DSB
double‐strand break
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papillomavirus
- HRR
homologous recombination repair
- MEM
minimum essential medium
- MMR
mismatch repair
- NER
nucleotide excision repair
- NHEJ
nonhomologous end‐joining
- PDX
patient‐derived xenografts
- RT
radiotherapy
- SRB
sulforhodamine B
- SSA
single‐strand annealing
- SSB
single‐strand break
- TCGA
The Cancer Genome Atlas
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a heterogeneous group of cancers divided into two major groups according to the presence or absence of high‐risk human papillomavirus (HPV). The majority of locally advanced HNSCC are treated with radiotherapy (RT) alone or in combination with chemotherapy independently of the HPV status.1 The overall survival rate of HNSCC patients has only improved marginally over the last 30 years with many patients facing local tumor recurrences, especially in the HPV‐negative group. The variation in RT sensitivity has a major clinical impact as RT resistant tumors show higher probability of local recurrences,2, 3 highlighting the need for novel radiosensitization approaches.
Emerging evidence indicates that targeted inhibition of DNA damage response (DDR) could modulate RT response and has the potential to increase the therapeutic window by increasing radiosensitization of tumors to a greater extent than normal tissues. This could be achieved by tailoring treatment to cancers lacking specific DDR pathways. However, the knowledge how to use DNA repair targeted agents in combination with RT is lagging behind the understanding how to use them as monotherapy.4, 5 Hence, a better understanding of the biology of the tumors and the mechanisms of RT response is needed.
Multiple studies demonstrated better local response rates in HPV‐positive HNSCC patients compared to HPV‐negative HNSCC patients, indicating that these HNSCC groups respond differently to RT.6 The role of HPV in dysregulation of DNA repair machinery is well established in context of its life cycle.7 However, less is known about the influence of HPV on DDR in cancer. Although no profound investigations on differences in DNA repair pathways between both groups of HNSCC was performed, several preclinical studies—ours included—ascribed increased RT response in HPV‐positive cancers due to defects in DDR pathways.8 Several studies showed that HPV‐positive HNSCC are characterized by decreased double‐strand break (DSB) repair capacity9, 10, 11, 12 probably because of malfunctions in nonhomologous end‐joining (NHEJ) and homologous recombination repair (HRR) pathways.8, 13, 14, 15 These observations strongly suggest that different approaches should be employed for radiosensitization of different groups of HNSCC patients.
Materials and Methods
Cell lines and reagents
UPCI‐SCC‐154 (RRID:CVCL_2230), UM‐SCC‐104 (RRID:CVCL_7712) and UM‐SCC‐47 (RRID: CVCL_7759) cell lines were cultured in Minimum Essential Medium (MEM) supplemented with 10% FBS, 1% l‐glutamine and 1% nonessential amino acids. UPCI‐SCC‐154 cell line was purchased from DSMZ. UM‐SCC‐47 and UM‐SCC‐104 were gifted by Dr T. Carey, University of Michigan. SQD9 (CVCL_D774), CAL‐27 (CVCL_1107) and NKI‐SC263 (CVCL_LI51) cell lines were gifted by Dr A. Begg, the Netherlands Cancer Institute and were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% FBS and 1% sodium pyruvate. All cell lines were authenticated using STR profiling by ATCC within the last 3 years. All cell culture media and supplements were purchased from Life Technologies (ThermoFisher Scientific, Waltham, MA). The experiments were performed with mycoplasma‐free cells.
Lentiviral shRNAs against luciferase or p16 (TRCN0000265833) were purchased from Sigma‐Aldrich (St. Louis, MO). Infected cells were selected by treatment with 4 μg puromycin (Sigma‐Aldrich) for 4 days. Scrambled siRNA (SIC001) and siRNA against LIG1 (siLIG1_2: 5′‐CAACUAUCAUCCCGUGGAA‐3′; siLIG1_3 5′‐GUUACAAUCCUGCCAAGAA‐3′) were purchased from Sigma‐Aldrich. siRNA experiments were performed as previously described.16
The inhibitors ABT‐888 (Abbott, Chicago, IL), NU7441 (KuDOS Pharmaceuticals, Cambridge, UK) were obtained from SelleckChem (Houston, TX). For in vitro experiments the inhibitors were dissolved in DMSO. For in vivo experiments the inhibitors were dissolved in 0.9% NaCl pH 4.0 for ABT‐888 or 40% PEG400 in 0.9% NaCl for NU7441. RT was delivered by linear accelerator (for in vitro: X‐rays, 6 MV photons; for in vivo: X‐rays, 16 MeV electrons, Varian Medical Systems, dose rate: 2.4 Gy/min) or Baltograph (for in vitro: X‐rays 199 kV photons, 15 mA, Balteau, dose rate: 3.7826 Gy/min).
CRISPR‐Cas9 screen
cRNAs were designed with CRISPR tool from Zhang lab (MIT, Cambridge, MA). To exclude possible off‐target effects, we used two cRNAs for each gene. Scrambled cRNA from IDT (IDTDNA, Coralville, IA) was used as a negative control. Transient transfections were performed with Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) according to the IDT Alt‐RT CRISPR‐Cas9 protocol. The optimized transfection efficiency assessed by BLOCK‐iT™ Fluorescent Oligo (Thermo Fisher Scientific) was about 80% for both cell lines. The indel efficiency was assessed by analyzing protein expression of DNA‐PK and PARP1. We found that cRNA‐Cas9 overexpression led to at least 40% decrease in expression of both proteins in either SCC154 or SQD9 cells (Supporting Information Fig. S1A).
Forty‐eight hours after transfection cells were treated either with 0 Gy (non‐irradiated controls) or with RT doses of 3 or 6 Gy. The survival rate was assessed 1 week after the treatment by a short‐term Sulforhodamine B (SRB) assay as previously described.16 The survival rate of the irradiated (3 or 6 Gy) cells expressing specific cRNA‐Cas9 was normalized to the non‐irradiated control. The survival ratio between cells treated with specific cRNAs and scrambled cRNA ratio was used to determine radiosensitization effect. Only genes for which both specific cRNAs led to decreased survival compared to control cRNA were considered as screen hits.
Colony formation assay
The cells were treated with drug (ABT‐888 and NU7441) 1–2 hr before exposure to increasing dose of RT (2–6 Gy). Then, 22–23 hr after RT, the cells were plated into 10 cm dishes with drug‐free medium. siRNA and cRNA‐Cas9 experiments were performed on 6 cm dishes. After 2–3 weeks cells were fixed with 2.5% glutaraldehyde in PBS and stained with 0.4% crystal violet. The colonies containing 50 cells or more were counted with ColCount colony counter (Oxford Optronix, Abington, UK).
Immunoblotting
Protein extracts were prepared in RIPA buffer containing protease and phosphatase inhibitors (Roche, Basel, Switzerland). Protein concentrations were determined by Bradford assay (Bio‐Rad, Hercules, CA) and 5–50 μg of proteins were subjected to SDS‐PAGE. Following antibodies were used: mouse monoclonal p16 (clone G175‐405, BD Pharmingen, San Diego, CA), mouse monoclonal vinculin (clone hVIN‐1, Sigma‐Aldrich), mouse monoclonal DNA‐PK (3H6, CST, Danvers, MA), rabbit polyclonal LIG1 (AV54307, Sigma‐Aldrich), rabbit polyclonal PARP1 (ab227244, Abcam, Cambridge, MA), rabbit polyclonal PARP2 (NB100‐185, Novusbio, Centennial, CO), rabbit polyclonal APE (#4128, CST), rabbit polyclonal FEN1 (# 2746, CST), antimouse IgG (#7076, CST) and antirabbit IgG (#7074, CST). Densitometry values were determined by correcting for corresponding loading controls from each loaded gel (ImageJ).
Immunofluorescence
Cells were plated on μClear 96‐well plates (Greiner Bio‐one) and fixed with 4% paraformaldehyde. After permeabilization with methanol, the cells were stained with primary antibodies against H2AX Ser139 (JBW301, Millipore, Burlington, MA) followed by staining with 488 Alexa Fluor secondary antibody (#4408, CST). The DNA was counterstained with DAPI (Sigma‐Aldrich). The immunofluorescence images were acquired using In Cell Analyzer 2000 (GE Healthcare, Chicago, IL).
Cell cycle analysis
Cells were fixed with 70% ethanol and stained with 10 μg/ml propidium iodide containing 100 μg/ml RNase A (Invitrogen). Cell cycle distribution was assessed by FACS (BD Facsverse, Piscataway, NJ).
Quantitative real‐time PCR
RNA extraction was performed using RNeasy MINI kit (Qiagen, Germantown, MD). cDNA was synthesized with SuperScript VILO cDNA synthesis kit (Life Technologies, Carlsbad, CA). The quantification of mRNA was performed on Light Cycler 480 (Roche) system using SYBR Green qPCR Mastermix (Roche). Gene expressions were calculated relative to the GEOMEAN of HMBS and TBP1. Following primers sequences were used:
XRCC1 (F:TTGGAGAAGGAGGAGCAGATA; R:GCTGAACTGCCCACCAG),
UNG (F:ACCGGATCCAGAGGAACA; R:CTTCTTCCAGCTCTCTCCAAAG),
PARP1 (F:GTACCACTTCTCCTGCTTCTG; R:CCGCTGTCTTCTTGACTTTCT),
PARP2 (F:AAACTCGTAGATGCCAGAGAC; R:CCTTCCTGGCATACCATCTT),
MBD4 (F:TGACCTCCGCAAAGAAGATG; R:GGGTTCTTGTAGCAAGGGATTA),
LIG1 (F:CCTAAAGACCTCCAAAGCAGAG; R:TTGGTCTGCTCTTCCTCCT),
HMGB2 (F:ATGTCCTCGTACGCCTTCT; R:CCATCTCTCCGAACACTTCTTG),
FEN1 (F:GGCTGGTGAAGGTCACTAAG; R:ACTGGGTGCATCAAGATAAGG);
APEX1 (F:CAGAGGCCAAGAAGAGTAAGAC; R:GATCTTGAGTGTGGCAGGTT);
HMBS (F:GGCAATGCGGCTGCAA; R:GGGTACCCACGCGAATCAC);
TBP (F:CGGCTGTTTAACTTCGCTTC; R:CACACGCCAAGAAACAGTGA).
Xenograft models
SCC154 or SQD9 cells were injected in each flank of female nu/nu NMRI mice and were performed according to the Ethical committee of KU Leuven (P141/2013 and P163/2017). Patient‐derived xenografts (PDXs) were generated by the Trace PDX platform (Leuven) with consent of Ethical committee of KU Leuven (P038/2015) and the medical ethics committee from University Hospitals Leuven (S54185).
The HNC019 (PDX019) model was generated from pretreatment tumor tissue from a 49‐year‐old male patient diagnosed with a moderately differentiated SCC located at the tongue. The HNC021 (PDX021) model was generated from a recurrent tumor from a 60‐year‐old male patient with a necrotic SCC located at the oropharynx. The patient had a recurrence after chemoRT treatment. HPV‐status was determined by HPV‐DNA using GP5+/GP6+ primers, DNA of SCC154 cells and cervix were used as positive control as described earlier.16 The mice were either treated with vehicle, DNA‐PK inhibitor NU7441 (10 mg/kg body weight) or PARP inhibitor ABT‐888 (25 mg/kg body weight), 2 hr before fractionated RT (2 Gy/fraction). All drug treatments were performed via i.p injections. SQD9 and PDX019 models were treated with a total dose of 10 Gy. SCC154 and PDX021 models were treated with total dose of 6 Gy. Tumor volumes were determined with caliper measurements, body weight and health of the mice were monitored daily during treatment and three times per week during follow‐up. MRI scans were taken before and after treatment with TurboRare T2 scan protocol. Images were analyzed and volumes were calculated by ITK snap software.
Immunohistochemistry
About 4 μm thick paraffin sections from formalin‐fixed paraffin‐embedded (FFPE) tumor tissues were stained for Ki67 (RM‐9106‐R7, ThermoScientific) and p16 (1/150, BD 51‐1325GR, BD Pharmingen™). EnVisionTM HRP antimouse or antirabbit (Dako, Carpinteria, CA) was used as secondary antibody. Counterstaining was performed with hematoxylin. Scoring of Ki67 was performed in 10 fields with a magnification of 10×.
Bioinformatics analysis
The cancer genome atlas (TCGA) HNSCC cancer samples were used for bioinformatic analysis. Alterations were determined by percentage of cancer harboring certain alterations in the DDR genes as was described in.17 Differences between HPV‐positive and HPV‐negative HNSCC were assessed by Fisher's exact test.
Statistics
For in vitro and in vivo experiments two‐tailed t‐test, two‐way ANOVA (normal distribution) and Kruskal–Wallis (nonparametric ANOVA) were used.
Results
Genomic and functional differences in DDR between HPV‐positive and HPV‐negative HNSCCs
To characterize DDR alterations in HPV‐positive and HPV‐negative HNSCCs, we performed pathway analysis on core DDR genes17 in the TCGA HNSCC samples. Although several DDR pathways were altered due to copy number variations (CNV) in both HNSCC groups, HPV‐positive HNSCCs showed higher number of alterations compared to HPV‐negative HNSCCs. The most striking difference between HPV‐positive and HPV‐negative HNSCCs was observed in mutation and methylation statuses of checkpoint related genes (Fig. 1 a). These results highlight the difference in DDR between HPV‐positive and HPV‐negative HSNCCs.
Figure 1.
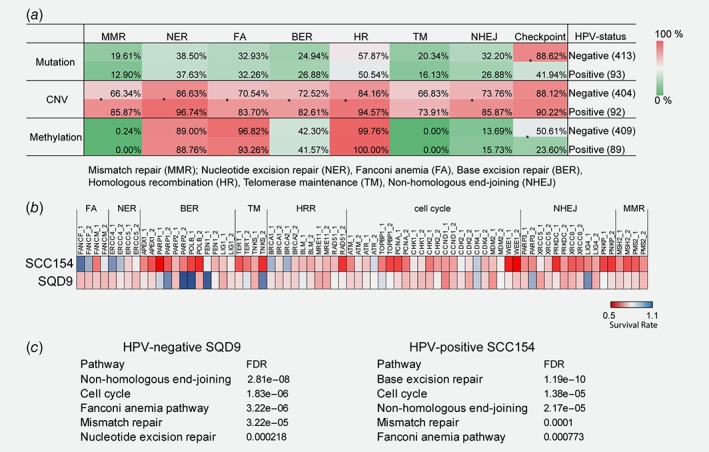
Genomic and functional differences in DDR of HPV‐positive and HPV‐negative HNSCCs. (a) Pathway enrichment analysis of the core DDR genes in the TCGA HNSCC samples. *p < 0.05 determined by Fisher's exact test. (b) CRISPR‐Cas9 screen using a library of 36 genes involved in DNA repair. A heatmap shows radiosensitization mediated by specific CRISPR‐Cas9 in SQD9 and SCC154 cells treated with 6 and 3 Gy, respectively. (c) KEGG pathway mapping of the screen hits analyzed by STRING. The order of enrichment is based on the false discovery rates (FDR).
To examine the radiosensitizing potential of the DDR gene depletion in HPV‐positive and HPV‐negative HSNCCs, we performed a CRISPR‐Cas9 screen targeting DDR genes (Supporting information Table S1A). We selected genes based on their drugability18 and their potential importance in RT response.19 To exclude possible off‐target effects, we used two cRNAs for each gene.
We first assessed whether DDR genes were essential for the survival of HNSCC cells by a SRB assay. Short‐term survival analysis of HPV‐negative SQD9 and HPV‐positive SCC154 cells demonstrated that CRISPR‐mediated knockdown of the majority of individual DDR genes did not alter cell survival and even showed slightly higher survival rate compared to the scrambled control cells. Only, indel of LIG4, a central component of nonhomologous end‐joining (NHEJ) repair, decreased the survival of HPV‐positive SCC154 cells (Supporting Information Fig. S1B). However, CRISPR‐mediated indel of the base excision repair (BER) and the NHEJ genes did not affect clonogenic growth of either HPV‐positive or HPV‐negative HNSCC cells (Supporting Information Fig. S1C). This is line with recent published functional genomic data from genome‐scale CRISPR/Cas9‐based loss‐of‐function screen, in which none of the selected BER and NHEJ related genes were identified as genetic dependencies.20
For the radiosensitization screen, HPV‐negative SQD9 and HPV‐positive SCC154 cells were treated with 6 or 3 Gy, respectively. The RT dose was chosen to have similar survival rates for HPV‐positive and HPV‐negative HNSCC cells (Supporting Information Fig. S1D). To assess the radiosensitizing effect of specific crRNAs, we performed a short‐term survival SRB assay. Only genes for which both cRNAs led to decreased survival compared to scrambled control cRNA were considered as screen hits. These hits were then analyzed by KEGG pathway mapping (Figs. 1 b and 1c).
In line with our hypothesis, we found that suppression of genes related to different DDR pathways showed differential RT responses in HPV‐negative and ‐positive HNSCC cells (Figs. 1 b and 1c). We found that inhibition of BER genes increased radiosensitization of HPV‐positive SCC154 cells, whereas inhibition of nucleotide excision repair (NER) genes resulted in more profound radiosensitization of HPV‐negative SQD9 cells. Inhibition of either mismatch repair (MMR) or NHEJ pathway related genes showed a strong radiosensitization potential in both groups. Knockout of cell cycle‐related genes also showed increased radiosensitization in both SQD9 and SCC154 cells, nonetheless there was only a slight overlap between the genes in both groups (Figs. 1 b and 1c). Altogether, these data confirm the genetic and functional differences in DDR between HPV‐positive and HPV‐negative HNSCCs.
The BER pathway inhibition radiosensitizes HPV‐positive HNSCCs
Next, we validated top DDR pathways identified in our screen (Fig. 1 c). The CRISPR screen suggested that inhibition of BER related genes radiosensitizes specifically HPV‐positive cells. Indeed, clonogenic assays demonstrated that indels of the BER‐related genes resulted in significant radiosensitization of SCC154, but not SQD9, cells (Supporting Information Fig. S1E).
To assess the role of the BER pathway in the RT response, we investigated the RT response in HPV‐positive and HPV‐negative cells after inhibition of PARP, an important component of the BER pathway. The PARP inhibitor ABT‐888 decreased colony formation of irradiated HPV‐positive cells, whereas showed only a minor radiosensitization effect in HPV‐negative cells (Fig. 2 a, Supporting Information Table S1B and S1C). Furthermore, the synergistic effects between RT and PARP inhibition were lower in the majority of HPV‐negative cells compared to HPV‐positive cells (Supporting Information Table S1D). Although ABT‐888 treatment alone resulted in increased levels of γH2AX foci, it did not affect the clonogenic growth of either HPV‐negative or HPV‐positive cells (Figs. 2 b, Supporting Information Fig. S2A). In contrast, the combination of ABT‐888 and RT resulted in higher levels of γH2AX retention and a slight increase in G2/M arrest of SCC154, but not SQD9 cells (Figs. 2 b and 2c). These results suggest that ABT‐888 significantly slowed down DNA repair after RT of HPV‐positive cells, whereas the combination treatment only slightly delayed DNA repair in HPV‐negative cells.
Figure 2.
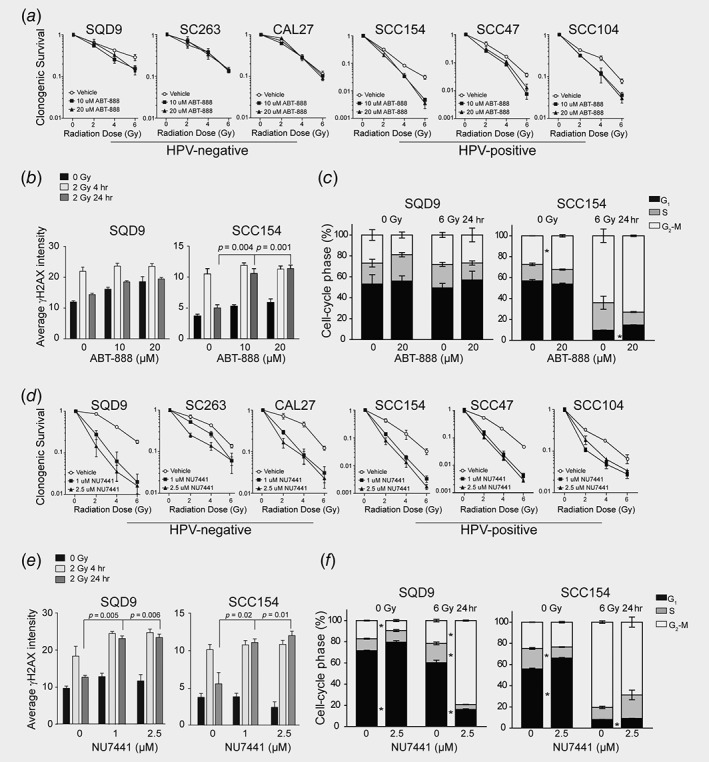
Targeting the BER and NHEJ pathways for radiosensitization of different HNSCC groups. SQD9 and SCC154 cells pretreated with DMSO, ABT‐888 or NU7441 were irradiated with the indicated RT doses. (a, d) Clonogenic cell survival is shown as mean ± SEM. relative to nonirradiated cells, n = 3. p values were calculated by two‐tailed t‐test and are shown in Supporting Information Table S1C. (b, e) Immunofluorescent analysis of γH2AX expression. Data are presented as mean ± SEM, n = 3. *p‐values <0.05 determined by two‐tailed t‐test. (c, f) Cell cycle distribution analysis. Data are presented as mean ± SEM, n = 3. *p‐values <0.05 determined by two‐tailed t‐test.
PARP inhibitors are believed to be especially effective in cancers deficient for HRR.21 In our previous study, we demonstrated that HPV‐positive HNSCC cells exhibited p16 mediated impairment of HRR.16 Therefore, we hypothesized that p16 mediated defects in the HRR could result in increased radiosensitivity of HPV‐positive cells to PARP inhibition. However, p16 suppression did not affect cell survival in response to the combination of RT and ABT‐888 (Supporting Information Fig. S3A), indicating that p16 did not contribute to radiosensitization of HPV‐positive cells to PARP inhibition.
Increased sensitivity to PARP inhibition is also observed in cells with high upregulation of the BER genes that serves as an indicator of their dependence on this pathway.13 The analysis of the TCGA database revealed that several components of the BER pathway were significantly upregulated specifically in HPV‐positive HNSCCs (Supporting Information Fig. S3B). Nonetheless, we did not observe a consistent difference in the expression of the majority of the BER genes between HPV‐positive and HPV‐negative cells. Only LIG1 was significantly upregulated on mRNA level and showed a trend for higher expression on protein level in HPV‐positive HNSCC cells (Supporting Information Fig. S3C and S3D). Higher expression of LIG1 in HPV‐positive HNSCC cells might lead to LIG1 dependency and higher sensitivity to the combination of ABT‐888 and RT. Consistently, we found that suppression of LIG1 expression abolished the radiosensitization effect of PARP inhibition in SCC154 cells (Supporting Information Fig. S3E). Together, our results suggest that higher sensitivity of HPV‐positive cells to the combination of RT and ABT‐888 could be correlated with increased expression of LIG1.
Inhibition of NHEJ radiosensitizes both HPV‐positive and HPV‐negative HNSCCs
The majority of the DSB induced by RT are repaired by NHEJ.19 The short‐term CRISPR‐based screen identified that most of the radiosensitization hits overlapped between HPV‐negative and HPV‐positive HNSCC cells belongs to the NHEJ pathway (Figs. 1 b and 1c). Clonogenic assays confirmed that indels of the NHEJ related genes by CRISPR‐Cas9 resulted in radiosensitization of both SCC154 and SQD9 cells (Supporting Information Fig. S1E).
Among the NHEJ genes, DNA‐PK‐encoded by PRKDC‐ is essential for effective DNA repair by the classical NHEJ. Therefore, we investigated the radiosensitization potential of the DNA‐PK inhibitor NU7441. As a single agent DNA‐PK inhibition has modest effects, but sensitizing effect to DNA damaging agents including RT has been documented in preclinical settings for other cancers.22 Consistently, the DNA‐PK inhibitor NU7441 alone did not show significant effect on clonogenic growth, but potentiated antitumor activity of RT in a synergistic manner in both groups of HNSCC cells (Fig. 2 d, Supporting Information Fig. S2B, Table S1B–S1D). NU7441 treatment induced persistent γH2AX foci 24 hr after RT, indicating DNA‐PK inhibition markedly impaired DNA repair in both groups of HNSCC (Fig. 2 e). The effect of DNA‐PK inhibition on RT response in HPV‐negative cells was also verified by G2/M cell cycle arrest 24 hr after RT (Fig. 2 f). On the other hand, DNA‐PK inhibition did not affect G2/M arrest in HPV‐positive cells as RT alone led to prolonged accumulation of SCC154 cells in the G2/M of the cell cycle (Fig. 2 f). Together, our results verify the crucial role of the NHEJ pathway in RT response of HNSCCs.
Preclinical validation of ABT‐888 and NU7441 as radiosensitizers for HNSCC
We next assessed radiosensitization potential of PARP and DNA‐PK inhibition using SCC154 and SQD9 xenograft models. Of note, HPV‐positive and HPV‐negative xenografts responded differently to RT treatment, which is consistent to previously published studies.9, 14, 23 HPV‐negative SQD9 xenografts showed tumor reduction only 2 weeks after the start of the treatment, whereas HPV‐positive SCC154 xenografts responded to RT immediately after the end of the treatment (Figs. 3 a, 3c, 3e and 3g). The absence of a clear regrowth of HPV‐negative xenografts and different treatment schedules hampers the possibility to confirm the difference in RT response between HPV‐positive and HPV‐negative xenografts side by side. Nonetheless, the fast RT response of HPV‐positive xenografts suggests higher radiosensitivity of these tumors.
Figure 3.
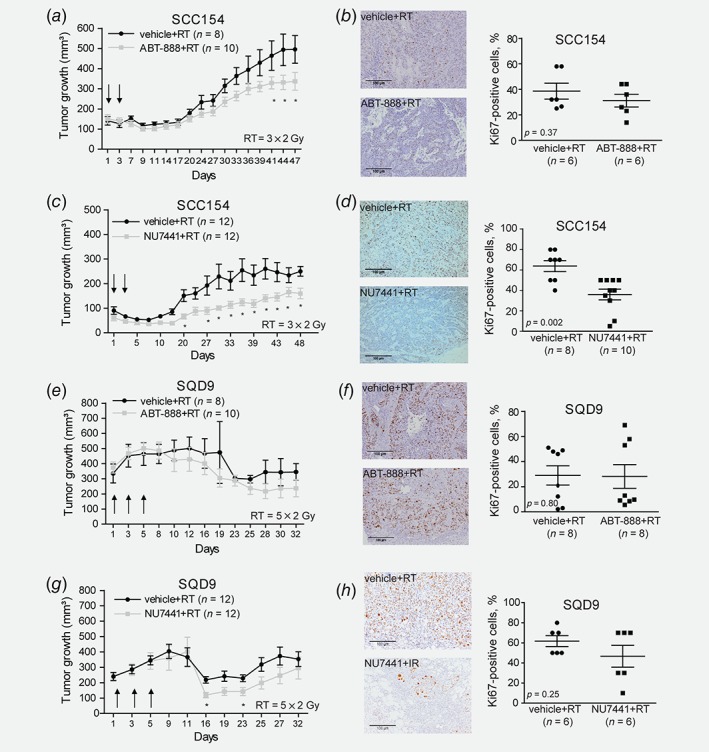
Preclinical validation of ABT‐888 and NU7441 in HPV‐positive and HPV‐negative HNSCC xenografts. (a, e) Tumor volume curves of SCC154 and SQD9 xenografts treated with Vehicle (0.9% NaCl pH 4.0) or ABT‐888 (25 mg/kg) after RT. Day 1 indicates the start of treatment. Data are presented as mean ± SEM. *p‐value <0.05 were determined by ANOVA. (b, f) Ki67 immunostaining of SCC154 and SQD9 xenografts treated with Vehicle (0.9% NaCl pH 4.0) or ABT‐888 (25 mg/kg) after RT. (c, g) Tumor volume curves of SCC154 and SQD9 xenografts treated with Vehicle (40% PEG400 in 0.9% NaCl) or NU7441 (10 mg/kg) after RT. Data are presented as mean ± SEM. *p‐values <0.05 were determined by ANOVA. Day 1 indicates the first day of the treatment. (d, h) Ki67 immunostaining of SCC154 and SQD9 xenografts treated with Vehicle (40% PEG400 in 0.9% NaCl) or NU7441 (10 mg/kg) after RT. *p‐values <0.05 were calculated by two‐tailed t‐test. Scale bar, 100 μm. n = numbers of tumors.
First, we assessed a radiosensitization potential of either ABT‐888 or NU7741 using HPV‐positive SCC154 xenograft model (Figs. 3 a and 3 c). The SCC154 xenografts did not show any reduction in tumor volume in response to either ABT‐888 or NU7741 treatment compared to vehicle‐treated xenografts (Supporting Information Figs. S4A and S4B). Although ABT‐888 reduced the tumor growth of irradiated HPV‐positive SCC154 xenograft from Day 41 till Day 47 (Fig. 3 a), PARP inhibition did not significantly decrease the number of Ki67‐positive cells (Fig. 3 b), suggesting only a mild effect of ABT‐888 in the radiosensitization of the SCC154 xenografts. In contrast, combining DNA‐PK inhibitor with RT reduced tumor volume compared to the RT alone from Day 3 till Day 20 and slowed down tumor regrowth from Day 27 till Day 48 (Fig. 3 c). Histological assessment of the tumor samples also showed that the combination of RT and DNA‐PK inhibition led to decreased number of Ki67‐positive cells (Fig. 3 d), confirming the reduction in tumor volume and growth delay seen in HPV‐positive SCC154 xenograft treated with the combination treatment.
The tumor growth analysis of the HPV‐negative SQD9 xenografts did not show any significant effect of ABT‐888 treatment through the whole course of monitoring the tumors except Day 21, which could be a technical variation due to caliper measurements (Supporting Information Fig. S4C). DNA‐PK inhibition in SQD9 xenograft inhibited the tumor growth from Day 11 till Day 16 (Supporting Information Fig. S4D). For the combination of ABT‐888 and RT, we did not observe an initial tumor volume reduction (Fig. 3 e), whereas the additive effect of NU7441 on RT in the SQD9 xenografts resulted in an initial tumor volume reduction from Day 16 to Day 23 (Fig. 3 g). The minimal regrowth after RT in ABT‐888‐treated mice makes it difficult to make a firm conclusion regarding the effect of ABT‐888 in the SQD9 model. However, the absence of an additive effect of NU7441 on RT in the regrowth period even in the presence of initial response, suggests that it is unlikely that ABT‐888 treatment would result in delayed regrowth of the irradiated SQD9 xenografts (Figs. 3 e and 3 g). In concordance with the tumor volume curves, histological assessment of the SQD9 tumor samples showed that the combination of RT and DNA‐PK inhibition decreased number of Ki67‐positive cells, whereas the combination of RT and PARP inhibition did not have an effect on Ki67 positivity (Figs. 3 f and 3 h).
To validate our findings in more clinically relevant models, we assessed the radiosensitization potential of DNA‐PK inhibition in PDX models from a primary (HNC019) and a recurrent (HNC021) tumor. Both models were HPV‐negative as detected by weak (<50% of the tumor cells) cytoplasmic p16 staining and HPV‐PCR (Supporting Information Figs. S4E and S4F). We found that the combination treatment reduced tumor growth compared to RT alone in both PDX models (Figs. 4 a and 4 d). The analysis of MRI scans confirmed that the combination treatment resulted in lower tumor volumes compared to RT groups, further confirming the radiosensitizing effect of the DNA‐PK inhibitor (Figs. 4 b and 4 e). Histological assessment of the tumor samples revealed that the combination of RT and DNA‐PK inhibition decreased the proliferation of HNSCC cells in both PDX models (Figs. 4 c and 4 f). Altogether, these results verify the radiosensitization effect of DNA‐PK inhibition.
Figure 4.
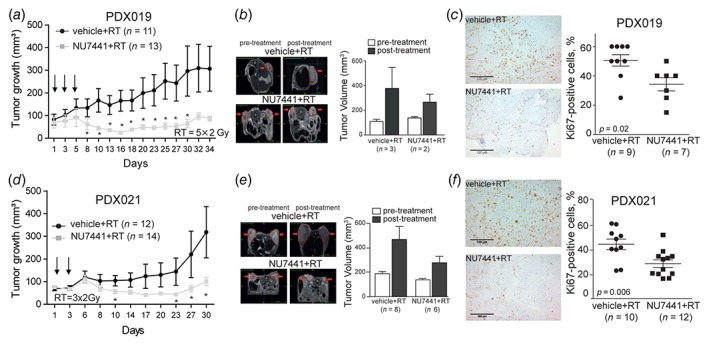
Preclinical validation of NU7441 as radiosensitizers in PDX models derived from primary and recurrent HNSCC patients. (a, d) Tumor volume curves of PDX019 and PDX021 treated with Vehicle (40% PEG400 in 0.9% NaCl) or NU7441 (10 mg/kg) after RT. Data are presented as mean ± SEM. *p‐value <0.05 were determined by Kruskal–Wallis. Day 1 indicates the start of treatment. (b, e) T2‐weighted MR images (left) and volumes of the tumors of PDX019 and PDX021 treated with Vehicle (40% PEG400 in 0.9% NaCl) or NU7441 (10 mg/kg) after RT. Tumors are delineated in red and shown by the arrows. Dataare presented as mean ± SEM. (c, f) Ki67 immunostaining of PDX019 and PDX021 treated with Vehicle (40% PEG400 in 0.9% NaCl) or NU7441 (10 mg/kg) after RT. *p‐values <0.05 were calculated by two‐tailed t‐test. Scale bar, 100 μm. n = numbers of tumors.
Discussion
The DNA repair capacity of irradiated cells provides a prominent mode of resistance and reduction in RT efficiency and can explain high local relapse rates. Targeting DNA repair processes could increase the therapeutic window and improve the treatment response. Here, we performed a CRISPR/CAS9 loss of function screen and identified the contribution of different DDR processes in RT response of HPV‐positive and HPV‐negative HNSCCs. We found that inhibition of the BER related genes improved the RT response predominantly of HPV‐positive HNSCCs, whereas inhibition of the NHEJ genes resulted in radiosensitization of both HPV‐positive and HPV‐negative HNSCC cells.
We assessed the preclinical potential of the BER and NHEJ pathways inhibitors in combination with RT in vitro and in vivo. We targeted the BER pathway with the PARP inhibitor ABT‐888. Although PARP inhibition has shown to enhance RT sensitization in several studies,24, 25, 26, 27, 28 the mechanism of action behind the enhanced sensitivity remains not fully understood. Previous studies reported that low HRR capacity of the HPV‐positive HNSCC could explain the sensitivity to PARP inhibition alone or in combination with RT.29, 30 Others have hypothesized that the reliance of HPV‐positive HNSCC on alternative EJ could provide a mechanistic explanation for the sensitivity to PARP inhibitors.15 In addition, dependency of PARP inhibition alone or in combination with RT is also reported to be independent of HPV‐status,31 but related to defects in HRR,32 impaired or DSB repair33 or increased ROS production.34
Here, we show that the enhanced sensitivity to PARP inhibition could be linked to higher activity of the BER pathway in HPV‐positive HNSCCs. High expression of selected BER and single‐strand break (SSB) related genes in HPV‐positive HNSCC cells was previously reported by Nickson et al.13 In contrast to their report, only LIG1 was significantly upregulated in all our HPV‐positive cell lines. The discrepancy can be explained by limited overlap in the assessed genes between our study and study of Nickson et al. In addition, we cannot exclude the possibility that expression of other BER proteins may be differentially expressed. High expression of LIG1 has also been described in the atypical molecular class of HNSCCs, which is characterized by a strong HPV signature.35 In addition to its role in the BER pathway, LIG1 is a key factor in single‐strand annealing (SSA) pathway.36 As this pathway plays a crucial role in DNA repair of HRR‐deficient tumors, it might explain the sensitivity of HPV‐positive HNSCC to the combination of PARP inhibition and RT. On other hand, LIG1 also plays a crucial role in the DNA replication process.37 Interestingly, Wurster et al.30 showed that PARP1 inhibition radiosensitizes HNSCC cells deficient in HRR by disabling the DNA replication, suggesting that higher sensitivity of HPV‐positive HNSCC to PARP inhibition could also be related to the inhibited DNA replication processes.
PARP inhibitors are currently licensed for high‐grade serous ovarian cancers harboring BRCA mutations, however, there are no clear criteria of how to select patients for PARP treatment for other solid cancers.5 Currently, several clinical studies are recruiting patients‐ including HNSCC patients for the combination of PARP inhibitors and RT without making any preselection. Our in vitro results suggest a difference in response to this combination treatment between HPV‐negative and HPV‐positive HNSCC. In vivo, we also observe a limited, but significant, reduction in tumor volumes of only HPV‐positive HNSCC treated with the combination of ABT‐888 and RT. However, absence of a clear regrowth in the HPV‐negative SQD9 xenografts, differences in treatment schedules and RT response between HPV‐positive and HPV‐negative xenografts, highlight the need for further assessment of the radiosensitizing effect of PARP inhibitors in a larger panel of xenografts from different HNSCC groups. The latter is essential for the establishment of predictive markers for combination therapies.
To target the NHEJ repair, we used the DNA‐PK inhibitor, NU7441. DNA‐PK is a key factor in the NHEJ pathway and several clinical studies show that the activity of DNA‐PK is correlated with cancer progression and therapy response. The clinical development of DNA‐PK inhibitors has been slow due to inadequate pharmacokinetic properties of the compounds.22 However, last generation DNA‐PK inhibitors with better pharmacokinetic properties and safety profiles shows promising results in preclinical settings when combined with RT and other DNA damaging agents.38 In line with these results, we found that the DNA‐PK inhibitor potentiates antitumor activity of RT in vitro and in vivo. Interestingly, DNA‐PK inhibition alone resulted in significant reduction of tumor volume only in HPV‐negative SQD9 model. However, the limited effect of DNA‐PK inhibition is previously documented by Zhao et al.39
We also assessed the preclinical potential of the combination treatment using clinically relevant PDX models of HNSCC. It should be noted that because of the limited availability and lower take rates of the HPV‐positive PDX models, we were not able to assess radiosensitization effects in the HPV‐positive PDX models. Nonetheless, we showed the enhancement of RT response by DNA‐PK inhibition in treatment of both naïve and recurrent PDXs. The latter is especially striking since recurrent HNSCC cancer patients are no longer amenable to curative therapy, resulting in a high morbidity and dismal survival.40 Combination of RT with DNA‐PK inhibition could be a feasible option for these patients. Importantly, the tested combination treatments did not result in excessive weight loss and distress of the mice. However, further pharmacological studies are necessary to assess the toxicity of the combination treatment. A number of novel DNA‐PK inhibitors have been recently entered clinical development, and one of them under evaluation in a phase I study in combination with RT in solid tumors (NCT02516813).
In summary, we found that inhibition of different DDR pathways differentially affects RT response in HPV‐negative and HPV‐positive HNSCCs, highlighting the importance of stratifying HNSCC patients for optimal radiosensitization approaches. This is especially important with the dramatic increase in the number of DDR inhibitors entering clinical trials in the last 10 years.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Table S1 (A) overview of DNA damage response related genes targeted by the CRISPR‐Cas9 screen. (B) Dose enhancement factors (DEFs) (C) p‐values calculated from the clonogenic assays (Figs. 2A and 2D) by two‐sided t‐test. (D) The most synergistic area score for the combination of RT and DNA repair inhibitors ABT‐888 (PARP inhibitor) and NU7441 (DNA‐PK inhibitor) assessed with the SynergyFinder (FIMM) using the ZIP method.
Acknowledgements
We thank Stijn Roden and Hilde Geeraerts for their technical assistance, Dr Esther Hauben for her help in the immunohistochemistry analysis, and the TRACE platform for their help with the PDX experiments. The work was supported by Kom op tegen Kanker (ZKD0061‐PS‐001), Fonds Wetenschappelijk Onderzoek (G0833.13N, G0A4116N), Stichting tegen Kanker (FAF‐C/2016/759), Emmanuel Van der Schueren doctoral grant from Kom op tegen Kanker.
Conflict of interest: The authors have declared that no conflict of interest exists.
Contributor Information
Anna Sablina, Email: anna.sablina@kuleuven.vib.be.
Sandra Nuyts, Email: sandra.nuyts@uzleuven.be.
Data availability
Data will be made available upon reasonable request.
References
- 1. Gregoire V, Langendijk JA, Nuyts S. Advances in radiotherapy for head and neck cancer. J Clin Oncol 2015;33:3277–84. [DOI] [PubMed] [Google Scholar]
- 2. Girinsky T, Bernheim A, Lubin R, et al. In vitro parameters and treatment outcome in head and neck cancers treated with surgery and/or radiation: cell characterization and correlations with local control and overall survival. Int J Radiat Oncol Biol Phys 1994;30:789–94. [DOI] [PubMed] [Google Scholar]
- 3. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011;11:239–53. [DOI] [PubMed] [Google Scholar]
- 4. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- 5. Pilie PG, Tang C, Mills GB, et al. State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 2018;16:81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer 2018;18:269–82. [DOI] [PubMed] [Google Scholar]
- 7. Wallace NA, Galloway DA. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol 2014;26:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dok R, Nuyts S. HPV positive head and neck cancers: molecular pathogenesis and evolving treatment strategies. Cancers (Basel) 2016;8:E41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kimple RJ, Smith MA, Blitzer GC, et al. Enhanced radiation sensitivity in HPV‐positive head and neck cancer. Cancer Res 2013;73:4791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rieckmann T, Tribius S, Grob TJ, et al. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother Oncol 2013;107:242–6. [DOI] [PubMed] [Google Scholar]
- 11. Arenz A, Ziemann F, Mayer C, et al. Increased radiosensitivity of HPV‐positive head and neck cancer cell lines due to cell cycle dysregulation and induction of apoptosis. Strahlenther Onkol 2014;190:839–46. [DOI] [PubMed] [Google Scholar]
- 12. Park JW, Nickel KP, Torres AD, et al. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol 2014;113:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nickson CM, Moori P, Carter RJ, et al. Misregulation of DNA damage repair pathways in HPV‐positive head and neck squamous cell carcinoma contributes to cellular radiosensitivity. Oncotarget 2017;8:29963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang L, Zhang P, Molkentine DP, et al. TRIP12 as a mediator of human papillomavirus/p16‐related radiation enhancement effects. Oncogene 2017;36:820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Q, Ma L, Jones T, et al. Subjugation of TGFbeta Signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end joining. Clin Cancer Res 2018;24:6001–14. [DOI] [PubMed] [Google Scholar]
- 16. Dok R, Kalev P, Van Limbergen EJ, et al. p16INK4a impairs homologous recombination‐mediated DNA repair in human papillomavirus‐positive head and neck tumors. Cancer Res 2014;74:1739–51. [DOI] [PubMed] [Google Scholar]
- 17. Knijnenburg TA, Wang L, Zimmermann MT, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep 2018;23:239–54.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pearl LH, Schierz AC, Ward SE, et al. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015;15:166–80. [DOI] [PubMed] [Google Scholar]
- 19. Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008;8:193–204. [DOI] [PubMed] [Google Scholar]
- 20. Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR‐Cas9 essentiality screens in cancer cells. Nat Genet 2017;49:1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what's next after PARP inhibitors? Nat Rev Clin Oncol 2018;15:564–76. [DOI] [PubMed] [Google Scholar]
- 22. Davidson D, Amrein L, Panasci L, et al. Small molecules, inhibitors of DNA‐PK, targeting DNA repair, and beyond. Front Pharmacol 2013;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sorensen BS, Busk M, Horsman MR, et al. Effect of radiation on cell proliferation and tumor hypoxia in HPV‐positive head and neck cancer in vivo models. Anticancer Res 2014;34:6297–304. [PubMed] [Google Scholar]
- 24. Brown JS, O'Carrigan B, Jackson SP, et al. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 2017;7:20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer 2011;105:1114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohn EC, Lee JM, Ivy SP. The HRD decision‐which PARP inhibitor to use for whom and when. Clin Cancer Res 2017;23:7155–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scott CL, Swisher EM, Kaufmann SH. Poly (ADP‐ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol 2015;33:1397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weaver AN, Cooper TS, Rodriguez M, et al. DNA double strand break repair defect and sensitivity to poly ADP‐ribose polymerase (PARP) inhibition in human papillomavirus 16‐positive head and neck squamous cell carcinoma. Oncotarget 2015;6:26995–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wurster S, Hennes F, Parplys AC, et al. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016;7:9732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pirotte EF, Holzhauser S, Owens D, et al. Sensitivity to inhibition of DNA repair by Olaparib in novel oropharyngeal cancer cell lines infected with human papillomavirus. PLoS One 2018;13:e0207934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verhagen CV, de Haan R, Hageman F, et al. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother Oncol 2015;116:358–65. [DOI] [PubMed] [Google Scholar]
- 33. Kotter A, Cornils K, Borgmann K, et al. Inhibition of PARP1‐dependent end‐joining contributes to Olaparib‐mediated radiosensitization in tumor cells. Mol Oncol 2014;8:1616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Q, Gheorghiu L, Drumm M, et al. PARP‐1 inhibition with or without ionizing radiation confers reactive oxygen species‐mediated cytotoxicity preferentially to cancer cells with mutant TP53. Oncogene 2018;37:2793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung CH, Parker JS, Karaca G, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004;5:489–500. [DOI] [PubMed] [Google Scholar]
- 36. Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double‐strand break. Trends Cell Biol 2016;26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howes TR, Tomkinson AE. DNA ligase I, the replicative DNA ligase. Subcell Biochem 2012;62:327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu FM, Zhang S, Chen BP. Role of DNA‐dependent protein kinase catalytic subunit in cancer development and treatment. Transl Cancer Res 2012;1:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Y, Thomas HD, Batey MA, et al. Preclinical evaluation of a potent novel DNA‐dependent protein kinase inhibitor NU7441. Cancer Res 2006;66:5354–62. [DOI] [PubMed] [Google Scholar]
- 40. Algazi AP, Grandis JR. Head and neck cancer in 2016: a watershed year for improvements in treatment? Nat Rev Clin Oncol 2017;14:76–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Table S1 (A) overview of DNA damage response related genes targeted by the CRISPR‐Cas9 screen. (B) Dose enhancement factors (DEFs) (C) p‐values calculated from the clonogenic assays (Figs. 2A and 2D) by two‐sided t‐test. (D) The most synergistic area score for the combination of RT and DNA repair inhibitors ABT‐888 (PARP inhibitor) and NU7441 (DNA‐PK inhibitor) assessed with the SynergyFinder (FIMM) using the ZIP method.
Data Availability Statement
Data will be made available upon reasonable request.