

Abstract
Introduction
In this study we characterized disease progression over 48 weeks among boys receiving deflazacort vs prednisone/prednisolone placebo arm treatment in two recent Duchenne muscular dystrophy (DMD) clinical trials.
Methods
Ambulatory boys with DMD receiving placebo in the phase 3 ataluren (N = 115) and tadalafil (N = 116) trials were included. The trials required at least 6 months of prior corticosteroid use and stable baseline dosing. Associations between corticosteroid use and 48‐week changes in ambulatory function were estimated using mixed models. Adjusted differences between corticosteroid groups were pooled in a meta‐analysis.
Results
In the meta‐analysis, deflazacort‐treated patients vs prednisone/prednisolone‐treated patients experienced, on average, lower declines of 28.3 meters on 6‐minute walk distance (95% confidence interval [CI], 5.7, 50.9; 2.9 seconds on rise from supine [95% CI, 0.9, 4.9 seconds]; 2.3 seconds on 4‐stair climb [95% CI, 0.5, 4.1 seconds]; and 2.9 [95% CI, 0.1, 5.8] points on the North Star Ambulatory Assessment linearized score).
Discussion
Deflazacort‐treated patients experienced significantly lower functional decline over 48 weeks.
Keywords: ambulatory function, deflazacort, Duchenne muscular dystrophy, meta‐analysis, prednisone/prednisolone
Abbreviations
- 6MWD
6‐minute walk distance
- ACT DMD
Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy
- CINRG
Cooperative International Neuromuscular Research Group
- DMD
Duchenne muscular dystrophy
- FOR DMD
Finding the Optimum Regimen for Duchenne Muscular Dystrophy
- MCID
minimal clinically important difference
- MMRM
mixed model with repeated measures
- NSAA
North Star Ambulatory Assessment
- PBO
placebo
- SES
socioeconomic status
1. INTRODUCTION
Approximately 1 in every 5000 live male births1 is affected by Duchenne muscular dystrophy (DMD), an inherited, X‐linked disease caused by mutations to the gene encoding dystrophin.2
The corticosteroids deflazacort and prednisone/prednisolone are standards of care for the treatment of DMD.3 Both drugs appear to improve muscle strength and slow disease progression,4, 5, 6, 7 and their increased use in early patient management is credited with changing the natural history of the disease.6, 8 Although prednisone/prednisolone is currently used off‐label to treat DMD, deflazacort was recently approved by the United States Food and Drug Administration for use in patients with DMD who are 5 years or older.
As only a few studies have involved both deflazacort and prednisone/prednisolone‐treated patients, there has thus far been limited investigations of their comparative efficacy on functional outcomes, particularly in the context of modern supportive care, physical therapy, and rigorous clinical trial evaluation. One recent study in the Cooperative International Neuromuscular Research Group (CINRG) natural history cohort9 reported a lower rate of ambulation loss among deflazacort‐treated patients in comparison to prednisone/prednisolone‐treated patients.6 However, analyses of placebo arm data from recently completed DMD clinical trials can provide additional information about the comparative efficacy of these drugs, as patients in trial placebo arms are required to be on stable corticosteroid dosing regimens at trial entry, and allowed to continue using stable doses of corticosteroids for the duration of the trial. In addition, trial patients receive a broad range of functional assessments, allowing for a more comprehensive investigation of the impacts of corticosteroids on different markers of disease progression.
In this study we compare disease progression rates between patients receiving these corticosteroids in the placebo arms of two recently concluded and independent phase 3 trials: the tadalafil DMD trial10 and the Ataluren Confirmatory Trial in DMD (ACT DMD).11 The aim of our study was to obtain a pooled estimate of the difference in progression rates, over a 48‐week period, between patients who were receiving deflazacort vs prednisone/prednisolone.
2. METHODS
2.1. Data sources
Individual patient data were used from placebo arm patients in a phase 3 trial of tadalafil for the treatment of DMD (https://clinicaltrials.gov/show/NCT01865084; data provided to the Collaborative Trajectory Analysis Project by Eli Lilly and Company).10 Published summary data were also obtained from placebo arm patients in a phase 3 trial of ataluren (https://clinicaltrials.gov/show/NCT01826487; ACT DMD).11 The trial data used in this study was received de‐identified from Eli Lilly and thus no institutional review board approval was required for our investigation.
The tadalafil DMD trial was a randomized, double‐blind, placebo‐controlled, phase 3 trial of tadalafil conducted at 63 sites in 15 countries, which enrolled ambulatory males with DMD aged 7 to 14 years who had baseline 6‐minute walk distance (6MWD) between 200 and 400 meters.10 Patients were randomly assigned to receive placebo, low‐dose tadalafil (0.3 mg/kg), or high‐dose tadalafil (0.6 mg/kg) daily for 48 weeks. A total of 116 patients were randomized to placebo and included in the present analysis.
ACT DMD was a randomized, double‐blind, placebo‐controlled, phase 3 trial of ataluren conducted at 54 sites in 18 countries, which enrolled ambulatory males aged 7 to 16 years with nonsense mutation DMD who had baseline 6MWD of at least 150 meters.11 Patients were randomly assigned to receive placebo or ataluren (40 mg/kg/d) for 48 weeks. A total of 115 patients were randomized to placebo and included in the present analysis.
In both trials, inclusion criteria required participants to have systemic deflazacort or prednisone/prednisolone use for at least 6 months before trial recruitment, with stable corticosteroid dosing for at least 3 months before the start of treatment. Patients continued their corticosteroid use during the trial period with stable dosing.
2.2. Functional outcomes
Functional assessments were conducted at baseline and every 12 weeks in the tadalafil DMD trial, and every 8 weeks in ACT DMD. Outcomes assessed in both trials included 6MWD,12 timed functional tests (supine to stand, 4‐stair climb, and 10‐meter walk/run),9 and the North Star Ambulatory Assessment (NSAA), a 17‐item functional scale specifically designed for ambulant boys with DMD.13, 14 For NSAA, the NSAA total score (range, 0–34) and the NSAA linearized score, a transformed version of the NSAA total score (range, 0–100), based on a Rasch analysis,15 were both calculated.
As some patients were not able to complete functional assessments at trial visits due to disease progression, imputations of 6MWD values and timed function test completion times were carried out in certain situations. Specifically, patients who became unable to perform a valid 6MWD from permanent loss of ambulation due to disease progression were assigned a 6MWD of zero at all visits after the date when such an event was reported. For ambulatory subjects who did not perform a valid test, the 6MWD was recorded as missing. Similarly, for patients who became unable to perform a timed function test due to disease progression, a value of 30 seconds was assigned for each visit at which the subject was no longer able to perform the test. For subjects who did not perform a timed function test for other reasons, such as a bone fracture, the relevant tests were considered as missing.
The NSAA total score in both trials was calculated as the sum of scores on the 17 test items making up the NSAA. For this analysis, if fewer than 13 of the 17 test items were performed for any patient, the total score for that patient was considered as missing. If between 13 and 16 test items were performed, the total score was calculated by rescaling the sum of the scores to reflect 17 test items, assuming the same average scores for the missing tests (ie, multiplying the sum of the scores in the number of test items performed by 17 / number of test items performed).11 The NSAA linearized score was computed in each trial based on a published scale transforming NSAA raw scores to NSAA linearized scores.13 No imputations were made for patients with missing values for NSAA linearized scores.
Handling of the missing data was consistent between deflazacort‐ and prednisone/prednisolone‐treated patients within each trial. In ACT DMD, multiple imputation was used to handle missing outcomes data.16 In the tadalafil DMD trial, a complete case analysis was used. All included patients had at least one follow‐up visit with nonmissing data for each outcome. At the visit level, the proportion of visits with missing data was less than 4% for all outcomes, except for the 10‐meter walk/run, which was missing in 14% of visits. Proportions missing were similar between steroid groups.
2.3. Statistical analysis
2.3.1. Individual patient data from Tadalafil DMD trial placebo arm
First, individual patient data from the tadalafil DMD trial placebo arm were analyzed to estimate disease progression rates in this population. Patients were classified as either deflazacort or prednisone/prednisolone users based on their corticosteroid use at trial enrollment. A mixed model with repeated‐measures (MMRM) analysis parallel to that used in a recent analysis15 of the ACT DMD placebo arm was used to model change in functional measures in the tadalafil DMD trial placebo arm. Specifically, the mean of the change from baseline for each functional measure at trial visits over time was modeled as a function of visit week (12, 24, 36, and 48 weeks), corticosteroid group (deflazacort vs prednisone/prednisolone), duration of prior corticosteroid use (≥6 to <12 months vs ≥12 months), age (<9 years vs ≥9 years), baseline 6MWD category (≥350 meters vs <350 meters), baseline value of the functional measure serving as the outcome, and interactions between visit week and each other characteristic.
The covariance among patients’ repeated measures over the 48‐week period was modeled using an unstructured covariance matrix. This assumes that the variances in the outcome measure may differ among visits, and estimates a unique correlation between every pair of study visits. Based on the fitted model, predicted means (least‐squares means) were obtained, and an estimate of the mean and standard error of the difference in 48‐week change between the deflazacort and prednisone/prednisolone groups was calculated.
2.3.2. Summary data from ACT DMD placebo arm
Disease progression rates among patients in the ACT DMD placebo arm were reported in a recent trial publication.16 Estimates of the 48‐week change in functional outcomes for patients in each corticosteroid group were extracted from this publication. Patients’ baseline characteristics and additional information on analysis specifications were also obtained from the ACT DMD study investigators.
2.3.3. Meta‐analysis of placebo arms
A fixed‐effects meta‐analysis using inverse variance weights was used to obtain combined estimates of disease progression rates from the tadalafil DMD trial and ACT DMD placebo arms. Pooled effect estimates and 95% confidence intervals for the difference in the 48‐week change between the deflazacort and prednisone/prednisolone groups were obtained for each functional outcome. Heterogeneity was assessed using the Cochran Q test and I 2 measure. No adjustment was applied for multiple comparisons.
2.3.4. Sensitivity analysis
To adjust for differences in steroid regimens between the deflazacort and prednisone/prednisolone groups, sensitivity analyses were performed for the analyses of change in the 6MWD. In particular, the MMRM analyses for the 48‐week change in 6MWD in each trial placebo arm were repeated with additional adjustments for the corticosteroid regimen at baseline (categorized as daily use vs non–daily use) and the interaction between corticosteroid regimen and visit week. The results adjusting for corticosteroid regimen in each trial placebo arm were then pooled using the fixed‐effects meta‐analysis approach just detailed.
3. RESULTS
3.1. Baseline characteristics
Baseline characteristics in each trial are summarized in Table 1. Patients receiving deflazacort were, on average, slightly older than those receiving prednisone/prednisolone. In both trial arms, the proportion of patients with at least 12 months of prior corticosteroid use and the proportion of patients receiving a daily regimen were higher among deflazacort than among prednisone/prednisolone‐treated patients. In both trial placebo arms, mean baseline 6MWD was similar between the deflazacort and prednisone/prednisolone groups.
Table 1.
Baseline characteristics in tadalafil DMD trial and ACT DMD placebo arms
| Tadalafil DMD trial placebo arm | ACT DMD placebo arm | |||||
|---|---|---|---|---|---|---|
| Total (N = 116) | Deflazacort (N = 58) | Prednisone/prednisolone (N = 58) | Total (N = 115) | Deflazacort (N = 53) | Prednisone/prednisolone (N = 62) | |
| Age (years) | 9.5 (1.8)* | 9.8 (2.0) | 9.1 (1.4) | 9.0 (1.7) | 9.2 (1.7) | 8.8 (1.6) |
| Corticosteroid duration before baseline, months [n (%)] | ||||||
| 6 to <12 months | 14 (12.1) | 6 (10.3) | 8 (13.8) | 19 (16.5) | 7 (13.2) | 12 (19.4) |
| ≥12 months | 102 (87.9) | 52 (89.7) | 50 (86.2) | 96 (83.5) | 46 (86.8) | 50 (80.6) |
| Daily corticosteroid use [n (%)] | 85 (73.3)* | 50 (86.2) | 35 (60.3) | 84 (73.0)* | 45 (84.9) | 39 (62.9) |
| 6MWD (meters) | 335.0 (49.6) | 343.5 (45.1) | 326.5 (52.8) | 362.7 (81.4) | 361.3 (87.7) | 363.9 (76.4) |
| NSAA total score | 20.2 (7.1) | 20.4 (6.7) | 20.0 (7.4) | 21.7 (8.1) | 20.7 (7.1) | 23.0 (9.0) |
| NSAA linearized score | 55.9 (14.8) | 56.0 (13.6) | 55.7 (16.1) | 60.0 (18.4) | 57.2 (15.4) | 63.3 (21.4) |
| Rise from supine (seconds) | 13.3 (10.0) | 13.3 (10.4) | 13.2 (9.7) | 9.8 (8.0) | 8.7 (7.7) | 10.7 (8.2) |
| 10‐meter walk/run (seconds) | 6.6 (1.9) | 6.6 (1.9) | 6.7 (2.0) | 6.8 (2.9) | 6.6 (3.2) | 7.1 (2.6) |
| 4‐stair climb (seconds) | 7.2 (5.8) | 7.1 (5.7) | 7.3 (6.0) | 6.5 (5.7) | 6.4 (6.9) | 6.5 (4.4) |
Note: Characteristics are presented as mean (standard deviation), unless otherwise specified.
Abbreviations: 6MWD, 6‐minute walk distance; ACT DMD, Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy; DMD, Duchenne muscular dystrophy; NSAA, North Star Ambulatory Assessment.
P < .05 for difference between deflazacort and prednisone/prednisolone groups.
Among patients on a daily dosing regimen, in the tadalafil DMD trial placebo arm, the mean dose among prednisone/prednisolone‐treated patients was 0.579 mg/kg/d (77.2% of the recommended dose) vs 0.655 mg/kg/d (72.8% of the recommended dose) among deflazacort‐treated patients. In the ACT DMD placebo, mean doses were 0.515 mg/kg/d (68.7% of the recommended dose) and 0.695 mg/kg/d (77.2% of the recommended dose) among prednisone/prednisolone‐ and deflazacort‐treated patients, respectively.16 Both trials required a stable dose of steroids over the 6 months prior to enrollment, but did not impose any requirements on frequency or dose.
3.2. Disease progression outcomes in each trial
For 6MWD outcomes at week 48, average declines in deflazacort‐treated patients were approximately 25 meters less in the tadalafil DMD trial placebo arm, and 32 meters less in the ACT DMD placebo arm compared with prednisone/prednisolone‐treated patients (Table 2). For timed function tests, compared with the prednisone/prednisolone group, those receiving deflazacort experienced smaller or comparable numerical declines in mean function (Table 2).
Table 2.
The 48‐week change in ambulatory function in deflazacort and prednisone groups in the tadalafil DMD trial and ACT DMD placebo arms
| Deflazacort (95% CI) | Prednisone/prednisolone (95% CI) | Difference* (95% CI) | P value | |
|---|---|---|---|---|
| 6MWD (meters) | ||||
| Tadalafil DMD trial PBO | −43.2 (−75.0, −11.4) | −68.0 (−98.2, −37.7) | 24.8 (−7.9, 57.4) | 0.14 |
| ACT DMD PBO | −39.0 (−68.9, −9.2) | −70.6 (−97.2, −44.0) | 31.6 (0.2, 62.9) | 0.05 |
| 4‐stair climb (seconds) | ||||
| Tadalafil DMD trial PBO | 3.8 (1.2, 6.4) | 5.4 (2.9, 7.8) | −1.6 (−4.2, 1.1) | 0.25 |
| ACT DMD PBO | 3.8 (1.5, 6.0) | 6.7 (4.7, 8.6) | −2.9 (−5.3, −0.5) | 0.02 |
| Rise from supine (seconds) | ||||
| Tadalafil DMD trial PBO | 5.4 (2.3, 8.5) | 8.8 (5.9, 11.6) | −3.4 (−6.5, −0.2) | 0.04 |
| ACT DMD PBO | 4.5 (2.0, 7.0) | 7.1 (4.9, 9.3) | −2.6 (−5.2, 0.0) | 0.05 |
| 10‐meter walk/run (seconds) | ||||
| Tadalafil DMD trial PBO | 3.4 (0.8, 6.0) | 2.8 (0.2, 5.3) | 0.6 (−2.0, 3.3) | 0.65 |
| ACT DMD PBO | 3.2 (1.3, 5.0) | 3.3 (1.6, 4.9) | −0.1 (−2.1, 1.9) | 0.93 |
| NSAA total score | ||||
| Tadalafil DMD trial PBO | −3.4 (−5.2, −1.6) | −4.6 (−6.3, −3.0) | 1.2 (−0.6, 3.0) | 0.19 |
| ACT DMD PBO | −3.4 (−4.8, −2.0) | −4.5 (−5.8, −3.2) | 1.1 (−0.4, 2.6) | 0.14 |
| NSAA linearized score | ||||
| Tadalafil DMD trial PBO | −8.3 (−12.6, −4.0) | −11.9 (−15.8, −7.9) | 3.6 (−0.9, 8.0) | 0.12 |
| ACT DMD PBO | −8.2 (−10.9, −5.4) | −10.6 (−13.2, −8.0) | 2.5 (−1.2, 6.2) | 0.19 |
Abbreviations: 6MWD, 6‐minute walk distance; ACT DMD, Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy; CI, confidence interval; PBO, placebo; NSAA, North Star Ambulatory Assessment.
Difference calculated as deflazacort − prednisone. Tadalafil DMD trial PBO estimates are from analyses of individual patient data. ACT DMD estimates are extracted from published results. For both trials, differences are based on least‐squares means of 48‐week change from baseline obtained from mixed model with repeated‐measures analyses.
3.3. Meta‐analysis of disease progression rates
Pooled estimates from the meta‐analysis gave similar results. In pooled estimates from the meta‐analysis, patients receiving deflazacort experienced significantly lower magnitudes of decline in function as assessed by 6MWD, rise from supine, 4‐stair climb, NSAA total score, and NSAA linearized score over 48 weeks compared with patients receiving prednisone/prednisolone (Table 3 and Figure 1). However, changes in 10‐meter walk/run time did not differ significantly between corticosteroid groups.
Table 3.
Meta‐analysis of difference* in 48‐week change in ambulatory function between deflazacort and prednisone/prednisolone groups
| Pooled estimate | SE | 95% CI | P value | Cochran Q | I 2 (%) | |
|---|---|---|---|---|---|---|
| 6MWD (meters) | 28.3 | 11.5 | (5.7, –50.9) | 0.01 | 0.09 | 0.00 |
| 4‐stair climb (seconds) | −2.3 | 0.9 | (−4.1, −0.5) | 0.01 | 0.54 | 0.00 |
| Rise from supine (seconds) | −2.9 | 1.0 | (−4.9, −0.9) | <0.01 | 0.13 | 0.00 |
| 10‐meter walk/run (seconds) | 0.2 | 0.8 | (−1.4, 1.8) | 0.85 | 0.18 | 0.00 |
| NSAA total score | 1.2 | 0.6 | (−0.01, 2.3) | 0.05 | 0.01 | 0.00 |
| NSAA linearized score | 2.9 | 1.5 | (0.1, 5.8) | 0.04 | 0.14 | 0.00 |
Abbreviations: 6MWD, 6‐minute walk distance; CI, confidence interval; NSAA, North Star Ambulatory Assessment; SE, standard error.
Difference calculated as deflazacort − prednisone.
Figure 1.
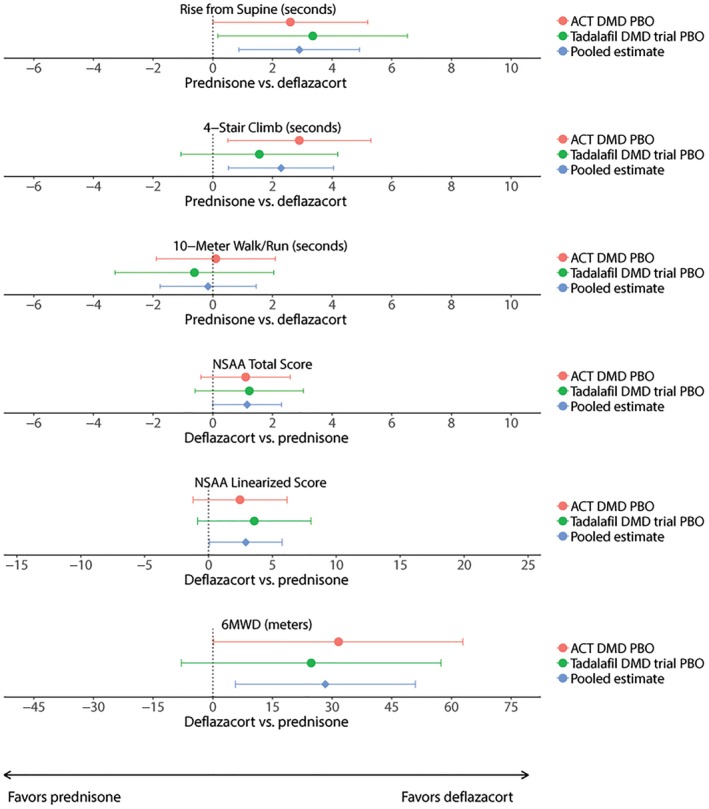
Forest plot shows differences in 48‐week change in ambulatory function between the prednisone and deflazacort groups. Abbreviations: 6MWD, 6‐minute walk distance; ACT DMD, Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy; NSAA, North Star Ambulatory Assessment; PBO, placebo
3.4. Sensitivity analysis
The greater preservation of 6MWD among deflazacort‐treated patients was observed even after adjusting for corticosteroid regimen in both trial placebo arms. After adjusting for regimen, declines in average 6MWD among deflazacort‐treated patients relative to prednisone/prednisolone‐treated patients were of a similar magnitude in the tadalafil DMD trial placebo arm (approximately 28 meters less) and the ACT DMD placebo arm (26 meters less). Pooling these results, the average 6MWD declined approximately 27 meters less among deflazacort‐treated patients than among prednisone/prednisolone‐treated patients (see Table S1 online).
4. DISCUSSION
Patients receiving deflazacort were found to experience significantly slower rates of decline over 48 weeks than those receiving prednisone/prednisolone on multiple measures of ambulatory function. Overall, the associations between steroid group and ambulatory measures were generally consistent in magnitude and direction across the two trials described in this meta‐analysis.
Recent research has sought to characterize the magnitudes of changes in functional measures indicative of meaningful clinical benefit for patients with DMD.13, 17, 18, 19 The pooled estimate of decline in 6MWD of approximately 30 fewer meters observed in deflazacort‐treated patients in these two trials is consistent with, or exceeds, the posited minimally clinically meaningful differences for 6MWD in patients with DMD, which ranges between 26.4 meters and 31.7 meters.18, 19 Similarly, the preservation in deflazacort‐treated patients of approximately 2 to 3 seconds on timed rise from supine and timed 4‐stair climb is generally in line with distribution‐based estimated minimal clinically important differences (MCIDs) for these measures, which are suggested to be around 2.2 seconds for timed 4‐stair climb and 3.6 seconds for timed rise from supine.18 A statistically significant 1 point relative preservation in the raw NSAA score was seen in deflazacort‐treated patients in comparison to prednisone/prednisolone‐treated patients. However, the observed preservation of approximately 3 more points in the linearized NSAA score with deflazacort treatment falls short of the 10‐point change that has been suggested as clinically meaningful for this measure.13 This proposed 10‐point MCID for the linearized NSAA was based on cross‐sectional data comparing patients on daily and intermittent steroid regimens where the differences between groups ranged from 5 points (at age 9 years) to 13.4 points (at age 10 years). However, the 1‐point change on the NSAA total raw score does indicate the relative preservation of one functional activity assessed by the NSAA, which at face value would be meaningful to a patient. Taken together, the magnitude of changes observed on these functional outcomes suggests that patients receiving deflazacort in these studies experienced clinically meaningful levels of slower disease progression on certain functional assessments.
The finding of slower disease progression among patients receiving deflazacort is consistent with the other studies9, 20, 21 that directly compared deflazacort and prednisone/prednisolone treatment in patients with DMD. One randomized trial, completed in 1995, showed that both corticosteroids were effective in preserving muscle strength relative to placebo, over a 12‐week period.20 In the trial, relative to 0.75 mg/kg/d prednisone, 0.9 mg/kg/d deflazacort was associated with improved muscle strength from week 12 to week 52, and numerically greater improvements from baseline to week 52.20 Another trial of 18 patients, completed in 2000, showed no difference in functioning between deflazacort and prednisone patients.21 More recently, a study in the CINRG natural history cohort9 showed both corticosteroids to be effective in preserving function relative to no treatment, but also noted a lower rate of loss of ambulation among deflazacort‐treated patients relative to prednisone/prednisolone.6, 22 Another recent investigation of longer term glucocorticoid use in CINRG also found that deflazacort‐treated patients, in comparison to prednisone/prednisolone‐treated patients, demonstrated an approximately 2‐year delay in median age at loss of ability to stand from supine, a 2.7‐year delay in median age at loss of ambulation, and a 2.7‐year delay in age at loss of hand‐to‐mouth function (defined as transition to a Brooke upper extremity function score of ≥5).6
There are several functional attributes and immunological effects that are the basis for the differential efficacy of deflazacort in comparison to prednisone. In a study of corticosteroid alterations of immune response to dystrophin in patients with DMD, Flanigan et al showed that deflazacort has a greater impact on decreasing interferon‐gamma levels than prednisone.23 This effect on interferon‐gamma leads to multiple immunomodulatory effects, including modulation of JAK–STAT pathways, nuclear factor‐κB levels, cytokine production, T‐cell differentiation, and a shift from a proinflammatory (M1) macrophage phenotype to a phenotype associated with repair and healing (M2, especially M2c).23, 24, 25, 26 Deflazacort also affects multiple gene expression pathways in DMD, including those involved in the activation of satellite cells, myogenesis, regeneration, adipogenesis, muscle growth, and tissue inflammation. Deflazacort upregulates CDH15, C‐MET, DLK1, FGF2, IGF1R, MYF5, MYF6, MYOD, and PAX7 expression toward normal values, and downregulates CD68, MYH8, and TNF‐α toward normal ranges.27
In terms of chemical attributes affecting bioavailability, deflazacort is a prednisolone analog with an oxazoline group at carbon 17 and has reduced lipid solubility vs prednisolone. Deflazacort is 40% protein bound and has no affinity for corticosteroid binding globulin (transcortin) and binds to plasma protein and blood cells instead, crossing the blood–brain barrier in very low concentrations in comparison to prednisone and prednisolone.28 As DMD progresses and the muscle tissue is replaced by fat, more prednisone or prednisolone would be expected to be taken up in the fatty tissues, whereas relatively more deflazacort would be bioavailable to target muscle fibers. This may explain the relative functional preservation in skeletal muscle endpoints with deflazacort. In addition, less deflazacort crosses the blood–brain barrier, which may impart an advantage with regard to behavioral side effects. With the approval and development of additional therapies for DMD, combination therapy with corticosteroids and dystrophin‐producing therapies, such as antisense oligonucleotides (eteplirsen), nonsense mutation read‐through mechanisms (ataluren), and potential other mechanisms, including gene therapies under development, warrants further study, as there is potential for additive benefits from different mechanisms of action.
An important strength of the meta‐analysis presented here is that it is based on data from two large, modern, high‐quality phase 3 clinical trials in DMD, both of which used standardized and validated clinical endpoints to assess functional change over time. In addition, consistent statistical approaches were used to measure average change in functioning over time in both studies.
This study also has some important limitations. As corticosteroid assignment was not randomized, results may be confounded by unobserved baseline differences between the deflazacort and prednisone/prednisolone groups. Nonetheless, adjustments were made for observed differences such as corticosteroid duration, corticosteroid regimen, age, and baseline 6MWD; the smaller declines observed in deflazacort patients in the respective trial populations were robust to these adjustments. As deflazacort was not commercially available in the United States until 2017, it is possible that, over the time frame of these trials, deflazacort use in the United States would have been more prevalent among patients from higher socioeconomic status (SES) families, who would generally be expected to have access to better supportive care. As markers of SES were not available in this study, an adjustment for this potential confounding factor could not be carried out. However, in both trials, patients from the United States made up a relatively small proportion of all placebo arm patients (28% and 32% of all patients in ACT DMD and tadalafil DMD trial placebo arms, respectively), and it appears unlikely that differences in these relatively small subsets of the population would be large enough to explain the overall consistent treatment benefit seen in the population. In general, deflazacort and prednisone/prednisolone use varied by region, and region‐specific differences in patient care may also have contributed to the observed differences in disease progression. Future analyses of pooled individual patient data will be able to investigate possible region‐specific differences, as well as other possible subpopulation differences in treatment patterns and outcomes.
Second, these analyses reflect differences over a 48‐week time horizon in a clinical trial setting; comparative effectiveness over longer periods of use in natural history settings warrant further study. Although recent studies in the CINRG cohort have suggested long‐term benefit of deflazacort on preservation of functional abilities,6, 22 further analyses in other natural history cohorts will be useful to confirm these findings, more precisely quantify long‐term functional benefit, and inform decisionmaking on the trade‐off between functional improvement and the risk of side‐effects associated with each corticosteroid.
As uncertainty still remains about the efficacy of specific corticosteroid regimens, a variety of dosing regimens are used in practice.29 Differences in dosing regimens were present within the corticosteroid groups in our study as well. Daily dosing regimens were more common in the deflazacort group in both studies, perhaps reflecting differences in tolerability between these steroids. In ACT DMD, 84% of deflazacort‐treated patients and 64% of prednisone/prednisolone‐treated patients were on a daily regimen at baseline, compared with 86% of deflazacort‐treated patients and 60% of prednisone/prednisolone‐treated patients in the tadalafil DMD trial. However, declines in 6MWD remained lower among deflazacort‐treated patients even after adjusting for differences in steroid regimen. Absolute daily doses were lower in prednisone/prednisolone‐treated patients, probably because the starting dose for this cohort of patients was lower (0.75 mg/kg/d) compared with the deflazacort group (0.9 mg/kg/d). However, the percentage of the recommended starting dose was actually higher for prednisone/prednisolone than deflazacort in the tadalafil study, but lower for prednisone/prednisolone in the ACT DMD study. Thus, dose reductions due to adverse events would not be a plausible explanation for the consistently lower rates of clinical progression favoring deflazacort across both studies.
No significant heterogeneity was detected across the two trials, supporting the use of a fixed‐effects meta‐analysis to pool the included data. However, with only two trials included, our study may not represent the variability in steroid associations that may be observed among a larger number of trials or data sources.
Last, this meta‐analysis pertains to only efficacy measures reported from the two trials in an ambulatory population. Although there are long‐term reports of upper limb functional preservation in deflazacort‐treated patients relative to prednisone/prednisolone treatment, upper limb and pulmonary endpoints were not assessed in these two trials conducted in ambulatory patients. Finally, the underlying clinical trials were not designed to systematically measure adverse effects of specific corticosteroid regimens.
Ongoing clinical trials, such as the Finding the Optimum Regimen for DMD (FOR DMD) trial (http://clinicatrials.gov identifier: NCT01603407), may provide definitive evidence for the efficacy of specific dosing regimens in the younger 4‐ to 7‐year‐old populations of children with DMD. Although this randomized clinical trial will yield valuable data from previously steroid‐naive patients, it may require longer follow‐up or an older population of patients with DMD, as assessed in the long‐term natural history studies, to compare the relative long‐term efficacy between deflazacort and prednisone, as these younger populations in the FOR DMD trial have less fatty replacement of muscle.
Combining individual patient data from the placebo arms of the multiple recently completed phase 2 and 3 trials in DMD10, 11, 30, 31 may be valuable in facilitating more granular assessments of the comparative efficacy of deflazacort and prednisone/prednisolone dosing regimens currently used in clinical practice. In the interim, the present meta‐analysis of summary data from two recent, high‐quality, phase 3 trial placebo arms complements the evidence supporting differences in functional change associated with steroid type in modern clinical practice.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
CONFLICT OF INTEREST
C.M.M. has served as a consultant for clinical trials for PTC Therapeutics, Inc, and outside the submitted work: BioMarin Pharmaceutical, Sarepta Therapeutics, Eli Lilly and Company, Pfizer, Inc., Santhera Pharmaceuticals, Cardero Therapeutics, Inc, Catabasis Pharmaceuticals, Capricor Therapeutics, Astellas, and FibroGen, Inc; serves on external advisory boards related to Duchenne muscular dystrophy for PTC Therapeutics, Inc., Eli Lilly and Company, Sarepta Therapeutics, Santhera Pharmaceuticals, and Capricor Therapeutics; and reports grants from the US Department of Education/National Institute on Disability and Rehabilitation Research, National Institute on Disability Independent Living and Rehabilitation Research, US National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases, US Department of Defense, and Parent Project Muscular Dystrophy US, during the conduct of the study. P.B.S. has served as an ad‐hoc advisory board member for AveXis, Biogen, Marathon, PTC Therapeutics, Inc, and Sarepta Therapeutics, but he has no financial interests in these companies. He has received research support from Ionis Pharmaceuticals and Biogen for their spinal muscle atrophy (SMA) studies, as well as from Cytokinetics for their SMA clinical trial; Sarepta for their DMD clinical trials; PTC Therapeutics for their DMD/ataluren trial; Pfizer for their DMD/myostatin clinical trial; Bristol‐Myers Squibb/Roche for their DMD/myostatin clinical trial; Sanofi Genzyme for their Pompe clinical trial; and Summit Therapeutics for their DMD clinical trial. B.T.D. has served as an ad‐hoc scientific advisory board member for AveXis, Biogen, Cytokinetics, Marathon Pharmaceuticals, PTC Therapeutics, Bristol‐Myers Squibb, Roche, and Sarepta Therapeutics, but he has no financial interests in these companies. He has received research support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the Slaney Family Fund for SMA, the Spinal Muscular Atrophy Foundation, CureSMA, and Working on Walking Fund; grants from Ionis Pharmaceuticals, Inc, for the ENDEAR, CHERISH, and CS1/CS2/CS10/CS12 studies; grants from Biogen for CS11, as well as from Cytokinetics, Sarepta Pharmaceuticals, PTC Therapeutics, FibroGen, and Summit Therapeutics. G.E., M.S., and S.W.P. are employees and stockholders of PTC Therapeutics. G.S., Z.Y., and J.S. are employees of Analysis Group. E.M. was an employee of Analysis Group, at the time this study was conducted. Analysis Group received funding from PTC Therapeutics for this research.
Supporting information
TABLE S1 The 48‐week change in 6MWD (meters) in deflazacort and prednisone/prednisolone groups in the tadalafil DMD trial and ACT DMD placebo arms, adjusting for corticosteroid regimen
ACKNOWLEDGMENTS
The authors are grateful to the patients for participating in the clinical assessments and for agreeing to make their data available for research. We also thank Eli Lilly and Company for contributing placebo arm data from the tadalafil DMD trial for this study. We are grateful to members of the Collaborative Trajectory Analysis Project (cTAP) for contributions to the interpretation of this research. Ibrahima Dieye and Madeline Jenkins, employees of Analysis Group, Inc, assisted with the development of tables and figures.
This study was conducted in collaboration with cTAP, a pre‐competitive coalition of academic clinicians, drug developers, and patient foundations formed in 2015 to overcome the challenges of high variation in clinical trials in DMD. cTAP has received sponsorship from Astellas (Mitobridge), Biophytis, BioMarin Pharmaceutical, Bristol‐Meyers Squibb, Catabasis Pharmaceuticals, FibroGen, Inc, Italfarmaco SpA, Marathon Pharmaceuticals, Pfizer, Inc, PTC Therapeutics, Roche, Sarepta Therapeutics, Shire, Solid Biosciences, Wave Life Sciences, Summit Therapeutics, plc, Parent Project Muscular Dystrophy, Charley's Fund, and CureDuchenne, a founding patient advocacy partner and provider of initial seed funding to the cTAP.
APPENDIX 1.
1.1.
ACT DMD Study Group consists of: C.M. McDonald, C. Campbell, R.E. Torricelli, R.S. Finkel, K.M. Flanigan, N. Goemans, P. Heydemann, A. Kaminska, J. Kirschner, F. Muntoni, A.N. Osorio, U. Schara, T. Sejersen, P.B. Shieh, H.L. Sweeney, H. Topaloglu, M. Tulinius, J.J. Vilchez, T. Voit, B. Wong, L.N. Alfano, M. Eagle, M.K. James, L. Lowes, A. Mayhew, E.S. Mazzone, L. Nelson, K.J. Rose, H.Z. Abdel‐Hamid, S.D. Apkon, R.J. Barohn, E. Bertini, C. Bloetzer, L.C. de Vaud, R.J. Butterfield, B. Chabrol, J.H. Chae, D.R. Jongno‐Gu, G.P. Comi, B.T. Darras, J. Dastgir, I. Desguerre, R.G. Escobar, E. Finanger, M. Guglieri, I. Hughes, S.T. Iannaccone, K.J. Jones, P. Karachunski, M. Kudr, T. Lotze, J.K. Mah, K. Mathews, Y. Nevo, J. Parsons, Y. Péréon, A.P. de Queiroz Campos Araujo, J.B. Renfroe, M.B.D. de Resende, M. Ryan, K. Selby, G. Tennekoon, and G. Vita.
Tadalafil DMD Study Group consists of: H. Abdel‐Hamid, S. Apkon, R. Barohn, E. Belousova, E. Bertini, J. Brandsema, C. Bruno, W. Burnette, R. Butterfield, B. Byrne, C. Campbell, J. Carlo, J.H. Chae, S. Chandratre, G. Comi, A. Connolly, I. De Groot, N. Deconinck, J. Dooley, A. Dubrovsky, J. Durigneux, E. Finanger, R. Finkel, L.M. Frank, N. Goemans, A. Harper, A. Hattori, O. Herguner, S. Iannaccone, J. Janas, Y.J. Jong, J. Kirschner, H. Komaki, N. Kuntz, W.T. Lee, E. Leung, J. Mah, K. Mathews, C.M. McDonald, E. Mercuri, H. McMillan, W. Mueller‐Felber, A. Lopez de Munain, A. Nakamura, E. Niks, K. Ogata, S. Pascual, E. Pegoraro, Y. Pereon, B. Renfroe, R.B. Sanka, J. Schallner, U. Schara, K. Selby, I.I. Sendra, L. Servais, E. Smith, S. Sparks, H. Topaloglu, R. Victor, J.J. Vilchez, M. Wicklund, E. Wilichoswki, and B. Wong.
McDonald CM, Sajeev G, Yao Z, et al. Deflazacort vs prednisone treatment for Duchenne muscular dystrophy: A meta‐analysis of disease progression rates in recent multicenter clinical trials. Muscle Nerve. 2020;61:26–35. 10.1002/mus.26736
The content of this study was presented as a poster at the International Society for Pharmacoeconomics and Outcomes Research 20th Annual European Congress, the 2018 Clinical Conference of the Muscular Dystrophy Association, the 2019 Annual Meeting of the American Academy of Neurology, and the 2019 Annual Meeting of the Child Neurology Society.
Refer to the Appendix for a complete listing of the ACT DMD Study Group and Tadalafil DMD Study Group participants.
Funding information Collaborative Trajectory Analysis Project (cTAP); PTC Therapeutics, Inc.
REFERENCES
- 1. Mendell JR, Shilling C, Leslie ND, et al. Evidence‐based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304‐313. [DOI] [PubMed] [Google Scholar]
- 2. Emery AE, Muntoni F, Quinlivan RC. Duchenne Muscular Dystrophy. Oxford: Oxford University Press; 2015. [Google Scholar]
- 3. Gloss D, Moxley RT 3rd, Ashwal S, Oskoui M. Practice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2016;86:465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4. Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016;5:CD003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McAdam LC, Mayo AL, Alman BA, Biggar WD. The Canadian experience with long‐term deflazacort treatment in Duchenne muscular dystrophy. Acta Myol. 2012;31:16‐20. [PMC free article] [PubMed] [Google Scholar]
- 6. McDonald CM, Henricson EK, Abresch RT, et al. Long‐term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391:451‐461. [DOI] [PubMed] [Google Scholar]
- 7. Ricotti V, Ridout DA, Scott E, et al. Long‐term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2013;84:698‐705. [DOI] [PubMed] [Google Scholar]
- 8. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDonald CM, Henricson EK, Abresch RT, et al. The Cooperative International Neuromuscular Research Group Duchenne Natural History study—a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve. 2013;48:32‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Victor RG, Sweeney HL, Finkel R, et al. A phase 3 randomized placebo‐controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology. 2017;89:1811‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:1489‐1498. [DOI] [PubMed] [Google Scholar]
- 12. McDonald CM, Henricson EK, Han JJ, et al. The 6‐minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:500‐510. [DOI] [PubMed] [Google Scholar]
- 13. Mayhew AG, Cano SJ, Scott E, et al. Detecting meaningful change using the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55:1046‐1052. [DOI] [PubMed] [Google Scholar]
- 14. Scott E, Eagle M, Mayhew A, et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. 2012;17:101‐109. [DOI] [PubMed] [Google Scholar]
- 15. Narayanan S, Souza M, Shao D, et al. Disease burden and treatment landscape in Duchenne muscular dystrophy (DMD) in the United States. Boston: International Society for Pharmacoeconomics and Outcomes Research; 2017. [Google Scholar]
- 16. Shieh PB, McIntosh J, Jin F, et al. Deflazacort vs prednisone/prednisolone for maintaining motor function and delaying loss of ambulation: a post hoc analysis from the ACT DMD trial. Muscle Nerve. 2018;58:639‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lynn S, Aartsma‐Rus A, Bushby K, et al. Measuring clinical effectiveness of medicinal products for the treatment of Duchenne muscular dystrophy. Neuromuscul Disord. 2015;25:96‐105. [DOI] [PubMed] [Google Scholar]
- 18. McDonald CM, Henricson EK, Abresch RT, et al. The 6‐minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henricson E, Abresch R, Han JJ, et al. The 6‐minute walk test and person‐reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically—meaningful changes over one year. PLoS Curr. 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griggs RC, Miller JP, Greenberg CR, et al. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87:2123‐2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonifati MD, Ruzza G, Bonometto P, et al. A multicenter, double‐blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23:1344‐1347. [DOI] [PubMed] [Google Scholar]
- 22. Bello L, Gordish‐Dressman H, Morgenroth LP, et al. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. 2015;85:1048‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flanigan KM, Campbell K, Viollet L, et al. Anti‐dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24:797‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu T, Zhang L, Joo D, Sun SC. NF‐kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thapa RJ, Basagoudanavar SH, Nogusa S, et al. NF‐kappaB protects cells from gamma interferon‐induced RIP1‐dependent necroptosis. Mol Cell Biol. 2011;31:2934‐2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wesemann DR, Benveniste EN. STAT‐1 alpha and IFN‐gamma as modulators of TNF‐alpha signaling in macrophages: regulation and functional implications of the TNF receptor 1:STAT‐1 alpha complex. J Immunol. 2003;171:5313‐5319. [DOI] [PubMed] [Google Scholar]
- 27. Jensen L, Petersson SJ, Illum NO, et al. Muscular response to the first three months of deflazacort treatment in boys with Duchenne muscular dystrophy. J Musculoskel Neuronal Interact. 2017;17:8‐18. [PMC free article] [PubMed] [Google Scholar]
- 28. Assandri A, Buniva G, Martinelli E, Perazzi A, Zerilli L. Pharmacokinetics and metabolism of deflazacort in the rat, dog, monkey and man. Adv Exp Med Biol. 1984;171:9‐23. [PubMed] [Google Scholar]
- 29. Griggs RC, Herr BE, Reha A, et al. Corticosteroids in Duchenne muscular dystrophy: major variations in practice. Muscle Nerve. 2013;48:27‐31. [DOI] [PubMed] [Google Scholar]
- 30. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50:477‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goemans N, Mercuri E, Belousova E, et al. A randomized placebo‐controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul Disord. 2018;28:4‐15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 The 48‐week change in 6MWD (meters) in deflazacort and prednisone/prednisolone groups in the tadalafil DMD trial and ACT DMD placebo arms, adjusting for corticosteroid regimen